Abstract
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only potentially curative treatment for the BM dysfunction seen in patients with Shwachman–Diamond syndrome (SDS). Historically, these patients have fared poorly with intensive conditioning regimens with increased regimen-related toxicity especially involving the heart and lungs. We report our institutional experience with a reduced-intensity-conditioning protocol in seven patients with SDS and BM aplasia or myelodysplastic syndrome/AML. The preparative regimen consisted of Campath-1H, fludarabine and melphalan. Four patients received matched related marrow and three received unrelated stem cells (two PBSCs and one marrow). All but one was 8 of 8 allele HLA matched. All patients established 100% donor-derived hematopoiesis. No patient in this cohort developed grades III–IV GVHD. One patient had grade II skin GVHD that responded to systemic corticosteroids and one had grade I skin GVHD, treated with topical corticosteroids. Two out of seven patients developed bacterial infections in the early post transplant period. Viral infections were seen in four out of seven patients and were successfully treated with appropriate antiviral therapy. All patients are currently alive. These data indicate that HSCT with reduced-intensity conditioning is feasible in patients with SDS and associated with excellent donor cell engraftment and modest morbidity.
Similar content being viewed by others
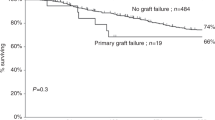

Introduction
Shwachman–Diamond syndrome (SDS) is a rare autosomal recessive disorder characterized by exocrine pancreatic insufficiency, skeletal abnormalities in the form of metaphyseal dysostosis and BM dysfunction manifested as cytopenias.1, 2, 3, 4, 5, 6 Additional clinical manifestations seen in some patients include short stature, variable immune dysfunction, delayed dentition and structural and functional abnormalities of the liver.3, 4, 7, 8 Patients with SDS are at an increased risk of developing aplastic anemia, myelodysplastic syndrome (MDS) and AML.3, 4, 7, 9, 10, 11, 12 In the largest reported patient series, 20% of cases developed pancytopenia and 6% progressed to MDS.4 Other authors have reported varying incidences of MDS ranging from 10 to 15% to as high as 44% of cases.3, 7, 10, 13 The risk of leukemic transformation in SDS patients is significant and increases with age, varying from 5% in childhood to nearly 24% as patients approach adulthood.13 Infections and thoracic dystrophy are the leading causes of morbidity and mortality during infancy, and the likelihood of long-term survival correlates most closely with the degree of BM dysfunction. Alter and Young14 calculated the projected median survival of SDS patients as more than 35 years, on the basis of a literature review. Survival is particularly reduced in patients who develop BM aplasia, MDS or acute leukemia, averaging 14 years in patients with aplastic anemia.10 The development of acute leukemia portends a poor prognosis as SDS patients do not respond well to chemotherapeutic intervention.
Hematopoietic stem cell transplantation (HSCT) is the only curative treatment for BM dysfunction associated with SDS. However, the timing of transplantation remains a subject of controversy, and the apparent lack of genotype–phenotype correlation makes selection of patients for early preemptive HSCT impossible. Additionally, patients with SDS tend to have increased toxicity with intensive conditioning regimens. Tsai et al.15 reported a case of fatal congestive heart failure following a Cy-containing-conditioning regimen. Other authors have described neurological complications,16 pulmonary complications and multiorgan failure with typical ablative regimens.17, 18
In this report, we describe our institutional experience with the use of a reduced-intensity-conditioning regimen consisting of Campath-1H, fludarabine and melphalan in seven patients with SDS, all of whom achieved excellent hematopoietic recovery and full donor chimerism with acceptable toxicity.
Methods
All patients were enrolled on an institutional review board-approved protocol at the Cincinnati Children's Hospital Medical Center and informed consent was obtained from the patient or their legal guardian.
Patients
Seven consecutive patients with SDS were transplanted at the Blood and Marrow Transplant Program of Cincinnati Children's Hospital Medical Center from April 2005 to July 2007. Patient demographics are described in Table 1. All patients had the diagnosis of SDS confirmed by genetic testing to document Shwachman–Bodian–Diamond syndrome (SBDS) mutations.
Liver dysfunction was noted prior to transplant in three of seven patients. One of these patients underwent a liver biopsy due to persistent elevation of liver enzymes that showed mild portal and periportal fibrosis, unchanged in comparison with a previous biopsy. Cardiac function was normal in all patients prior to transplant though trivial mitral and/or aortic regurgitation was noted in three out of seven patients. Patient number 1 had bronchiolitis obliterans of the lower lobe of the left lung as a consequence of repeated pulmonary infections during childhood. Patient number 7 had bronchopulmonary dysplasia and chronic pulmonary insufficiency as a consequence of thoracic dysplasia as part of the SDS phenotype. She had pulmonary hypertension in the neonatal period but pretransplant echocardiogram (ECHO) was normal.
The indication for HSCT was worsening cytopenias with increasing transfusion dependence and/or the appearance of clonal hematopoeisis in six of seven patients. Patient number 6 had AML at the time of presentation to our institution. She was treated with cytarabine and L-asparaginase in a modified Capizzi regimen prior to transplant.
Stem cell sources
Stem cell sources and cell doses are described in Table 2. Four of seven patients received marrow from a sibling donor. One of these patients (Patient number 4) received stored umbilical cord blood (UCB) together with marrow from the same sibling donor, collected because the UCB cell dose was low. High-resolution HLA typing was performed at HLA-A, B, C and DRB1 for all cases. Stem cells were 8 of 8 allele matched in all but one case. Patient number 6 received PBSCs from an unrelated donor, mismatched at a single antigen at the DRB1 locus.
Preparative regimen
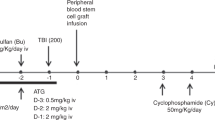
The conditioning regimen consisted of Campath-1H (10 mg on day 1, 15 mg on day 2 and 20 mg on day 3) i.v. on 3 successive days (between days −28 and −19), after a test dose (3 mg over 2 h, given not more than 7 days prior to starting conditioning). In this treatment plan, we administered the campath-1H on days −21, −20 and −19 to ensure that negligible levels of Campath-1H remained in the patients at the time of donor stem cell infusion. The intent was to intensely immunosuppress the recipient pretransplant (in addition to the fludarabine and melphalan used as part of the conditioning) to improve the likelihood of engraftment of donor stem cells. Fludarabine (30 mg/m2 per day) was given for 5 days on days −8 to −4. Melphalan was given at 140 mg/m2 on day −3. Doses were modified for recipients <10 kg as follows: Campath-1H 10 mg on 3 consecutive days (days −21 to −19), fludarabine 1 mg/kg and melphalan 4.7 mg/kg.
GVHD prophylaxis
CYA and MTX were used for GVHD prophylaxis unless the stem cell source was UCB, when methylprednisolone 1 mg/kg was used in place of MTX. CYA was commenced on day −2 at a dose of 2.5 mg/kg per dose every 12 h given i.v. Trough CSA levels were measured two times weekly with dose adjustments as needed until therapeutic levels were achieved (250–350 ng/ml) and weekly thereafter. Therapeutic levels were maintained until day +100. Patients were switched to oral CYA at the time of discharge from hospital if they could tolerate the drug orally. From day +100 to 6 months post transplant, CSA was tapered in patients without significant acute or chronic GVHD by approximately 5–10% per week. MTX was given at a dosage of 10 mg/m2 on day +1 and 7.5 mg/m2 per day on days +3 and +6. Methylprednisolone was used until day +28 and then tapered slowly by day +56.
Patient number 6 initially received GVHD prophylaxis with mycophenolate mofetil and CSA. However, CSA was changed to tacrolimus due to intolerance.
Supportive care
All patients were hospitalized at the Blood and Marrow Transplant Unit at Cincinnati Children's Hospital Medical Center until evidence of myeloid engraftment was seen. Patients were housed in single rooms ventilated with high efficiency air filtration systems. G-CSF (5 μg/kg per day) was given s.c. or i.v. starting day +7 for PBSC or BM recipients and on day +1 for UCB recipients until ANC was>1500 per 100 ml for 3 consecutive days. Intravenous immunoglobulin replacement was given every 3 weeks until patients were weaned off immunosuppression and for the first 6 months post transplant when cord blood was the stem cell source. Antimicrobial prophylaxis was as follows: voriconazole or liposomal amphotericin (10 mg/kg weekly) as antifungal prophylaxis; acyclovir for herpes simplex virus -seropositive patients; i.v. pentamidine for Pneumocystis carinii prophylaxis until engraftment. Trimethoprim/sulfamethoxazole was substituted as clinically indicated after hematologic recovery (ANC>1500 per 100 ml) and continued for a minimum of 1-year post transplant. Other antimicrobial prophylaxis was continued for 6 months post transplant, or until immunosuppression was discontinued, whichever was later. CMV, EBV and adenovirus surveillance was instituted 7 days after stem cell infusion, and treatment was commenced preemptively if the CMV test was positive, with continued monitoring for response.
Hematological reconstitution
Neutrophil engraftment was defined as the day on which the ANC rose to⩾500 cells per 100 ml for 3 consecutive days after the postpreparative regimen neutrophil nadir. Platelet engraftment was defined as the first day on which the platelet count rose to⩾50 000 per 100 ml over a 7-day interval without transfusion support. Chimerism assessment by variable number of tandem repeats (VNTR) analysis (or XY FISH in sex-mismatched donor–recipient pairs) and/or cytogenetic studies was performed approximately 4 weeks post transplant, and initially every 2–4 weeks thereafter, then every 3 months or as clinically indicated.
Results
Engraftment
Hematopoietic recovery was prompt in all cases, and all patients have stable full donor chimerism, with no evidence of late loss of donor cell engraftment (Table 3). Myeloid engraftment occurred at a median of 14 days (range, 11–15 days) and platelet recovery at a median of 33 days (range, 14–68 days).
GVHD
No patient in this cohort developed grades III–IV GVHD. One patient had grade II skin GVHD that responded to systemic corticosteroids and one had grade I skin GVHD, treated with topical corticosteroids. Both these patients received PBSCs from an unrelated donor. No patient needed or received donor lymphocyte infusions.
Survival and regimen-related toxicity
All seven patients are surviving with a median follow-up of 548 days (range, 93–920 days). Patient number 1 entered transplant with bronchiolitis obliterans of the lower lobe of the left lung. Bronchoalveolar lavage done during the pretransplant period grew Pseudomonas aeruginosa that resolved following appropriate antibiotic therapy. He was hospitalized for left lower lobe pneumonia on day 170 post transplant that responded to antibacterial therapy directed at P. aeruginosa. This patient also had persistent emesis and secretory diarrhea. Gastrointestinal endoscopy revealed mild chronic duodenitis with focal cryptitis and apoptosis believed to be the consequence of prior infection, as CMV was detected by in situ hybridization and symptoms resolved following treatment with ganciclovir. This patient also had evidence of EBV antigenemia by PCR that responded to a single dose of rituximab. Additional bacterial infections occurred in three out of seven patients. There were two patients with documented bacteremia, one gram-negative and the other with oxacillin-resistant Staphylococcus aureus and one with a urinary tract infection (Table 3). Adenoviral diarrhea was seen in two patients whereas one patient had enteroviral diarrhea, all of which resolved with supportive care alone. Patient number 6 had evidence of disseminated adenoviral infection 5 months post transplant that resolved after treatment with cidofovir. This emphasizes the need for close monitoring and early treatment of infection, particularly viral infections with sensitive methods early in the post transplant period.
Biochemical evidence of renal insufficiency occurred in two out of six patients that responded to fluid administration. No cardiac toxicity was seen.
Discussion
In this report we describe a case series of patients with the rare disorder, SDS treated with a reduced-intensity-conditioning regimen prior to allogeneic hematopoeitic stem cell transplantation. Our patient population showed prompt myeloid engraftment with full donor chimerism in all cases. Regimen-related toxicity was modest with no patients developing grades III–IV GVHD, and all patients are currently alive.
Hematopoietic stem cell transplantation is the only known curative treatment for the hematological abnormalities seen in Shwachman–Diamond syndrome. The available literature on HSCT in SDS patients is limited and consists mainly of case reports.19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 Vibhakar et al.30 recently reviewed the published experience with HSCT in SDS patients and reported a total of 28 patients, including their own. All but four patients received ablative conditioning regimens containing CY with or without TBI/TLI. Most patients received unrelated BM as a source of stem cells, though Vibhakar et al. reported three cases where UCB was used successfully as a source of stem cells. At the time of reporting, 17 of these patients were alive, that is, mortality approached 40%. More than 50% of the patients died in the early post transplant phase of cardiopulmonary complications. Similarly, in a recent review of the European experience with HSCT in SDS patients, Cesaro et al.17 reported an overall treatment-related mortality of 35.5% at 1 year. Interestingly, they found a significantly higher mortality rate in patients receiving a TBI-conditioning regimen (67 vs 20% for TBI vs non-TBI-containing regimen, P=0.03).
Though the mechanism is unclear, patients with SDS seem to have a predilection for increased cardiac toxicity with CY-containing-conditioning regimens.15, 23, 28, 31 Savilahti and Rapola31 reported significant cardiac dysfunction in patients with SDS even without exposure to CY. In their series of 16 Finnish patients, 8 had cardiac abnormalities on necropsy including cardiac fibrosis and areas of necrosis. Clinical reports also suggest that patients with SDS are more susceptible to transplant-related toxicity as compared to patients with other myelodysplastic disorders like Kostmann's syndrome and juvenile myelomonocytic leukemia.32, 33 Dror and Freedman34 have demonstrated that the BM mononuclear cells from patients with SDS show an increased propensity for apoptosis mediated by hyperactivation of the Fas-signaling pathway. The same authors have also reported decreased telomere length in the marrow-derived mononuclear cells from patients with SDS.35 It is possible, although currently unexplored, that similar mechanisms are important in the increased susceptibility to organ toxicity with intensive conditioning regimens seen in SDS.
The significant regimen-related toxicity observed during HSCT in past reports of HSCT for SDS has led to recent interest in reduced-intensity preparative regimens that might ameliorate cardiac and pulmonary toxicities. Sauer et al.36 reported three patients with SDS and BM failure transplanted using a regimen consisting of fludarabine, treosulfan and melphalan. All three patients engrafted rapidly with 100% donor chimerism. Although two of the patients tolerated the regimen with minimal toxicity, one patient died on day 98 secondary to idiopathic pneumonitis syndrome. The first two patients had the common 183_184 TA to CT mutation in the SBDS gene, whereas the third patient who died carried a c.297-300delAAGA deletion, leading the authors to speculate on whether genotype is predictive of treatment-related toxicity.
Attempts have been made to predict the clinical phenotype from the genetic mutation but no correlation has been found thus far between the hematologic or skeletal manifestations and the genotype in the small numbers of patients that were studied.37, 38, 39 However, with widespread availability of genetic testing, it may be possible to collect this data in a prospective fashion to see if there is a correlation between genotype and outcome, particularly treatment-related toxicity. However, such efforts are likely to be limited due to the rarity of SDS.
In summary, our data indicate that transplantation of children with SDS using reduced-intensity conditioning is feasible and associated with modest morbidity. Increased understanding of the genetic and biochemical basis for this disorder, and prospective careful data collection will hopefully allow optimization of therapy for this complex group of patients.
References
Shwachman H, Diamond L, Oski F, Khaw K . The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr 1964; 65: 645–663.
Bodian M, Sheldon W, Lightwood R . Congenital hypoplasia of the exocrine pancreas. Acta Paediatr 1964; 53: 282–293.
Aggett PJ, Cavanagh NPC, Matthew DJ, Pincott JR, Sutcliffe J, Harries JT . Shwachman's syndrome: a review of 21 cases. Arch Dis Child 1980; 55: 331–347.
Ginzberg H, Shin J, Ellis L, Morrison J, Ip W, Dror Y et al. Shwachman syndrome: phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J Pediatr 1999; 135: 81–88.
Berrocal T, Simón MJ, al-Assir I, Prieto C, Pastor I, de Pablo L et al. Shwachman–Diamond syndrome: clinical, radiological and sonographic aspects. Pediatr Radiol 1995; 25: 289–292.
Robberecht E, Nachtegaele P, Van Rattinghe R, Afschrift M, Kunnen M, Verhaaren R . Pancreatic lipomatosis in the Shwachman–Diamond syndrome. Identification by sonography and CT-scan. Pediatr Radiol 1985; 15: 348–349.
Mack DR, Forstner GG, Wilschanski M, Freedman MH, Durie PR . Shwachman syndrome: exocrine pancreatic dysfunction and variable phenotypic expression. Gastroenterology 1996; 111: 1593–1602.
Dror Y, Ginzberg H, Dalal I, Cherepanov V, Downey G, Durie P et al. Immune function in patients with Shwachman–Diamond syndrome. Br J Haematol 2001; 114: 712–717.
Huijgens P, van der Veen E, Meijer S, Muntinghe O . Syndrome of Shwachman and leukemia. Scand J Haematol 1977; 18: 20–24.
Smith OP, Hann IM, Chessells JM, Reeves BR, Milla P . Haematological abnormalities in Shwachman–Diamond syndrome. Br J Haematol 1996; 94: 279–284.
Dokal I, Rule S, Chen F, Potter M, Goldman J . Adult onset of acute myeloid leukaemia (M6) in patients with Shwachman–Diamond syndrome. Br J Haematol 1997; 99: 171–173.
Dror Y, Freedman MH . Malignant myeloid transformation with isochromosome 7q in Shwachman–Diamond syndrome. Leukemia 1998; 12: 1591–1595.
Donadieu J, Leblanc T, Bader Meunier B, Barkaoui M, Fenneteau O, Bertrand Y, et al., French Severe Chronic Neutropenia Study Group. Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica 2005; 90: 45–53.
Alter BP, Young NS . Shwachman Diamond syndrome. In: Nathan DG, Orkin SH (eds). Nathan and Oski's Hematology of Infancy and Childhood, 5th edn, vol 1 WB Saunders: Philadelphia, 1997, pp 276–278.
Tsai PH, Sahdev I, Herry A, Lipton JM . Fatal cyclophosphamide-induced congestive heart failure in a 10-year-old boy with Shwachman–Diamond syndrome and severe bone marrow failure treated with allogeneic bone marrow transplantation. Am J Pediatr Hematol Oncol 1990; 12: 472–476.
Okcu F, Roberts WM, Chan KW . Bone marrow transplantation in Shwachman–Diamond syndrome: report of two cases and review of the literature. Bone Marrow Transplant 1998; 21: 849–851.
Cesaro S, Oneto R, Messina C, Gibson BE, Buzyn A, Steward C, et al., EBMT Severe Aplastic Anaemia and Paediatric Diseases Working Party. Haematopoietic stem cell transplantation for Shwachman–Diamond disease: a study from the European Group for blood and marrow transplantation. Br J Haematol 2005; 131: 231–236.
Donadieu J, Michel G, Merlin E, Bordigoni P, Monteux B, Beaupain B et al. Hematopoietic stem cell transplantation for Shwachman–Diamond syndrome: experience of the French neutropenia registry. Bone Marrow Transplant 2005; 36: 787–792.
Barrios N, Kirkpatrick D, Regueira O, Wuttke B, McNeil J, Humbert J . Bone marrow transplant in Shwachman Diamond syndrome. Br J Haematol 1991; 79: 337–338.
Smith OP, Chan MY, Evans J, Veys P . Shwachman–Diamond syndrome and matched unrelated donor BMT. Bone Marrow Transplant 1995; 16: 717–718.
Arseniev L, Diedrich H, Link H . Allogeneic bone marrow transplantation in a patient with ShwachmanDiamond syndrome. Ann Hematol 1996; 72: 83–84.
Faber J, Lauener R, Wick F, Betts D, Filgueira L, Seger RA et al. Shwachman–Diamond syndrome: early bone marrow transplantation in a high risk patient and new clues to pathogenesis. Eur J Pediatr 1999; 158: 995–1000.
Fleitz J, Rumelhart S, Goldman F, Ambruso D, Sokol RJ, Pacini D et al. Successful allogeneic hematopoietic stem cell transplantation (HSCT) for Shwachman–Diamond syndrome. Bone Marrow Transplant 2002; 29: 75–79.
Cesaro S, Guariso G, Calore E, Gazzola MV, Destro R, Varotto S et al. Successful unrelated bone marrow transplantation for Shwachman–Diamond syndrome. Bone Marrow Transplant 2001; 27: 97–99.
Hsu JW, Vogelsang G, Jones RJ, Brodsky RA . Bone marrow transplantation in Shwachman–Diamond syndrome. Bone Marrow Transplant 2002; 30: 255–258.
Park SY, Chae MB, Kwack YG, Lee MH, Kim I, Kim YS et al. Allogeneic bone marrow transplantation in Shwachman–Diamond syndrome with malignant myeloid transformation. A case report. Korean J Intern Med 2002; 17: 204–206.
Cunningham J, Sales M, Pearce A, Howard J, Stallings R, Telford N et al. Does isochromosome 7q mandate bone marrow transplant in children with Shwachman–Diamond syndrome? Br J Haematol 2002; 119: 1062–1069.
Mitsui T, Kawakami T, Sendo D, Katsuura M, Shimizu Y, Hayasaka K . Successful unrelated donor bone marrow transplantation for Shwachman–Diamond syndrome with leukemia. Int J Hematol 2004; 79: 189–192.
Grewal SS, Barker JN, Davies SM, Wagner JE . Unrelated donor hematopoietic cell transplantation: marrow or umbilical cord blood? Blood 2003; 101: 4233–4244. E-pub 9 January 2003.
Vibhakar R, Radhi M, Rumelhart S, Tatman D, Goldman F . Successful unrelated umbilical cord blood transplantation in children with Shwachman–Diamond syndrome. Bone Marrow Transplant 2005; 36: 855–861.
Savilahti E, Rapola J . Frequent myocardial lesions in Shwachman syndrome. Eight fatal cases among 16 Finnish patients. Acta Paediatr Scand 1984; 73: 642–651.
Ferry C, Ouachée M, Leblanc T, Michel G, Notz-Carrére A, Tabrizi R et al. Hematopoietic stem cell transplantation in severe congenital neutropenia: experience of the French SCN register. Bone Marrow Transplant 2005; 35: 45–50.
Donadieu J, Stephan JL, Blanche S, Cavazzana-Calvo M, Baruchel A, Herbelin C et al. Treatment of juvenile chronic myelomonocytic leukemia by allogeneic bone marrow transplantation. Bone Marrow Transplant 1994; 13: 777–782.
Dror Y, Freedman MH . Shwachman–Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood 2001; 97: 3011–3016.
Thornley I, Dror Y, Sung L, Wynn RF, Freedman MH . Abnormal telomere shortening in leucocytes of children with Shwachman–Diamond syndrome. Br J Haematol 2002; 117: 189–192.
Sauer M, Zeidler C, Meissner B, Rehe K, Hanke A, Welte K et al. Substitution of cyclophosphamide and busulfan by fludarabine, treosulfan and melphalan in a preparative regimen for children and adolescents with Shwachman–Diamond syndrome. Bone Marrow Transplant 2007; 39: 143–147.
Kuijpers TW, Alders M, Tool AT, Mellink C, Roos D, Hennekam RC . Hematologic abnormalities in Shwachman Diamond syndrome: lack of genotype–phenotype relationship. Blood 2005; 106: 356–361.
Mäkitie O, Ellis L, Durie PR, Morrison JA, Sochett EB, Rommens JM et al. Skeletal phenotype in patients with Shwachman–Diamond syndrome and mutations in SBDS. Clin Genet 2004; 65: 101–112.
Kawakami T, Mitsui T, Kanai M, Shirahata E, Sendo D, Kanno M et al. Genetic analysis of Shwachman–Diamond syndrome: phenotypic heterogeneity in patients carrying identical SBDS mutations. Tohoku J Exp Med 2005; 206: 253–259.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bhatla, D., Davies, S., Shenoy, S. et al. Reduced-intensity conditioning is effective and safe for transplantation of patients with Shwachman–Diamond syndrome. Bone Marrow Transplant 42, 159–165 (2008). https://doi.org/10.1038/bmt.2008.151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bmt.2008.151
Keywords
This article is cited by
-
Approach Toward Germline Predisposition Syndromes in Patients with Hematologic Malignancies
Current Hematologic Malignancy Reports (2022)
-
Long-term outcome after allogeneic hematopoietic stem cell transplantation for Shwachman–Diamond syndrome: a retrospective analysis and a review of the literature by the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation (SAAWP-EBMT)
Bone Marrow Transplantation (2020)
-
First experience of hematopoietic stem cell transplantation treatment of Shwachman–Diamond syndrome using unaffected HLA–matched sibling donor produced through preimplantation HLA typing
Bone Marrow Transplantation (2017)
-
Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes
Bone Marrow Transplantation (2015)
-
Congenital neutropenia: diagnosis, molecular bases and patient management
Orphanet Journal of Rare Diseases (2011)
