
In this article, two arguments will be provided, based on both clinical data and mathematical insights on the dynamics of the leukemic cell population, to highlight the substantial effect that imatinib also seems to have on the leukemic stem cells (LSCs). The hypothesis proposed here is that imatinib may be able to affect the LSCs in the same way it affects all other leukemic cells, that is, by inhibiting their proliferation and consequently causing them to reach their life span and die out. This is, indeed, the general consensus on the effect that the drug has on leukemic non-stem cells.1, 2, 3, 4 In fact, the different lifespans in the hierarchical structure of the leukemic cell population can explain the seemingly different (short-term) outcomes observed among the various types of leukemic cells under imatinib, rather than a differential effect of the drug on them. The latter has been a typical point of view expressed in the literature on chronic myeloid leukemia (CML), see, for example, Sloma et al.2 I believe this represents an example of the usefulness of combining mathematical modeling with the experimental and clinical data to obtain a deeper understanding of the biological system under consideration and its dynamics.
CML is a cancer of the white blood cells characterized by a chromosomal translocation between chromosomes 9 and 22 known as the ‘Philadelphia chromosome’, which creates the fusion gene BCR–ABL. The resulting protein, a tyrosine kinase, yields growth factor independence, increased proliferation, genetic instability and suppression of apoptosis in leukemic cells.3 Imatinib (Gleevec/Glivec; formerly STI571, Novartis, East Hanover, NJ, USA) is a potent inhibitor of the BCR–ABL tyrosine kinase and today is the standard first-line therapy for patients with newly diagnosed CML in chronic phase. This drug acts by blocking the ATP-binding site of BCR–ABL, thus inhibiting the enzyme activity and its downstream signaling (see Hochhaus et al.3 and the references therein). Imatinib has proven to be very effective in reducing the total leukemic cell burden, as measured by cytogenetic analysis. Data coming from the International Randomized Study of Interferon vs STI571 Trial (IRIS) for patients with newly diagnosed CML in chronic phase show that, under imatinib, the estimated rate of a major cytogenetic response at 18 months is 87.1%, whereas the estimated rate of a complete cytogenetic response at 18 months is 76.2%.5 This large decline in the leukemic burden can also be observed using reverse transcriptase-PCR, a more sensitive methodology that measures the levels of BCR–ABL transcripts. For example, the data in Michor et al.4 show that on average, after about 3 months from the start of the treatment, a 100-fold decline in the leukemic cell population is reached among patients.
There is evidence, however, indicating that imatinib may not be able to completely eradicate the disease, even in patients responding well to the therapy and for whom the vast majority of leukemic cells died in the first months of the treatment. One of the most convincing arguments in favor of this hypothesis is given by the following observation. After prolonged treatment, if imatinib is removed from these patients, the disease will often relapse and will quickly reach levels at or beyond the pretreatment baseline.4, 6 Note that these relapses may be observed in patients where there is no evidence of drug resistance development, the presence of which could otherwise explain the events. Indeed, after the relapse occurs, the patient will typically respond well to the reintroduction of imatinib,6 an impossible event if the CML clone causing the relapse were to be drug resistant. What then is causing these relapses? Clearly, some leukemic burden must still be present, notwithstanding imatinib. In fact, a minimal residual disease remains usually detectable by reverse transcriptase-PCR even after many years of treatment.7 Importantly, it has been shown that this residual disease is associated with the presence of primitive LSCs.1, 2
Evidence, both in vitro and in vivo, seems to indicate that this small sub-population of LSCs is insensitive, that is, unresponsive to imatinib.1, 2, 4 This appears to be a rather common, established point of view in the CML literature.2 A mathematical model,4 which includes among its assumptions the hypothesis that LSCs cannot be depleted, has been occasionally referred to support this hypothesis. To the contrary, it is this author's opinion that, given the data available today, the substantial effect that imatinib also has on the LSCs should be recognized. To support my statement, I will provide the following two arguments, based on both clinical data and mathematical insights on the dynamics of the leukemic cell population.
-
1)
Data coming from the IRIS Study show that generally the disease is kept under control by imatinib if the treatment is not discontinued, and as long as there is no development of drug resistance.3 This observation seems to imply that, under treatment, the residual population of LSCs is not growing in number. If it did grow, then we would also expect to observe the growth of the associated minimal residual disease, on average, among patients. However, the opposite occurs: clinical data indicate that the incidence of patients under imatinib with undetectable BCR–ABL levels (complete molecular response (CMR)) grows, on average, with time.6, 7 Although the relationship between the LSC compartment and the minimal residual disease is not completely understood, there should be a direct connection between the two. That the LSC compartment may have stopped growing during treatment should imply that imatinib has also been able to affect the LSCs, given that the LSC compartment was certainly growing before treatment began, otherwise there would be no disease, and probably going through many symmetric renewals, as we estimated in Tomasetti et al.8
-
2)
A stronger argument is the following. Note that long-lived drug resistance will only be caused by those long-lived cells that have the ability of self-renewal, that is, the stem-like cells. Thus, for the problem of drug resistance, we may focus only on them.8 Assume now that, contrary to intuition, the dynamics of the minimal residual disease, as measured by cytogenetic and molecular analyses, does not reflect at all the dynamics of the LSC compartment and that the LSCs are still dividing and growing in number even under imatinib. Then we should be able to observe cases of patients relapsing at any given time during treatment, because of the development of drug resistance due to the random point mutation mechanism, which is always acting on each cell division. Actually, if we assume a growing LSC population, then these occurrences should even increase with time. Now, it has been estimated that it takes approximately 5–7 years for CML to reach detection size.4 Thus, if random point mutations causing drug resistance could happen during treatment, due to the continuing divisions of LSCs, then we should see various cases of patients having relapses well after 5 years from the start of the therapy, and even increasingly so, until when possibly all patients under study have relapsed, as can be calculated using the formula in Tomasetti et al.8 However, this is again in stark contrast with the available data.3 As we can see in Figure 1, relapses due to drug resistance seem to stop occurring ∼5 years into the treatment. Note how the ‘curve’ for the various events (which include relapses) peaks at around 2 years into treatment and then approaches zero at 4–5 years. Importantly, only a relatively small fraction of patients did have a relapse due to drug resistance (less than 15%).3 This curve is far from the shape we would observe for the case where the LSCs were able to proliferate during treatment, a scenario that is depicted in Figure 2, where the expected relapses—using the formula in Tomasetti et al.8 with realistic parameters’ values—are plotted against the actual clinical data. Note that the bad fit of the estimated relapses to the actual data, for the first years, is due to the fact that, for simplicity, the estimate used did not include the variability, among patients, given by the time of disease detection, initial tumor load and its growth rate. Independently of this limitation, the essential difference between the hypothesis of drug-insensitive LSCs and the data is clearly captured in the long -term outcome. Thus, if the LSC compartment is not growing under imatinib, could it be that the number of LSCs stabilizes after the treatment starts (for example, due to the inability of imatinib to deplete the LSCs or due to a saturation level) rather than contracting? Using the previously mentioned formula,8 it can be shown that for the case where the populations were not contracting, we would have a small percentage of the patients under treatment relapsing every year during the study (unless the LSCs were to be eternal). The fact that this is also contrary to the evidence provided by the available clinical data (Hochhaus et al.3 and later reports from the IRIS Study), indicates that, overall, the LSCs should not be dividing under imatinib, but rather should slowly die out, otherwise drug resistance would still be able to develop.
Figure 1 Figure 2 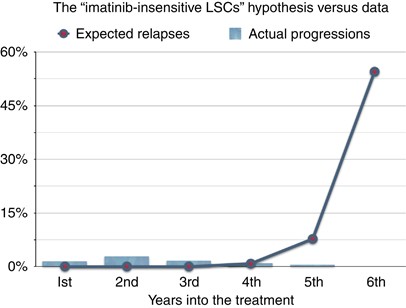
Data from the IRIS Study (bars) versus estimated annual rates of relapses due to drug resistance (line with dots).


To summarize, the above two arguments seem to indicate that, overall, the LSC compartment is not growing under treatment, because imatinib has been able to affect these cells by blocking their proliferation. Actually, they also provide some evidence for an LSC compartment that is slowly shrinking in size under imatinib. If imatinib were able to block the LSCs from proliferating, these cells should not be able to do anything but reach their lifespan and die, causing the LSC compartment to slowly die out. This seems to be supported by the clinical data, if there is any relationship between the surviving LSC population at a given point in time and the amount of Philadelphia-positive cells and BCR–ABL levels found at that time. In fact, the data clearly indicate that the percentage of patients under imatinib who are in complete cytogenetic response and/or CMR is generally increasing over time.7 Furthermore, relapses due to drug resistance stop occurring, a reflection of the fact that the LSC population is not proliferating anymore but rather shrinking over time. Importantly, the potential ability of imatinib to slowly induce the elimination even of the LSC population is also suggested by recent data demonstrating that a certain percentage of patients, who achieve and keep CMR, have been able to discontinue imatinib without relapsing.6
The hypothesis proposed here is, therefore, that imatinib may be able to affect the LSCs in the same way, at least qualitatively, it affects all other leukemic cells. Indeed, the ability of imatinib to inhibit proliferation of the leukemic cells consequently causing them to slowly die out is in accordance with the general consensus in the medical literature regarding the effect that the drug has on leukemic non-stem cells.1, 2, 3, 4 Thus, this hypothesis simply extends the effects of imatinib also to include the LSC compartment. After all, it has not been biologically explained why a targeted drug, which is able to bind to the ATP-binding site of BCR–ABL in a leukemic cell, would not be able to have the same effect on its mother stem cell. This fact also explains why point mutations in the BCR–ABL domain can cause resistance to imatinib. If imatinib were not able to bind to the ATP-binding site of BCR–ABL in a LSC, we would not have a distinction between drug-resistant LSCs and not-resistant LSCs, and, thus, there would not exist a problem of long-term drug resistance caused by random genetic mutations.
Note that this hypothesis does not contradict the experimental evidence indicating the apparent inability of imatinib to eliminate the LSCs within a certain time frame.
Indeed, the observed decline of the leukemic cells is due to imatinib blocking their proliferation rather than killing them: the leukemic cells can then only reach their lifespan and die. Given that all blood cells are relatively short-lived (days to several months, depending on the type of cell), with the exception of the stem cells, all such cells will die out relatively quickly, leaving only the long-lived LSCs as a residue. Therefore, the different lifespans of the various types of leukemic cells can potentially explain what leads to different (short-term) outcomes in the experimental observations, rather than assuming a differential effect of the drug on the various types of leukemic cells. In fact, by simple calculations, we can grossly estimate that on average, among patients, the LSCs are able to survive for at least 10 years. This large difference in the lifespan among the cells of the blood hierarchical system is sufficient to explain the observed experimental and clinical data. We leave to a future work a more precise estimation, which would have the potential to indicate the average time needed to eradicate the disease using imatinib. In mathematical terms, the average dynamics of the LSC compartment before treatment could be described by the equation S′(t)=(L0(1−a−2b)−D0)S(t) (see Tomasetti et al.8 for a detailed explanation), the next compartment (say progenitors) by N ′1(t)=(L0(a+2b))S(t)−(L1+D1) N1(t), whereas all other compartments by equations of the form N ′i(t)=2Li−1 Ni−1(t)−(Li+Di) Ni(t), i=2,…,n, where n+1 is the total number of compartments in the hematopoietic system. The effect of imatinib would then be to reduce to very low values (possibly zero) the division rates Li of all types of cells. There is also the possibility that imatinib could even increase the death rates Di of the leukemic cells (for example, by attenuating the apoptosis inhibition caused by BCR–ABL), but clear evidence is lacking.
In conclusion, imatinib seems to affect the LSCs in the same way it affects all other leukemic cells; however, the different lifespans of the various types of leukemic cells lead to different (short-term) outcomes in the experimental and clinical observations. This hypothesis, which has been formulated on the basis of experimental evidence, clinical data and mathematical insights on the dynamics of CML, seems to explain the current clinical data on CML better than previous assumptions, which appear to contradict at least some of the evidence available today on CML. The proposed hypothesis is indeed able to both elucidate the reasons behind the quick contraction in the leukemic burden occurring after starting the treatment, due to cells with relatively short lifespan whose proliferation has been inhibited, as well as to explain why the disease is not completely eliminated typically at least for some years. This is due to the surviving LSCs, which are not killed but rather are very slowly dying out under the inhibitory effect of imatinib. It would also explain why there is no clear evidence of relapses due to drug resistance caused by random point mutations originated during treatment. Furthermore, if the treatment is discontinued before the LSC sub-population is completely eradicated, then a relapse will be observed, caused by the surviving LSCs resuming their cycling activity, while otherwise a potentially complete cure would be the expected outcome.6
References
Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002; 99: 319–325.
Sloma I, Jiang X, Eaves AC, Eaves CJ . Insights into the stem cells of chronic myeloid leukemia. Leukemia 2010; 24: 1823–1833.
Hochhaus A, O’Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009; 23: 1054–1061.
Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL et al. Dynamics of chronic myeloid leukaemia. Nature 2005; 11: 1029–1035.
O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F et al. Imatinib compared with Interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348: 994–1004.
Mahon F-X, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010; 24: 1823–1833.
Kantarjian H, O’Brien S, Shan J, Huang X, Garcia-Manero G, Faderl S et al. Cytogenetic and molecular responses and outcome in chronic myelogenous leukemia. Cancer 2008; 112: 837–845.
Tomasetti C, Levy D . Role of symmetric and asymmetric division of stem cells in developing drug resistance. Proc Natl Acad Sci USA 2010; 107: 16766–16771.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tomasetti, C. A new hypothesis: imatinib affects leukemic stem cells in the same way it affects all other leukemic cells. Blood Cancer Journal 1, e19 (2011). https://doi.org/10.1038/bcj.2011.17
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2011.17
