
Abstract
Background:
Novel treatment strategies in Ewing sarcoma include targeted cellular therapies. Preclinical in vivo models are needed that reflect their activity against systemic (micro)metastatic disease.
Methods:
Whole-body magnetic resonance imaging (WB-MRI) was used to monitor the engraftment and dissemination of human Ewing sarcoma xenografts in mice. In this model, we evaluated the therapeutic efficacy of T cells redirected against the Ewing sarcoma-associated antigen GD2 by chimeric receptor engineering.
Results:
Of 18 mice receiving intravenous injections of VH-64 Ewing sarcoma cells, all developed disseminated tumour growth detectable by WB-MRI. All mice had lung tumours, and the majority had additional manifestations in the bone, soft tissues, and/or kidney. Sequential scans revealed in vivo growth of tumours. Diffusion-weighted whole-body imaging with background signal suppression effectively visualised Ewing sarcoma growth in extrapulmonary sites. Animals receiving GD2-targeted T-cell therapy had lower numbers of pulmonary tumours than controls, and the median volume of soft tissue tumours at first detection was lower, with a tumour growth delay over time.
Conclusion:
Magnetic resonance imaging reliably visualises disseminated Ewing sarcoma growth in mice. GD2-retargeted T cells can noticeably delay tumour growth and reduce pulmonary Ewing sarcoma manifestations in this aggressive disease model.
Similar content being viewed by others


Main
Ewing sarcoma is a cancer of bone and soft tissue that is characterised by a molecular rearrangement of the EWS gene on chromosome 22 with an ets-type gene (Delattre et al, 1992). Survival of patients with localised disease has improved substantially by modern multimodal treatment regimens (Paulussen et al, 1998), but primary multifocal disease or disseminated relapse often remains incurable (Ladenstein et al, 2010). Novel treatment strategies aim to eliminate residual microscopic disease remaining after standard therapy and include molecularly targeted drugs and antibodies (Erkizan et al, 2009; Juergens et al, 2011) as well as immunological therapies (Mackall et al, 2008; Kailayangiri et al, 2012). One approach is based on the genetic engineering of T cells with recombinant chimeric antigen receptors (CARs) directed against the ganglioside antigen GD2 that is overexpressed on many Ewing sarcomas (Kailayangiri et al, 2012). GD2-specific CARs consist of the antigen-binding domain of a GD2-specific antibody linked to T-cell receptor-signalling domains and mediate efficient cytolytic T-cell interaction with GD2-expressing tumour cells both in vitro and in vivo (Rossig et al, 2001; Pule et al, 2008).
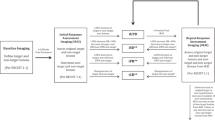
In vivo preclinical data are often generated in localised tumour models, although the capacity of novel therapies to prevent the outgrowth of subcutaneous tumours in mice inadequately reflects their activity against systemic (micro)metastatic disease. To study the biology of systemic disease and to develop novel therapies including translational validation, preclinical in vivo models are needed that recapitulate the biology of multifocal disease and tumour dissemination. In accordance with the known haematological spread pattern of Ewing sarcoma, intravenous transplantation of single-cell suspensions of Ewing sarcoma cells into highly immunodeficient mice was found to efficiently establish multifocal sarcomas in many of the animals (Vormoor et al, 2001). The use of systemic disease models as preclinical tools for monitoring the antitumour activity of novel therapies requires visualisation of tumour growth in the living animal. Here, we used whole-body (WB) imaging techniques, including both standard magnetic resonance imaging (MRI) and diffusion-weighted WB imaging with background signal suppression (DWIBS) to follow-up the engraftment and systemic spread of human Ewing sarcoma xenografts in mice and to address the therapeutic efficacy of adoptive T-cell transfer against disseminated tumour cells in vivo.
Materials and methods
Cell lines
The Ewing sarcoma cell line VH-64 was a gift from Frans van Valen at the Institute of Experimental Musculoskeletal Medicine of University of Muenster, Germany. It was originally established from a malignant pleural effusion of a patient with Ewing sarcoma of the metatarsal bone. The identity of the cell line throughout the experiments was confirmed by repeated short tandem repeat profiling. VH-64 cells were cultured in collagen-coated 25 cm2 tissue culture flasks in RPMI 1640 medium (Invitrogen, Darmstadt, Germany), supplemented with 10% heat-inactivated foetal calf serum (Thermo Fisher, Bonn, Germany) and 2 mM L-glutamine and maintained at 37 °C and 5% CO2. The retroviral packaging cell lines Phoenix-ampho and FLYRD 18 were provided by Gary P Nolan (Stanford University School of Medicine, Stanford, CA, USA) and E Vanin (Center for Cell and Gene Therapy, Houston, TX, USA), respectively.
Xenogeneic NOD/scid mouse model of Ewing sarcoma
Mouse experiments were approved by the animal care committee of the local government (Bezirksregierung Muenster, Az. 87-51.04.2010.A117). NOD/scid mice (8–10-week old; Charles River Laboratories, Sulzfeld, Germany) were irradiated with a single dose of 3.5 Gy from a linear accelerator 1 day before transplantation. We chose VH-64 cells for this study, as the engraftment, dissemination, and in vivo proliferation of this cell line is highly representative of Ewing sarcoma xenografts (Vormoor et al, 2001). Single-cell suspensions of 2 × 106 tumour cells in 0.2 ml of medium were injected into the tail veins. All experimental manipulations were performed under sterile conditions in a laminar flow chamber. Animals developing clinical signs of disease (weight loss, shaggy fur, bent spine) were killed.
Whole-body MRI of mice
High resolution MR data sets were acquired on a clinical 3 Tesla WB MR system (Achieva, Philips Medical Systems, Best, the Netherlands) equipped with standard gradient coils (gradient strength 40 mT m−1, slew rate 200 mT m−1 ms−1). A dedicated small animal solenoid receiver coil (Philips Research, Hamburg, Germany) with an inner diameter of 40 mm and an integrated heating system to regulate the body temperature of the animals was used for signal reception. The MRI protocol consisted of the following sequences: T2 TSE sagittal (TR 1732 ms, TE 60 ms, FOV 70 (tail to head) × 30 (anterior-posterior) mm, reconstruction matrix 560 × 240, number of signal averages 4, slice thickness 1 mm, number of slices 27, scan time 3 : 34.8 min), T2 TSE axial (TR 2854 ms, TE 60 ms, FOV 32 (right to left) × 32 (anterior-posterior) mm, reconstruction matrix 288 × 288, NSA 4, slice thickness 1.5 mm, number of slices 45, scan time 05 : 53.9 min), STIR TSE sagittal (TR/TI 2445/200 ms, TE 68 ms, FOV 70 (tail to head) × 29 (anterior-posterior) mm, reconstruction matrix 512 × 214, NSA 14, slice thickness 1 mm, number of slices 25, scan time 07 : 59.3 min), DWIBS axial (TR/TI 16470/240 ms, TE 87 ms, FOV 50 (tail to head) × 50 (anterior-posterior) mm, reconstruction matrix 256 × 256, multi-shot EPI, EPI factor 9, number of b-factors 2 (0;1000), NSA 1, slice thickness 0.8 mm, gap 0.2 mm, number of slices 87, scan time 08 : 30.6 min; Takahara et al, 2004). Zero-filling was used to calculate the reconstruction matrix. Apparent diffusion coefficient maps were calculated from the native diffusion images with the built-in software tools of the MRI scanner. All sequences were acquired with the same geometry to assure comparability of the various sequences. First, MRIs were performed between 20 and 24 days after tumour cell transplantation independent of the development of disease symptoms. Up to four follow-up MRIs were performed to monitor tumour growth. Time points (TPs) were designated as follows: TP0, tumour cell transplantation; TP1, initial scan, median 20 days (range 20–24) after TP0; TP2, median 30 days (range 27–31) after initial scan; TP3, median 34 days (range 34–39) after initial scan; TP4, 41 days after initial scan. During examination, the mice were anaesthetised with isoflurane. Precautions to reduce the stress associated with general anaesthesia and imaging included maintenance and monitoring of the body temperature, transport to the imaging facility in suitable transport boxes, reduction of total imaging time to 1 h, and limitation to one imaging session per week. No contrast agent was given, and images were generated without respiratory gating. Two experienced radiologists independently evaluated all acquired images. Tumours were measured on the slice with their largest extent in T2 sagittal images in two directions (x, y), with an intraclass correlation of 0.958 between both investigators. Tumour volume estimation was done by manually tracing the tumours on each slice image, and then adding all the voxel volumes within the boundaries of the region of interest. Total volumes of individual tumours were calculated using Amira Software, Visage Imaging, Berlin, Germany.
Histopathology and immunohistochemistry
To confirm the presence of Ewing sarcoma xenografts, lungs, kidneys, and macroscopically detected bone and soft tissue tumours were examined by light microscopic examination and by immunohistochemistry. Tissues were fixed in 4% buffered formalin, processed and embedded in paraffin. Four-to five-micrometer sections were prepared and stained with haematoxylin and eosin or with the anti-human CD99 monoclonal antibody TÜ12 (BD Pharmingen, Heidelberg, Germany).
Production of recombinant retrovirus, transduction and expansion of T cells
Generation of the GD2-specific CAR 14.G2a-CD28ζ has been described in detail in previous publications from our group (Rossig et al, 2001; Altvater et al, 2006; Kailayangiri et al, 2012). The receptor contains the single-chain antibody domain of the monoclonal anti-GD2 antibody 14.G2a (Mujoo et al, 1989), the transmembrane domain of CD8α, and the intracellular domains of CD28 and TCR ζ. Retroviral supernatant was generated and used to transduce in vitro OKT-3/anti-CD28 antibody-preactivated human T cells, as previously described (Kailayangiri et al, 2012). To superexpand the gene-modified T cells, 2 × 107 cells were transferred to gas-permeable culture devices with 500 ml capacity (Wilson Wolf Manufacturing, New Brighton, MN, USA) in 400 ml of medium containing IL-2 (50 U ml−1) on day 5 after the prestimulation.
T-cell therapy
Following transplantation of 2 × 106 VH-64 tumour cells into the tail veins of NOD/scid mice, experimental cohorts of mice were given intravenous injections of 1 × 107 14.G2a-CD28ζ-transduced human T cells, whereas control cohorts received non-transduced T cells. In a first cohort of 10 mice (5 treated and 5 control mice), T-cell transfer was given on days 4, 10, 15, and 22 after tumour inoculation. A subsequent cohort of another eight mice (four treated and four control mice) received a total of six T-cell therapies on days 1, 3, 6, 9, 12, and 15. Intraperitoneal injections of human recombinant IL-2 were given twice weekly to all mice to sustain T-cell survival within the non-human environment. Mice receiving four and six injections were analysed together. Tumour development was monitored using WB-MRI as described above.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics 21 for Windows (IBM Corporation, Somers, NY, USA) and SAS software, Version 9.3 of the SAS System for Windows (SAS Institute Inc., Cary, NC, USA). P-values were interpreted in Fisher’s sense, representing the metric weight of evidence against the respective null hypothesis of no effect, and were considered noticeable in case P⩽0.05 and highly noticeable in case P⩽0.01. The intraclass correlations coefficient (ICC) was calculated as a measure of inter-rater agreement. Continuous data are reported as median, minimum, and maximum. Categorical variables are described as absolute and relative frequencies. Relationships between categorical variables were compared via contingency tables, χ2-, and Fisher’s exact test, where appropriate. Exact χ2-goodness-of-fit test was applied to compare the allocation of tumour numbers between cohorts. Pairwise group comparison for tumour numbers and tumour volumes were performed using exact non-parametric Mann–Whitney U-tests. Wilcoxon-signed rank tests were applied to detect the statistically noticeable changes between two TPs. Kaplan–Meier analyses and log-rank tests were performed to compare event-free and overall survival between treated and control cohorts. Multivariable, mixed model analyses were conducted to analyse the impact of potentially explanatory variables on target parameters.
Results
Whole-body MRI visualises disseminated tumour growth after intravenous injection of xenogeneic Ewing sarcoma cells in mice
High quality images in mice were obtained using a clinical 3 Tesla MRI scanner without respiratory triggering and without the need for contrast-enhancing agents. Among the first cohort of mice (n=18) receiving intravenous injections of 2 × 106 VH-64 cells, all 18 mice developed disseminated tumours detectable by WB-MRI within 20–31 days (median of 27 days). All engrafted mice had lung tumours (100%). Sixteen mice had additional tumour manifestations in extrapulmonary sites: 55.6% of mice (10 of 18) had bone or bone marrow tumours in the lower extremities, pelvis and/or vertebral column, and 27.8% (5 of 18) had soft tissue tumours (Figure 1A). Moreover, 72.2% (13 of 18) of mice had tumours in the kidney, and three of these also had tumours within the suprarenal gland. One of five female mice and none of 13 male mice had an abdominal tumour, potentially representing an ovarial manifestation. The highest numbers of tumours were found in the lungs, with a median of 19.5 (range 1–60) per mouse (Figure 1B). Interobserver agreement was consistent with an overall ICC of 0.915 for tumour numbers and 0.967 when excluding lung tumours. The median volume of tumours at first detection varied with the tumour site (Figure 1C). Soft tissue and kidney tumours were detectable at relatively small median sizes of 8.94 mm3 (range 2.53–25.05 mm3) and 8.15 mm3 (1.24–40.3 mm3), respectively, whereas bone tumours were relatively large upon first detection (median 33.11 mm3, range 13.49–117.9 mm3). Lung tumours were detectable at very small volumes of <1 mm3. Dissection and histological analysis confirmed the presence of CD99+ small, blue, round cell tumours in bones, lungs, and kidneys in all examined cases (Figure 1D).

Visualisation of disseminated growth of Ewing sarcoma xenografts in mice by WB-MRI. VH-64 cells (2 × 106) were injected into the tail veins of 18 NOD/scid mice, followed by WB-MRI after 3 weeks, and then at weekly intervals. T2 axial sequences were used for detection of lung tumours, and T2 axial, sagittal, and STIR sequences were used for detection of manifestations at all other sites. (A) Numbers of mice developing detectable tumour manifestations at the indicated locations. (B) Total numbers of tumours per individual tumour site. (C) Boxplots of median tumour volumes at first detection at the various locations. The lines indicate the median sizes, and boxes represent the 25th and 75th percentiles. Circles symbolise outliers, asterisks show extreme values. (D) Light microscopy of haematoxylin and eosin (HE)-stained sections (left panel) and immunohistochemistry analysis by huCD99 staining (right panel) of tumours from the indicated organ and tissue sites. Altogether, the Ewing sarcoma origin was confirmed by histology and/or immunohistochemistry in four lungs with multiple lesions, five bone lesions (three in legs, two in pelvis and vertebral column, respectively), and four renal tumours.
Sequential WB-MRI allows to monitor tumour growth in vivo
Sequential WB-MRI scans were performed until the mice had to be killed. T2 sagittal WB sequences revealed in vivo growth of tumours at all sites (Figure 2A), with heterogeneous kinetics. Soft tissue tumours increased in sizes at relatively slow rates, potentially owing to the early detection of very small tumours. As reliable volumetry of individual lung tumours is complicated by their diffuse infiltrative character, pulmonary involvement was monitored by combining volumes of all individual lung tumours for each lung. Moreover, lung tumours were quantified by numbers in axial images (Figure 2B). A continuous increase of the numbers of lung tumours both per lung and per mouse was found in follow-up scans (Figure 2B and C).

The kinetics of disseminated tumour growth in xenografted mice. (A) The volumes of individual tumours were determined in T2 axial (lung tumours) and T2 sagittal (all others) WB sequences in NOD/scid mice at time points 1, 2, 3, and 4. Extrapulmonary tumour manifestations were categorised by localisation to leg bones, skeletal trunk bones (pelvis, vertebral column), kidneys and/or suprarenal gland, and soft tissue. For lungs, each line corresponds to one mouse, as volumes of all tumour lesions were combined for each affected lung. For all other sites, each line reflects one individual tumour. Lung tumour volumes increased between TPs 1 and 2 (P<0.001) and TPs 2 and 3 (P<0.001). Femur/tibia tumours and pelvic tumours increased between TPs 1 and 2 (P<0.001 and P=0.031, respectively). Kidney tumours increased between TPs 1 and 2 (P<0.001), 2 and 3 (P<0.001), and 3 and 4 (P<0.001). (B) Lung tumours were counted in T2 axial WB sequences in NOD/scid mice at TPs 1, 2, 3, and 4. Each line represents one mouse. In addition, boxplots represent the tumour number of the evaluable mice at each TP. Brackets represent statistically noticeable differences (P⩽0.05) of the Wilcoxon-signed rank tests. Circles symbolise outliers; asterisks show extreme values. (C) Sequential MRI scans of one individual mouse demonstrate the progressive pulmonary involvement over time. The number and size of metastases that are detected as hyperintense manifestations in the dark lung in axial T2 images increases over time. Photodocumentation (to the right) of mice upon autopsy confirms the MRI findings, showing white-blueish metastases within the reddish lung tissue.
Diffusion-weighted WB imaging with background signal suppression allows for functional detection of Ewing sarcoma manifestations and growth in mice
Whole-body diffusion-weighted MRI with background body signal suppression is a functional imaging technique. Tumours are visualised based on the restricted diffusion of the malignant tissue architecture (Takahara et al, 2004). In our cohort of 18 mice, effective background signal suppression was obtained, except for fluids in the bladder, renal pelvis, and intestinal tract. Diffusion-weighted WB imaging with background signal suppression effectively visualised disseminated Ewing sarcoma growth in bones, retroperitoneal organs, and soft tissues, but not in lungs where susceptibility artefacts and respiratory motions cause signal loss (Wang et al, 2012). Sequential DWIBS demonstrated increases of tumour volumes over time (Figure 3A). Four tumours (three bone tumours, one kidney tumour) were visualised by DWIBS before they appeared in MR T2 sequences. To determine the sensitivity and specificity of DWIBS, standard T2-weighted sagittal MRI sequences were defined as standard. Interobserver agreement on the presence of individual tumours by DWIBS was 92%. Limited specificity of DWIBS was found for renal manifestations (Figure 3B), explained by signal artifacts in the vicinity of liquid-filled spaces. Tumours of the suprarenal gland were not detected by DWIBS, potentially owing to their close proximity to the diaphragm (Kwee et al, 2008). Moreover, DWIBS failed to detect the pelvic bone lesions detected on T2 MRI sequences in 3 of 13 examinations. Tumours undetectable by DWIBS were smaller than those reliably detected with both techniques (Figure 3C). All in all, these results demonstrate feasibility of detection of disseminated Ewing sarcoma growth in a small animal model using DWIBS sequences.

Whole-body MRI with DWIBS of Ewing sarcoma-engrafted mice. Sequential WB-MRI and DWIBS scans were performed in the cohort of 18 mice starting 3 weeks after tumour inoculation. (A) T2 sagittal MRI sequences (left and right panel) and DWIBS (central panel) images of a single mouse at sequential examinations performed 20–41 (TPs 1–4) days after tumour inoculation, as indicated. The left and right panels illustrate the growth of a kidney and a femur tumour, respectively. Images of DWIBS performed at the same time points demonstrate the two tumour manifestations in the femoral bone (larger arrow) and kidney (smaller arrow). T2 sagittal sequences were used to define the presence of tumour manifestations. The following regions were separately analysed for the presence or absence of tumours: right and left lower limbs, pelvic bones/vertebral column, left and right kidneys and suprarenal glands, and soft tissues. (B) Sensitivity, specificity, and positive (ppV) and negative predictive values (npV) of DWIBS were calculated for each localisation in comparison with standard T2 sagittal sequences. Numbers in brackets represent 95% confidence intervals. (C) Boxplots of median tumour volumes at all sites detectable and non-detectable by DWIBS, assessed by volumetry on T2 sagittal MR images. The bracket represents the result of the exact Mann–Whitney U-test. Circles symbolise outliers; asterisks show extreme values.
In vivo antitumour activity of GD2-retargeted T cells against established tumour xenografts
To demonstrate the therapeutic efficacy of adoptive T-cell transfer against disseminated tumour cells, further cohorts of mice were intravenously injected with 2 × 106 VH-64 cells/mouse, followed by four (n=5) to six (n=4) transfusions of 1 × 107 14.G2a-CD28ζ gene-modified human GD2-specific T cells (group A, n=9). Control mice (group B, n=9) received analogous injections of non-transduced T cells. Analytic end points were the number of mice developing tumour manifestations and the numbers and volumes of tumours at the individual sites. Two control mice failed to develop Ewing sarcomas. All mice with lung tumours also had extrapulmonary tumour manifestations. Abdominal tumours that in female mice potentially represent ovarial manifestations were found in 2 of 6 male and in 10 of 12 female mice. As these tumours could not be assigned to specific organs, they were excluded from the analysis.
The numbers of mice developing tumours and the numbers of tumours at extrapulmonary localisation sites were not noticeably different between treated and untreated mice (Figure 4A and B). Mice receiving gene-modified T-cell therapy had lower numbers of pulmonary tumours than control mice (Figure 4B). Soft tissue tumours at first detection had lower volumes in the treatment group (Figure 4C). Differences at further sites did not reach statistical significance. All tumour-engrafted mice in both groups had progressive disease over time (Figure 4D). For comparisons of tumour growth between the two groups, multivariable analysis according to a generalised linear-mixed model was performed to account for the complexity of the experimental system and to include the effects of time and localisation. For all localisations and TPs combined, the estimated tumour volume was reduced by 7.95 mm3 in the treatment group, but the difference was not statistically noticeable (P=0.0965; Table 1). As tumour localisation itself had a noticeable influence on tumour volume (P<0.0001), we next compared tumour growth at individual localisations. At non-pulmonary sites, the maximum sizes of tumours over the entire follow-up were not noticeably different between mice receiving T-cell therapy and non-treated control mice, with the exception of reduced sizes of femoral and tibial bone tumours at the TP 4 (P=0.056; n=7 (A), n=2 (B)). Noticeable differences between groups were identified for lung tumours (Figure 4E): Mice treated with GD2-redirected T cells had a growth delay of lung tumours, with both lower numbers and smaller volumes (Figure 4B and E). No overall survival (as defined in the figure legend) advantage was found for mice receiving T-cell therapy using Kaplan–Meier analysis (Figure 4F). Thus, GD2-retargeted T cells cannot completely prevent disseminated tumour growth in this systemic disease model, but are active to delay and reduce pulmonary disease manifestations.

MRI documentation of in vivo activity of 14.G2a-28 ζ T cells against disseminated tumour xenografts. A cohort of nine mice were intravenously injected with 2 × 106 VH-64 cells per mouse, followed by intravenous transfer of 1 × 107 human 14.G2a-28ζ gene-modified T cells (group A). Control mice (n=9) received analogous injections of non-transduced T cells (group B). Mice were followed up by sequential T2 sagittal and axial, STIR and DWIBS WB-MRI scans. Tumour numbers were assessed at the time of termination. (A) Total numbers of mice of groups A and B with at least one tumour at indicated tumour sites. (B) Total numbers of tumours at the indicated tumour sites in groups A and B. The bracket represents a statistically highly noticeable result (P⩽0.01) of the exact χ2-goodness-of-fit test. (C) Boxplots of median tumour volumes at first detection at the various locations in groups A and B. Brackets represent statistically noticeable differences (P⩽0.05) between the groups determined by exact Mann–Whitney U-tests. (D) Growth of tumours at the individual sites in group A (upper panel) and group B (lower panel) over time. Volumes of lung tumours per mouse were combined and presented as the total lung tumour burden per mouse in mm3. For all other localisations, each line corresponds to one individual tumour. (E) Boxplots of median numbers (left panel) and volumes (right panel) of lung tumours at the indicated TPs. Brackets indicate differences between the groups determined by exact Mann–Whitney U-tests. At TPs 1 and 2, group A consisted of nine mice, and group B of seven mice. Owing to drop-out by termination or unexpected death during anaesthesia, the sizes of groups were reduced to n=4 (group A and n=3 (group B) at TP 3. Circles symbolise outliers; asterisks show extreme values. (F) Kaplan–Meier curves of overall survival after tumour cell injection in groups A and B, and P-value of the log-rank test. Overall survival is defined by killing of the animals, as explained in the Materials and Methods section or by unexpected death of few animals during the anaesthesia.
Discussion
Paediatric bone and soft tissue sarcomas tend to spread to and recur in lungs as well as in the bone and bone marrow. Multifocal disease is the hardest challenge to further improving cure rates. For the development of novel therapies and their clinical translation, in vivo preclinical models are needed that adequately mimic the dissemination pattern of these diseases and allow for non-invasive monitoring of tumour growth and response to treatment.
Here, we have established a small animal imaging model of systemic Ewing sarcoma growth using MRI techniques. Compared with the more broadly applied bioluminescence small animal imaging techniques, MRI directly reflects high resolution sarcoma staging in human patients and thus facilitates clinical translation of our findings.
Intravenous injection of human Ewing sarcoma cells into immunodeficient mice reliably established the disease in the majority of mice within <5 weeks, confirming previous experience (Vormoor et al, 2001). Compared with the routine histology of specific organs and macroscopically affected tissues in deceased mice, sequential whole-body imaging allows more comprehensive detection of various tumour manifestations. Specifically, small tumours were detected at sites not included into routine examination in the previous study (Vormoor et al, 2001). Importantly, the non-invasive nature of MRI further allows for follow-up investigations of tumour growth, both during the natural course of the disease and upon therapeutic intervention. Indeed, MRI provided a highly reproducible representation of clinical Ewing sarcoma development in mice and revealed systemic treatment efficacy of GD2 CAR gene-modified T cells. Our model is easy to apply and has translational potential within a wider scale for novel therapeutic developments against this malignant disease.
Besides their use in small animal models, non-invasive whole-body imaging tools are clinically needed for accurate tumour staging and assessment of treatment response. MRI is one of the most important diagnostic tools in the bone and soft tissue tumours where it provides excellent anatomical information (Lang et al, 1998a, 1998b). Whole-body MRI is currently evaluated for the initial staging of patients with malignant solid tumours including Ewing sarcomas (Haubold-Reuter et al, 1993; Walker et al, 2000; Lauenstein et al, 2002). Its specific value lies within the early detection of bone and bone marrow disease (Eustace et al, 1997; Steinborn et al, 1999; Altehoefer et al, 2001; Lauenstein et al, 2002; Hargaden et al, 2003; Ghanem et al, 2006). MRI further allows for volume-based assessment of response to cancer therapy. A limitation is that it does not assess the metabolic activity of remaining tumour tissue. Diffusion-weighted MRI was introduced as a non-invasive technique providing functional information for both staging and response monitoring of solid tumours (Roth et al, 2004; Thoeny et al, 2005; Moffat et al, 2006; Schubert et al, 2006; Vandecaveye et al, 2009; Wang et al, 2009; Oka et al, 2010). In first preclinical studies in Ewing sarcomas, diffusion-weighted imaging effectively predicted early response of single subcutaneous tumours to chemotherapy in mice before the onset of morphological changes (Reichardt et al, 2009), and in a mouse model of ovarian cancer, DWIBS had the potential to detect peritoneal tumour dissemination (Lee et al, 2013). In pilot clinical studies, WB-MRI with DWIBS was found useful for detecting metastatic tumours especially in the bone (Nakanishi et al, 2007; Manenti et al, 2012; Sommer et al, 2012). Here, to the best of our knowledge, we are the first to demonstrate the feasibility of DWIBS for the staging of multifocal Ewing sarcoma in small animals. With the known limitation of DWIBS not detecting pulmonary metastases, the technique provided a rapid overview of extrapulmonary disease manifestations. The future value of the DWIBS technique in sarcoma staging may lie within providing complementary non-invasive functional tissue information for extrapulmonary sites of the disease, while avoiding exposure to ionising radiation.
Among novel experimental cancer therapies, adoptive transfer of T cells expressing recombinant antigen-specific CARs holds high promise. In patients with lymphoid malignancies, CAR-reengineered T cells mediated unequivocal clinical responses even against bulky tumours, functionally persisted in vivo, and established immunological memory (Kalos et al, 2011; Louis et al, 2011; Porter et al, 2011). However, results from individual clinical studies are variable (Brentjens et al, 2011; Kalos et al, 2011; Porter et al, 2011; Savoldo et al, 2011; Kochenderfer et al, 2012), and the reasons for the compelling activity of this therapy in some clinical studies but not in others are unclear. Besides the design of the individual CARs and the nature of their target antigen, host factors are thought to contribute to determining CAR treatment responses (Dudley et al, 2002). Although in our study, GD2 CAR gene-modified T cells were not effective to prevent the disease, they had noticeable antitumour activity especially against pulmonary dissemination. Thus, our findings support further development and translational application of cellular therapies in Ewing sarcoma. A potential reason for the limited antitumour activity of the reengineered T cells is the high number of disseminated tumour cells present in the mouse organism at the time of T-cell transfer. Lower numbers of tumour cells reflecting minimal residual disease are more probably to be eradicated by this and other experimental interventions, but are also less reliable to engraft the disease in all mice. High-sensitivity imaging, as established here, may allow to detect tumour manifestations mediated by smaller numbers of tumour cells and mimic minimal disseminated disease more accurately than current models. To more adequately reflect the spectrum of local progression and organ and tissue dissemination of the disease, future experiments will include additional cell lines and low-passage cultures.
The challenge now is to maximise the activity of the reengineered T cells, for example, by manipulation of costimulatory domains and optimal CAR design. Transfer of tumour antigen-specific T cells in the setting of advanced disease may be inadequate to control the disease. This is also supported by recently published in vivo experiments using T cells with native receptor specificity for EWS-FLI-1, which despite clear antitumour activity could not halt disease progression (Evans et al, 2012). Thus, combined approaches with cytotoxic therapies may be more adequate to exploit the benefit of T-cell therapies. Moreover, recent insights into the components of the inflammatory microenvironment that contributes to local progression in Ewing sarcoma suggest that combinations with chemokine targeting may improve the efficacy of T-cell strategies (Berghuis et al, 2012). Although xenograft models carry limitations for immunological treatment manoeuvers as they do not completely represent the immunological in vivo complexity of interactions between host, tumour, and treatment, a comparative assessment of the antitumour activity of various constructs in our mouse model may help to choose the most promising constructs for clinical translation.
Change history
06 August 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Altehoefer C, Ghanem N, Hogerle S, Moser E, Langer M (2001) Comparative detectability of bone metastases and impact on therapy of magnetic resonance imaging and bone scintigraphy in patients with breast cancer. Eur J Radiol 40: 16–23.
Altvater B, Pscherer S, Landmeier S, Niggemeier V, Juergens H, Vormoor J, Rossig C (2006) CD28 co-stimulation via tumour-specific chimaeric receptors induces an incomplete activation response in Epstein-Barr virus-specific effector memory T cells. Clin Exp Immunol 144: 447–457.
Berghuis D, Schilham MW, Santos SJ, Savola S, Knowles HJ, Dirksen U, Schaefer KL, Vakkila J, Hogendoorn PC, Lankester AC (2012) The CXCR4-CXCL12 axis in Ewing sarcoma: promotion of tumor growth rather than metastatic disease. Clin Sarcoma Res 2: 24.
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M (2011) Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118: 4817–4828.
Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, De Jong P, Rouleau G (1992) Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359: 162–165.
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854.
Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, Uren A, Toretsky JA (2009) A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med 15: 750–756.
Eustace S, Tello R, DeCarvalho V, Carey J, Wroblicka JT, Melhem ER, Yucel EK (1997) A comparison of whole-body turboSTIR MR imaging and planar 99mTc-methylene diphosphonate scintigraphy in the examination of patients with suspected skeletal metastases. AJR Am J Roentgenol 169: 1655–1661.
Evans CH, Liu F, Porter RM, O'Sullivan RP, Merghoub T, Lunsford EP, Robichaud K, Van Valen F, Lessnick SL, Gebhardt MC, Wells JW . EWS-FLI-1-targeted cytotoxic T-cell killing of multiple tumor types belonging to the Ewing sarcoma family of tumors. Clin Cancer Res 18: 5341–5351 (2012).
Ghanem N, Altehoefer C, Kelly T, Lohrmann C, Winterer J, Schafer O, Bley TA, Moser E, Langer M (2006) Whole-body MRI in comparison to skeletal scintigraphy in detection of skeletal metastases in patients with solid tumors. In Vivo 20: 173–182.
Hargaden G, O'Connell M, Kavanagh E, Powell T, Ward R, Eustace S (2003) Current concepts in whole-body imaging using turbo short tau inversion recovery MR imaging. AJR Am J Roentgenol 180: 247–252.
Haubold-Reuter BG, Duewell S, Schilcher BR, Marincek B, von Schulthess GK (1993) The value of bone scintigraphy, bone marrow scintigraphy and fast spin-echo magnetic resonance imaging in staging of patients with malignant solid tumours: a prospective study. Eur J Nucl Med 20: 1063–1069.
Juergens H, Daw NC, Geoerger B, Ferrari S, Villarroel M, Aerts I, Whelan J, Dirksen U, Hixon ML, Yin D, Wang T, Green S, Paccagnella L, Gualberto A (2011) Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol 29: 4534–4540.
Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, Titze U, Leuchte K, Landmeier S, Hotfilder M, Dirksen U, Hardes J, Gosheger G, Juergens H, Rossig C (2012) The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer 106: 1123–1133.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3: 95ra73.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA (2012) B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119: 2709–2720.
Kwee TC, Takahara T, Ochiai R, Nievelstein RA, Luijten PR (2008) Diffusion-weighted whole-body imaging with background body signal suppression (DWIBS): features and potential applications in oncology. Eur Radiol 18: 1937–1952.
Ladenstein R, Potschger U, Le Deley MC, Whelan J, Paulussen M, Oberlin O, van den Berg H, Dirksen U, Hjorth L, Michon J, Lewis I, Craft A, Jurgens H (2010) Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28: 3284–3291.
Lang P, Johnston JO, Arenal-Romero F, Gooding CA (1998a) Advances in MR imaging of pediatric musculoskeletal neoplasms. Magn Reson Imaging Clin N Am 6: 579–604.
Lang P, Wendland MF, Saeed M, Gindele A, Rosenau W, Mathur A, Gooding CA, Genant HK (1998b) Osteogenic sarcoma: noninvasive in vivo assessment of tumor necrosis with diffusion-weighted MR imaging. Radiology 206: 227–235.
Lauenstein TC, Goehde SC, Herborn CU, Treder W, Ruehm SG, Debatin JF, Barkhausen J (2002) Three-dimensional volumetric interpolated breath-hold MR imaging for whole-body tumor staging in less than 15 minutes: a feasibility study. AJR Am J Roentgenol 179: 445–449.
Lee HJ, Luci JJ, Tantawy MN, Lee H, Nam KT, Peterson TE, Price RR (2013) Detecting peritoneal dissemination of ovarian cancer in mice by DWIBS. Magn Reson Imaging 31: 227–234.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118: 6050–6056.
Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, Tesso M, Long LM, Grindler D, Merino M, Kopp W, Tsokos M, Berzofsky JA, Helman LJ (2008) A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res 14: 4850–4858.
Manenti G, Ciccio C, Squillaci E, Strigari L, Calabria F, Danieli R, Schillaci O, Simonetti G (2012) Role of combined DWIBS/3D-CE-T1w whole-body MRI in tumor staging: comparison with PET-CT. Eur J Radiol 81: 1917–1925.
Moffat BA, Chenevert TL, Meyer CR, Mckeever PE, Hall DE, Hoff BA, Johnson TD, Rehemtulla A, Ross BD (2006) The functional diffusion map: an imaging biomarker for the early prediction of cancer treatment outcome. Neoplasia 8: 259–267.
Mujoo K, Kipps TJ, Yang HM, Cheresh DA, Wargalla U, Sander DJ, Reisfeld RA (1989) Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res 49: 2857–2861.
Nakanishi K, Kobayashi M, Nakaguchi K, Kyakuno M, Hashimoto N, Onishi H, Maeda N, Nakata S, Kuwabara M, Murakami T, Nakamura H (2007) Whole-body MRI for detecting metastatic bone tumor: diagnostic value of diffusion-weighted images. Magn Reson Med Sci 6: 147–155.
Oka K, Yakushiji T, Sato H, Hirai T, Yamashita Y, Mizuta H (2010) The value of diffusion-weighted imaging for monitoring the chemotherapeutic response of osteosarcoma: a comparison between average apparent diffusion coefficient and minimum apparent diffusion coefficient. Skeletal Radiol 39: 141–146.
Paulussen M, Ahrens S, Burdach S, Craft A, Dockhorn-Dworniczak B, Dunst J, Frohlich B, Winkelmann W, Zoubek A, Jurgens H (1998) Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9: 275–281.
Porter DL, Levine BL, Kalos M, Bagg A, June CH (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725–733.
Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu EL, Gee AP, Mei Z, Yvon E, Weiss HL, Liu H, Rooney CM, Heslop HE, Brenner MK (2008) Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14: 1264–1270.
Reichardt W, Juettner E, Uhl M, Elverfeldt DV, Kontny U (2009) Diffusion-weighted imaging as predictor of therapy response in an animal model of Ewing sarcoma. Invest Radiol 44: 298–303.
Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK (2001) Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer 94: 228–236.
Roth Y, Tichler T, Kostenich G, Ruiz-Cabello J, Maier SE, Cohen JS, Orenstein A, Mardor Y (2004) High-b-value diffusion-weighted MR imaging for pretreatment prediction and early monitoring of tumor response to therapy in mice. Radiology 232: 685–692.
Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G (2011) CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest 121: 1822–1826.
Schubert MI, Wilke M, Muller-Weihrich S, Auer DP (2006) Diffusion-weighted magnetic resonance imaging of treatment-associated changes in recurrent and residual medulloblastoma: preliminary observations in three children. Acta Radiol 47: 1100–1104.
Sommer G, Wiese M, Winter L, Lenz C, Klarhofer M, Forrer F, Lardinois D, Bremerich J (2012) Preoperative staging of non-small-cell lung cancer: comparison of whole-body diffusion-weighted magnetic resonance imaging and (18)F-fluorodeoxyglucose-positron emission tomography/computed tomography. Eur Radiol 22: 2859–2867.
Steinborn MM, Heuck AF, Tiling R, Bruegel M, Gauger L, Reiser MF (1999) Whole-body bone marrow MRI in patients with metastatic disease to the skeletal system. J Comput Assist Tomogr 23: 123–129.
Takahara T, Imai Y, Yamashita T, Yasuda S, Nasu S, Van CM (2004) Diffusion weighted whole body imaging with background body signal suppression (DWIBS): technical improvement using free breathing, STIR and high resolution 3D display. Radiat Med 22: 275–282.
Thoeny HC, De KF, Chen F, Vandecaveye V, Verbeken EK, Ahmed B, Sun X, Ni Y, Bosmans H, Hermans R, van OA, Marchal G, Landuyt W (2005) Diffusion-weighted magnetic resonance imaging allows noninvasive in vivo monitoring of the effects of combretastatin a-4 phosphate after repeated administration. Neoplasia 7: 779–787.
Vandecaveye V, De KF, Verslype C, Op de BK, Komuta M, Topal B, Roebben I, Bielen D, Roskams T, Nevens F, Dymarkowski S (2009) Diffusion-weighted MRI provides additional value to conventional dynamic contrast-enhanced MRI for detection of hepatocellular carcinoma. Eur Radiol 19: 2456–2466.
Vormoor J, Baersch G, Decker S, Hotfilder M, Schafer KL, Pelken L, Rube C, Van VF, Jurgens H, Dockhorn-Dworniczak B (2001) Establishment of an in vivo model for pediatric Ewing tumors by transplantation into NOD/scid mice. Pediatr Res 49: 332–341.
Walker R, Kessar P, Blanchard R, Dimasi M, Harper K, DeCarvalho V, Yucel EK, Patriquin L, Eustace S (2000) Turbo STIR magnetic resonance imaging as a whole-body screening tool for metastases in patients with breast carcinoma: preliminary clinical experience. J Magn Reson Imaging 11: 343–350.
Wang H, Sun X, Chen F, De KF, Yu J, Landuyt W, Vandecaveye V, Peeters R, Bosmans H, Hermans R, Marchal G, Ni Y (2009) Treatment of rodent liver tumor with combretastatin a4 phosphate: noninvasive therapeutic evaluation using multiparametric magnetic resonance imaging in correlation with microangiography and histology. Invest Radiol 44: 44–53.
Wang N, Zhang M, Sun T, Chen H, Huang Z, Yan L, Wang L, Li F, Chen Z (2012) A comparative study: diffusion weighted whole body imaging with background body signal suppression and hybrid Positron Emission Computed Tomography on detecting lesions in oncologic clinics. Eur J Radiol 81: 1662–1666.
Acknowledgements
This work was supported by Grant #109566 from Deutsche Krebshilfe (to CR) and a grant from the University of Muenster Faculty of Medicine ‘Innovative Medizinische Forschung (IMF)’ program (to BA and CR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
HK is an employee of Philips Healthcare, the manufacturer of the MRI equipment used in this work. The remaining authors declare no conflicts of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Liebsch, L., Kailayangiri, S., Beck, L. et al. Ewing sarcoma dissemination and response to T-cell therapy in mice assessed by whole-body magnetic resonance imaging. Br J Cancer 109, 658–666 (2013). https://doi.org/10.1038/bjc.2013.356
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.356
Keywords
This article is cited by
-
CAR T cells as micropharmacies against solid cancers: Combining effector T-cell mediated cell death with vascular targeting in a one-step engineering process
Cancer Gene Therapy (2023)
-
Longitudinal imaging of T cell-based immunotherapy with multi-spectral, multi-scale optoacoustic tomography
Scientific Reports (2020)
-
Pilot Trial of FANG Immunotherapy in Ewing's Sarcoma
Molecular Therapy (2015)
