
Abstract
The liver is a tolerogenic organ with exquisite mechanisms of immune regulation that ensure upkeep of local and systemic immune tolerance to self and foreign antigens, but that is also able to mount effective immune responses against pathogens. The immune privilege of liver allografts was recognized first in pigs in spite of major histo-compatibility complex mismatch, and termed the “liver tolerance effect”. Furthermore, liver transplants are spontaneously accepted with only low-dose immunosuppression, and induce tolerance for non-hepatic co-transplanted allografts of the same donor. Although this immunotolerogenic environment is favorable in the setting of organ transplantation, it is detrimental in chronic infectious liver diseases like hepatitis B or C, malaria, schistosomiasis or tumorigenesis, leading to pathogen persistence and weak anti-tumor effects. The liver is a primary site of T-cell activation, but it elicits poor or incomplete activation of T cells, leading to their abortive activation, exhaustion, suppression of their effector function and early death. This is exploited by pathogens and can impair pathogen control and clearance or allow tumor growth. Hepatic priming of T cells is mediated by a number of local conventional and nonconventional antigen-presenting cells (APCs), which promote tolerance by immune deviation, induction of T-cell anergy or apoptosis, and generating and expanding regulatory T cells. This review will focus on the communication between classical and nonclassical APCs and lymphocytes in the liver in tolerance induction and will discuss recent insights into the role of innate lymphocytes in this process.
Similar content being viewed by others


The liver immune system
Immune regulation in the liver is for a large part controlled by unique populations of conventional but also unconventional APCs that can react in a spatiotemporally regulated way that enables fine-tuned modulation of local and systemic tolerance and immunity.1, 2, 3, 4, 5 In contrast to conventional APCs, like dendritic cells (DCs), these unconventional APC populations consist of Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs) and even hepatocytes that express only low levels of major histo-compatibility complex (MHC)-I/MHC-II and co-stimulatory molecules in the steady-state hepatic environment.2
Furthermore, the quantitative and qualitative composition of immune cells in the liver differs markedly from secondary lymphoid organs, like lymph nodes, spleen or peripheral blood. The CD8+/CD4+ ratio (3.5:1) of hepatic T cells is reversed compared with a ratio of 1:2 for CD8+/CD4+ cells found in peripheral blood, lymph nodes and spleen. There is an increased proportion of CD3+CD4+CD8+ and CD3+CD4−CD8− T cells in the liver, 15% of T cells express the γδ−TCR (T-cell receptor, compared with 2.7% in spleen), up to 50% of the liver-resident lymphocytes are natural killer (NK) cells in humans and the liver supports an unusually high frequency of natural killer T (NKT) cells.3, 5, 6, 7 In the liver, the majority of DCs display an immature phenotype, which in contrast to DCs in secondary lymphoid organs induces tolerogenic deviation rather than immunity accompanied by high IL-10 and low IL-12 secretion.2 IL-10, which is also produced by KCs, and regulatory T cells (Tregs), has a pivotal, non-redundant role in controlling hepatic inflammation: IL-10 deficiency or depletion exacerbates hepatic immune-mediated liver damage and abrogates tolerance induction.8, 9, 10
The liver tolerance effect
The liver tolerance effect was first recognized by Calne in 1969, when porcine liver allo-transplantation protected secondary transplanted organs from the same donor from rejection.11 The liver is indispensable for the maintenance of oral tolerance toward nutrients, gut-derived bacterial metabolites, lipopolysaccharides from the cell walls of Gram-negative bacteria and cellular debris that enters the liver via the portal vein from the intestine.2, 12 Also, metabolic transformation in the liver parenchyma leads to intrahepatic generation of food antigens and neo-antigens/neo-adducts that challenge immune ignorance.2 The feeding of antigens while bypassing the hepatic circulation by a portacaval shunt impairs development of oral tolerance. In agreement with the liver’s function as an inductor of systemic tolerance, the injection of donor cells into the vein before transplantation of multiple organs from the same donor yields extended graft survival and maintenance of microchimerism.13, 14, 15 Regulatory immune cells and conventional and unconventional APCs (T cells, DCs, KCs, LSECs) ensure that tolerance is maintained under homeostatic conditions, but still, potent ad hoc immune responses to combat infections can be initiated. The downstream effectors of the conventional and unconventional APCs are Tregs, and where appropriate, their action and function is contextualized.
Antigen-presenting cells in the liver and their function in tolerance
Liver sinusoidal endothelial cells
The blood passing through the liver enters the hepatic circulation via the sinusoids. The sinusoids are lined by highly specialized LSECs that form a physical barrier between the intraluminal space and the subendothelial space of Dissé. Here, the HSCs are located (Figure 1). LSECs interact intensively with passenger leukocytes (Figure 2) and are involved in hepatic leukocyte recruitment.

Schematic representation of the microanatomy of the liver sinusoids and their cellular composition. The hepatocytes are separated from the sinusoidal blood flow by the liver sinusoidal LSECs that create the Space of Dissé and shield the hepatocytes from sinusoidal blood flow. Between the LSECs and the hepatocytes, hepatic HSCs are interspersed. In the sinusoidal lumen, KCs and passenger leukocytes are located. Note that T cells can form intimate contacts with microvilli from hepatocytes, but also LSECs or KCs, which enables priming of T cells in the liver. HSCs, hepatic stellate cells; KCs, Kupffer cells; LSECs, liver sinusoidal endothelial cells.

Electron microscopic analyses of liver sinusoids. (a) Transmission electron microscopic image of a lymphocyte (L) within the intrahepatic sinusoidal lumen (S); original magnification × 12 000; e=LSEC; H=hepatocyte. (b) Intrasinusoidal leukocyte, scanning microscopic image (s.e.m.); note that its cytoplasmic extensions exhibit a similar diameter compared with the sinusoidal fenestrations; original magnification × 10 000. (c) s.e.m. of microvilli from a hepatocyte (H) protruding into the sinusoidal lumen (S); original magnification × 40 000. (d) s.e.m. of LSECs; original magnification × 15 000. (e) Higher magnification s.e.m. picture, showing a liver sieve and visibility of the hepatocyte’s microvilli underneath the endothelial layer; original magnification × 20 000. Note the absence of the basal lamina between the LSECs and hepatocytes. Reproduced from Warren et al.126 with permission.
In contrast to canonical leukocyte recruitment by non-hepatic vasculature via selectin-ligand-selectin-mediated tethering, leukocyte recruitment in the sinusoids relies on the constitutive expression of CD54 (ICAM-1), CD106 (VCAM-1), vascular adhesion protein-1 (VAP-1), CD44 and hyaluronan.16, 17 Of note, hepatic neutrophil adhesion in the systemic inflammatory response syndrome (SIRS) and endoxemia, is selectin- and integrin-β2-independent. Instead, it depends on hyaluronic acid-serum-derived hyaluronan-associated protein-(SHAP)-complex and CD44 interactions between LSECs that express hyaluronan and bind SHAP, and CD44+ neutrophils.18, 19 This process is coordinated independently by toll-like receptor 4 (TLR4) activation in LSECs and KCs,20 leading to increased production of tumor-necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β), which promotes endothelial hyaluronic acid expression and facilitate adhesion of activated CD44+ T cells, NK cells and myeloid cells.21 Under homeostatic conditions, TLR4 signaling, mediated by the constant flow of LPS arriving from the gut, is pivotal for the efficient antigen-independent entrapment and elimination of activated CD8+ T cells.22, 23
LSECs do not only regulate immune responses via selective recruitment of leukocytes, they also interact and activate both naïve CD4+ and CD8+ T cells. The outcome of such interaction is tolerance rather than immunity. As a major hepatic scavenger cell population, LSECs are as efficient in antigen uptake and processing as DCs.24 Expression of the endocytic mannose receptor, scavenger receptor, Fcγ receptor IIb and lymph node sinusoidal endothelial cell C-type lectin (LSECtin)25, 26 ensures highly efficient receptor-mediated endocytosis in LSEC. Although the expression of the mannose receptor, MHC class II and the co-stimulatory molecules CD80/CD86 on LSECs are kept at low levels due to high local concentrations of the immunosuppressive cytokine IL-10.27 LSECs are able to induce cytokine production in naive CD4+ T cells, and the resulting activated CD4+ T cells do not posses Th1 effector function; due to lack of IL-12 during priming, they convert into IL-10 and IL-4 producing cells,28 that may have suppressive properties without expressing classical Treg markers like Foxp3 and CD25.29 In addition to the activation of naive CD4 T cells, LSECs exert immune modulating effects on various effector CD4 T-cell populations. Stimulation of Th1 and Th17 cells by LSECs inhibits their secretion of interferon-γ (IFN-γ) and IL-17 although their proliferation is not affected.30 This immune suppressive and tolerogenic effect is mediated by LSEC-derived IL-10 and enhanced PD-1/PD-L1 signaling.30 In addition, LSECs can promote expansion of IL-4 producing Th2 cells.31 The tolerogenic function of LSECs was also highlighted recently by the observation that nanoparticle-based autoantigen targeting to LSECs in a mouse model of experimental autoimmune encepahalomyelitis (EAE) produced protection from EAE, even if the disease was already established.32 Hence, the induction of antigen-specific Tregs by LSECs provides a promising approach to control autoimmunity. Also, LSECs utilize the Notch-signaling pathway to induce IL-10 expression in Th1 cells with acquisition of in vivo suppressive function.33 LSECs constitutively express ligands of the delta-like and Jagged family and interaction with LSECs triggers expression of the Notch target genes hes-1 and deltex-1 in Th1 cells, which suppresses their pro-inflammatory properties and constitutes a self-limiting, anti-inflammatory pathway that can prevent autoimmunity.33, 34, 35
Another particular effect that is elicited by LSECs during their interaction with CD4+ T cells, is the imprinting of a gut tropism phenotype, that is, CD4+ T cells acquire integrin α4β7 and CC-chemokine receptor 9 (CCR9) expression.36 This LSEC-induced expression of gut homing molecules in CD4+ T cells is dependent on all-trans retinoic acid (RA).37 LSECs express the enzymes retinaldehyde dehydrogenase (RALDH1) 1 and 4 for conversion of vitamin A into all-trans RA.36 RA is supplied by HSCs, in direct proximity to LSECs in the space of Dissé. CCR9 as well as α4β7 integrin are key determinants in lymphocyte homing to the gut, where nearly all intestinal intraepithelial and lamina propria lymphocytes express CCR9 and α4β7 integrin.38 α4β7 integrin+ T cells bind to mucosal vascular addressin cell adhesion molecule-1 (MAdCAM-1)-expressing endothelium in the gut, and are further attracted by the CCR9-ligand CCL25 produced by intestinal epithelial cells.39 This imprinting suggests that regulatory or immune-modulated CD4+ T cells migrate to the gut after their encounter with LSECs in the liver, and thus contribute to the upkeep of hepatic homeostasis. During primary sclerosing cholangitis (PSC) or in patients with inflammatory bowel disease, however, both CCL25 and MAdCAM-1 can be expressed ectopically by LSECs themselves, thus extending the action radius of pathogenic, possibly auto-reactive T cells to the liver.38 This is especially interesting with respect to the emerging importance of the gut–liver axis in systemic immune regulation and probably in the diversion of hepatic tolerance toward (auto-)immune diseases (see below). In contrast, LSECs interaction with CD8+ T cells does not induce gut tropism, but rather results in the deletion of effector cells and skewing of naive CD8+ T cells toward a particular differentiation state resembling central memory T cells.40, 41 Similar to DCs, LSECs can efficiently cross-present antigens to CD8+ T cells. However, contact of naive CD8+ cells with LSECs upregulated PD-L1 expression on LSECs, but not of any co-stimulatory molecules. This effect was specifically observed in LSECs, and not in DCs, and resulted in a matured LSEC phenotype (PD-L1high/CD80/86low) that is required for the generation of LSEC-primed CD8+ T cells.42 The priming of naive CD8+ T cells by LSECs that cross-present low abundant circulating antigens in the absence of inflammation results in a refractory state after initial expansion.42, 43, 44 This is characterized by reduction in inflammatory cytokine production, such as IFN-γ, but also depends on the Ag concentration and duration of Ag exposure. At low Ag concentrations, initial T-cell proliferation and stimulation is observed, followed by attrition of cytokine release.24, 45 This unresponsiveness is controlled by the co-inhibitory receptor programmed cell-death-1 (PD-1) and its ligand PD-L1, where PD-L1 expression on LSECs represses IL-2 production in CD8+ T cells. After sustained co-inhibitory signaling over time, development of LSEC-primed T cells cannot be overridden by additional CD28 co-stimulation.45 High antigen load, however, induces the development of cytolytic effector cells,24 most likely due to such high IL-2 production after strong TCR signaling leading to upregulation of CD25 and initiation of effector T-cell development.
Classically LSEC-primed CD8+ T cells acquire a memory-like phenotype and, unlike their CD4+ counterparts, they express the lymphoid addressin CD62L and do not migrate to the gut, but return to the secondary lymphoid organs.46 After reactivation, they elicit protective immunity in infectious inflammation as CTLs, and produce IFN-γ and eradicate pathogens.47 In this way potential pathogens that gained access to the liver under non-inflammatory conditions, may still be eradicated in the case of an infection.
Not only can LSECs skew CD4+ and CD8+ T-cell responses directly, but they can suppress the activity of neighboring APCs, such as DCs, that would otherwise be capable of inducing T-cell immunity.48, 49 This so-called vetoing function exerted by LSECs depends on physical contact between the APCs and the LSECs, which markedly reduced co-stimulatory molecule expression and IL-12 secretion by DCs, leading to stunted CD8+ T-cell priming.48 The method of vetoing to avoid sufficient T-cell priming in the liver seems a general one, as hepatic HSCs also contact-dependently prevent CD8+ T-cell activation by non-HSC APCs.49
In addition to their tolerogenic properties, however, LSECs can fulfill pro-inflammatory roles for example in sepsis, where upregulation of PD-L1 on LSECs and PD-1 on KCs leads to exacerbation of endothelial damage by increased leakage and edema.50 This PD-1/PD-L1-dependent interaction between KCs and LSECs impedes endothelial function, and therefore promotes acute-on liver failure in sepsis.50
Tolerogenic function of KCs
In addition to LSECs, the liver-resident macrophages, the KCs, have a non-redundant role in antigen uptake, tolerance induction and pathogen clearance. Central to their macrophage function, they phagocytose particulate material, and eliminate dead-cell debris and pathogens. KCs patrol the sinusoids but do not actively migrate into the liver parenchyma to scavenge pathogens. They constitute 80% of the whole body macrophage population, and ~35% of the non-parenchymal liver cells.12 KCs express the macrophage markers CD68 and F4/80 as well as the TLRs 2, 3 and 4, and low to intermediate levels of CD11b and Ly6C.2, 51 KCs hold a principal role as sentinels purging pathogens and pathogen-derived products. They take up significant amounts of bacteria or endotoxins, but under steady-state conditions, they do not induce inflammation but rather diversion towards tolerance. As a result, depletion of KCs, but not splenectomy, results in fatal outcome of infections with Listeria monocytogenes, Brucella burgdorferi or Staphylococcus aureus in mice, accompanied with enhanced bacterial dissemination and reduction in hepatic bacterial uptake.52, 53, 54 This is a result of impaired bacterial clearance, which could link the observation that patients with advanced cirrhosis, end-stage liver disease and acute liver failure are prone to acquire infections and develop sepsis or a SIRS55, 56 with impaired KC function in those diseases. Under physiological conditions, KCs are constantly exposed to LPS (ranging from 100 pg/ml to 1 ng/ml) in the portal venous blood that is derived from the splanchnic circulation.28 In response to LPS or endotoxins KCs can produce both anti-inflammatory and immunosuppressive factors, such as IL-10, nitric oxide, transforming growth factor-β (TGF-β), or the arachidonic acid metabolite prostaglandin E2 (PGE2) and pro-inflammatory cytokines, especially TNF-α, IL-1β and IL-6, illustrating their dichotomous role in liver defense and inflammation.57, 58, 59, 60
In their physiological steady state, KCs exhibit an anti-inflammatory M2-like macrophage polarization, which is known from the context of helminthic infections or allergies.61 Although M2-like polarization is anti-inflammatory in the context of prominent M1-polarized inflammation in acute tissue injury or bacterial infections, it can still contribute to propagation of inflammation in allergies, helminth infections or sustain tumor growth.61 Hence, looking at M2-polarized inflammatory responses with relation to tolerance induction, it should rather be considered as a counterpart to type 1-based inflammation, and with its contribution to tolerance induction depending on the local cytokine micromilieu. LPS and TLR4 signaling promote M2-like polarization of KCs, which promotes endotoxin tolerance and protects from liver injury by preventing pro-inflammatory cytokine production and the attraction of Treg to the liver.62 Of note, M2-polarized KCs apparently can also promote selective apoptosis of classical, M1-polarized KCs in alcoholic hepatosteatitis (ASH), a process that involves the paracrine induction of arginase, an M2 marker, via IL-10 secretion in the inflammatory local microenvironment,63 thereby acting to restore homeostasis. Apart from these anti-inflammatory effects, antigen engulfment by KCs can directly contribute to the induction of local but also systemic tolerance: KCs can elicit CD4+ T-cell arrest of passenger leukocytes and T-cell proliferation, and the secretion of IL-10 induces expansion and activation of CD4+CD25+Foxp3+ Treg.64 KCs are able to present engulfed antigens to effector cells to prevent intrahepatic dissemination of B. burgdoferi. This effect appears to be dependent on the local production of reactive oxygen species, and ligation of pattern recognition receptors with their cognate ligands, bacterial antigens and cell-wall products, presence of double-stranded RNA and signals provided by the local microenvironment.64, 65 Furthermore, it was indicated that ingestion of Borrelia burgdorferi by KCs leads to aggregation and activation of iNKT cells that are in close contact with the KCs via KC-iNKT contact formation via CD1d.53
Besides these antigen-presenting capacities, KCs can express both immune-modulatory PD-L1 and PD-1: PD-L1 expression on KCs was demonstrated in the context of hepatocellular carcinomas, leading to reduction of PD-1+ T-cell effector function;66 similarly, PD-L1 is upregulated on KCs in autoimmune hepatitis,67 and in hepatitis B virus (HBV) infection, modulates PD-L1–PD-1 interactions between KCs, LSECs and effector T cells differentially modulates disease activity.68 In the chronic stage of HBV, the reduction of PD-L1 expression by KCs may reduce PD-1-mediated effector cell exhaustion, which aids to control viral replication.68 Intrahepatic PD-1 and PD-L1 levels correlate with the degree of inflammation in HBV.69 Therefore, first therapeutical attempts to block PD-L1 expression in hepatic myeloid cells are made: using lipoid nanoparticle-assisted delivery of PD-L1 siRNAs in the model of murine cytomegaly virus infection, a successful reduction of immune suppression via the PD-1/PD-L1 axis was observed.70 Here, increased NK and CD8+ T-cell intrahepatic accumulation, increase in effector function, and enhanced CD8+ T-cell-mediated viral clearance were observed. Moreover, Fas-L expression on KCs that is stimulated by gut-derived endotoxins can lead to T-cell apoptosis in vitro.71 This process is accompanied by KC-derived reactive-oxygen production that triggers a transcriptional program to enhance Fas-L expression and thus enable induction of apoptosis in permissive cells.71 Hence, KCs per se constitute an immune regulatory cell population that is involved in the direct elimination of activated T cells in the liver. Previously, it has been described that IFN-γ induces upregulation of Fas-L on KCs, and can lead to elimination of activated T cells in rats.72 Also, in liver transplantation, graft-derived KCs of accepted grafts were shown to upregulate Fas-L, as well as capable of regulating IL-10 and TGF-β production allogeneic mixed lymphocyte reactions in vitro.73, 74 These data indicate that KCs have a concrete regulatory role in the modulation of liver tolerance.
A widely used model to study tolerance induction and immune-mediated hepatitis in mice is the Concanavalin A hepatitis model.75 ConA hepatitis resembles autoimmune hepatitis as ConA-mediated liver injury is mediated by CD4+ T cells, NKT cells and KC.9, 75, 76, 77, 78, 79 Cytokines inducing liver damage are TNF-α, IFN-γ, IL-12 and IL-18.80, 81, 82 IL-10 is a powerful, immune suppressive counter regulator that exerts tolerogenic and hepatoprotective effects in this model, and Treg and KC-derived IL-10 are crucial for the development of tolerance toward ConA after repeated injection.8, 9 Furthermore, Heymann et al. recently demonstrated that KCs are central in mediating T-cell arrest and induction of tolerance to scavenged particulate material by expansion of Foxp3+CD25+IL-10+ Tregs.64 This particle-associated tolerance induction also protected mice from extrahepatic tissue damage in experimental models of nephritis.64 Importantly, the role of hepatic tolerance in the development of glomerulonephritis was highlighted, when liver-borne protection failed in chronic liver injury and fibrosis,64 where KCs lost their “tolerogenic signature”,64 and subsequently, antigen-specific CD4+ T cells remained immunogenic and were not converted into Tregs.64
DCs
Another cell population with scavenger function are DCs; however, liver-resident DCs have low endocytotic capacities and poorly stimulate T cells, but on the other hand, secrete high levels of IL-10, which discriminates them from DCs in extrahepatic tissues.2, 83, 84 Hepatic DCs exhibit an immature phenotype with low MHC-II and barely detectable co-stimulatory molecule expression. Also, in mice, four DC subsets can be identified: the two classical DC subsets, CD103+ and CD103- DCs, the plasmacytoid B220+ DCs and hepatic DC precursors.2, 85, 86 The liver is enriched in pDCs. Hepatic DCs can mediate immune tolerance either by direct T-cell depletion, inducing T-cell anergy or inducing and expanding Treg.84, 87 Hepatic DCs express higher levels of IL-10 but low levels of IL-12, and can exert tolerogenic properties via production of anti-inflammatory PGE2, which upregulates the immune suppressive metabolite indoleamine dioxygenase in DCs and enhances their IL-10 secretion and capacity to induce Treg.2, 83, 84, 88 Although abundantly present, hepatic pDCs secrete lower levels of type I interferon after CpG stimulation when compared with their splenic counterparts.89 This could be due to their expression of high levels of the NLR-family member nucleotide-binding oligomerization domain 2 (NOD2), which is known to down-modulate responses to microbial products and dampen TLR signaling.89, 90 Engagement of NOD2 by its ligand interferes with TLR4 and TLR9 signaling in pDCs, resulting in reduced secretion of pro-inflammatory cytokines (IL-6, IL12p70, TNF-α and IFN-γ). Simultaneously, NOD2 signaling increases PD-L1 expression on pDCs resulting in their reduced capacity to stimulate T-cell proliferation.89 Physical interaction between Tregs and DCs appears to be required to impose cell contact-dependent suppression by CTLA-4 and LFA-1, whereby Tregs actively inhibit DC maturation (that is, upregulation of CD80/CD86), even in the presence of DC-maturing cytokines (GM-CSF, TNF-α, IFN-γ, type I IFNs) and LPS.91 This has also been observed in hepatocellular carcinomas, where hepatic CD4+CD25+ Treg induce downregulation of CD80/86, and reduce TNF-α and IL-12 secretion by DCs and thus promote a tolerogenic phenotype.92 In acute colitis provoked by dextran sodium sulfate, reprogramming of hepatic DCs induces a Th1 response, and compromises their tolerogenic function. In this model, liver inflammation is accompanied by expansion of mononuclear cells in the liver, where classical DCs increase, as well as levels of IFN-γ and TNF-α, and the number of potentially Treg-inducing pDCs declines, leading to liver inflammation.93 Similarly, in ConA-induced immune-mediated hepatitis, tolerogenic CCR9+ pDCs are decreasing, while hepatic infiltration of CCR9+ macrophages increases and exacerbates liver injury.94 CCR9 has been identified as a marker for tolerogenic pDCs that mediate immunosuppression in acute graft-versus-host disease.95 When Treg-inducing capacities of CCR9+ and CCR9- pDCs are compared, CCR9- pDCs were significantly less effective in inducing regulatory T cells and thus inhibiting antigen-specific immune responses. In addition to their maturation and activation status, the capacity of hepatic DCs to present antigen also depends on their lipid contents; in murine and human liver, a “lipid-based dichotomy”131 was found specific to hepatic DCs.96 Interestingly, DCs carrying a high lipid load are pro-inflammatory, secreted higher levels of pro-inflammatory cytokines (TNF-α, IFN-γ, IL-2 and IL-6) and could activate T cells, NK cells and NKT cells. They also expressed higher levels of CD54, CD40, CD80/CD86, as well as CD1d, when compared with DCs with low lipid content. Congruently, hepatic DCs with a low lipid load induced Treg, anergy to cancer and oral tolerance.96 This observation points toward an interesting link to fatty liver disease and non-alcoholic steatohepatitis (NASH)/ASH.
HSCs
HSCs or Ito cells are located in the abluminal sinusoidal space of Dissé. In adult mice, they are derived from bone-marrow precursors and constitute ~5–8% of the non-parenchymal hepatic cell population.97, 98 HSCs store 80% of the total body vitamin A in intracytoplasmic lipid droplets, and regulate sinusoidal blood flow according to their functional and structural kinship with pericytes.99, 100 Upon their activation, for example, after infection with hepatitis virus C, HSCs metabolize vitamin A and all-trans RA, and differentiate into extracellular matrix-producing myofibroblasts that are central effectors in hepatic fibrosis and cirrhosis.101, 102 HSCs express antigen-presenting molecules for both peptides and lipids (MHC-I/MHC-II, CD1c, CD1d), as well as co-stimulatory molecules, such as CD86. Although they are capable of antigen (cross)presentation, it is at much lower efficiency compared with other APCs.2 In line with this, HSCs can interact with T cells and induce CD4+ T-cell effector responses.103 In addition, they also function as immunological bystanders that produce TGF-β and RA for induction of Treg.103, 104, 105, 106 As already described for LSECs, HSCs also possess the capability to veto priming of naive CD8+ T cells in a cell–cell contact and CD54-dependent fashion.49 After contact with activated T cells or exposure to IFN-γ, HSCs induce expression of PD-L1, which subsequently promotes attenuation of T-cell responses by increasing apoptosis.107
Although various reports support cell-autonomous antigen-presenting function in HSCs, LSECs, KCs and also hepatocytes, a recent report suggests that intensive exchange of MHC class I molecules from HSCs to liver DCs and KCs, but also from HSCs to LSECs by a mechanism called trogocytosis.108 Trogocytosis describes a mechanism in which membrane fragments that contain antigen-presenting molecules, or co-stimulatory or co-inhibitory molecules are transferred between cells.109 Trogocytosis was shown to occur between T cells and APCs within the immunological synapse to modulate inhibition or amplification of immune responses.110 Trogocytosis apparently occurs between HSCs and liver DCs and KCs, prompting speculations whether in the liver this mechanism is used to facilitate antigen presentation and modulation of immune responses that result in hepatic tolerance or inflammation. Also, LSECs were shown to acquire MHC class I molecules from HSCs, which may function to increase the visibility of antigens present in HSCs to CD8+ T cells passing through the sinusoids.108, 111
Hepatocytes
Hepatocytes exert antigen-presenting properties, which are unique for parenchymal cells.112, 113 Hepatocytes express low levels of MHC-I molecules under homeostatic conditions, which may activate CD8+ T cells and contribute to viral clearance and allograft rejection. In an allogenic transplantation model, hepatocytes induce initial proliferation and transient activation of naïve CD8+ T cells, but fail to promote long-term survival, leading to early elimination of allo-reactive CD8+ T cells.114
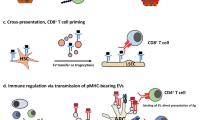
These two scenarios demonstrate that the thresholds of antigen presentation by hepatocytes matter in the upkeep of the balance between a tolerogenic versus an effector immune response: once the initial hepatocellular antigen load is low, an effector CD8+ T-cell response is initiated and contrary, if the antigens are presented in high abundance, CD8+ T-cell exhaustion and silence are the consequence, and T cells express high levels of PD-1,47, 115 (Figure 3). Furthermore, autoreactive CD8+ T cells can actively invade hepatocytes and are subsequently degraded in the lysosomal compartment, a process called emperilopoiesis, which is believed to contribute to hepatic tolerance (Figure 3).116

Liver-mediated T-cell priming and hepatocyte-T-cell interactions as tools for tolerance induction. (a) and (b) depending on the antigen load and the density of ligands presented by hepatocytes, priming of T cells can result in either activation and expansion, and initiation of an effector response, when the antigen density is low, or (a) T-cell anergy and exhaustion are induced when the antigen-load is high (b). (c) An alternate mechanism to induce peripheral tolerance by hepatocyte-T-cell interaction is emperiopolesis, where autoreactive T cells are invading hepatocytes and are thus eliminated in the hepatocytic lysosomal compartments.116
In addition, hepatocytes can express low levels of MHC-II molecules, which can be further upregulated under inflammatory conditions in chronic liver disease, and after stimulation with IFN-γ.117, 118, 119 Hepatocytes from injured or inflamed livers can actively induce IL-10-producing, CD4+ T cells in a Notch signaling-dependent fashion117 (Figure 4).

Differences in the outcome of T-cell priming between conventional APCs in the lymph nodes and nonconventional APCs, such as hepatocytes. (a) In secondary lymphoid organs, DC-mediated T-cell priming results in T-cell expansion and activation of their effector function. In the case of cytolytic T cells, naive CD8+ T cells expand after antigen-specific stimulation, exhibit prolonged survival indicated by up-regulated bcl-xL expression, express inflammatory cytokines such as IFN-γ and CD107a (LAMP1 as markers of their cytotoxic activity. Bcl-xL is a member of the Bcl-2 family of apoptosis regulators and enhances apoptosis protection and prolongs survival.200 (b) Naive CD8+ T-cell priming in the liver by hepatocytes leads to death by neglect, a mechanism leading to premature death of T cells. This demonstrates that hepatocytes can induce antigen-specific activation and proliferation of naive CD8+ T cells independent of co-stimulatory signals; premature cell death of liver-primed T cells can be prevented by CD28 cross-linking.201 Death by neglect is a pivotal mechanism to induce peripheral tolerance to hepatic antigen recognition. (c) After ConA treatment, TGF-β and IL-10 are produced, and induce tolerance via Foxp3+ Treg induction; Treg-derived IL-10 and TGF-β also contribute to the upkeep of tolerance;9 (d) Hepatocytes express the Notch ligand Jagged-1 in inflammation after induction of ConA hepatitis;118 CXCR3+ Tregs are recruited to the liver by enhanced CXCL9/CXCL10 expression in ConA hepatitis, and are converted into IL-10+Foxp3+ Tregs, which also express Notch1.130 This conversion depends on the presence of IFN-γ. Bcl-2, B-cell lymphoma-2.
In this way, hepatocytes may help to return to an immunological equilibrium after liver injury. More interestingly, hepatocytes from healthy, non-injured livers but also injured livers induce Foxp3 positive Tregs, again in a Notch-dependent fashion as Treg induction was abolished by γ-secretase inhibition, which prevents Notch cleavage at the cell surface.118 In addition, TGF-β, which can be abundantly produced by KCs, significantly enhances hepatocyte-induced generation of Foxp3+ Tregs.118 Thus, usage of the Notch-signaling pathway to suppress inflammatory cells or induce regulatory cells seems a more general feature of liver parenchymal cells, which may open up therapeutic possibilities to modulate the liver immune system toward tolerance or immunity.
TREGS
The tolerogenic potential of the liver includes conversion of conventional T cells to Tregs. Tregs, that express high levels of the high-affinity IL-2R alpha chain (CD25) as well as the forkhead-winged helix transcription factor, Foxp3+, are instrumental in the upkeep of peripheral tolerance toward auto-antigens and in liver infection:120, 121 in neonatally thymectomized Pd1−/− mice that lack Tregs, AIH was suppressed by Treg transfer from wild-type mice.121 In this model, production of autoantibodies (for example, ANA, anti-nuclear antibody and antibodies against hepatic antigens) was observed, authentically resembling AIH in humans.
Furthermore, Kido et al.121 demonstrated that hepatitis induction depended on autoreactive CD4+ T cells. Tregs are either generated in the thymus (nTregs) or in inflammatory microenvironments in the periphery (iTregs). In the gut, the conversion of conventional CD4+ T cells into iTregs depends on TGF-β and RA produced by CD103+ DCs.122, 123 In the liver, such DCs have not been identified, but the factors necessary for iTreg induction are abundantly expressed. TGF-β is produced by KCs, HSCs and to a lesser extent by LSECs. Moreover, vitamin A conversion into RA is prominently controlled by HSCs, and LSECs, with LSECs as the major inducers of CD4+CD25+Foxp3+ iTregs.124 This conversion depends on their ability to retain latent TGF-β on their cell surface via latency-associated peptide and the anchor molecule Glycoprotein-A-repetitions predominant (GARP=LRRC32).
Furthermore, LSEC-induced antigen-specific Tregs limit experimental autoimmune encephalomyelitis (EAE) in a mouse model, where experimental multiple sclerosis is induced by injection of myelin-basic protein (MBP); this, indeed, leads to increase of MBP-specific Tregs to counteract inflammation.124 In a recent report, the pivotal role of LSECs in Treg conversion was highlighted.32 By nanoparticle-assisted targeting of auto-antigens to LSECs, EAE onset was prevented, which was dependent on Treg expansion after nanoparticle administration.32 Ectopic expression of MBP in hepatocytes also prevents EAE due to the TGF-β-dependent induction of antigen-specific Foxp3+ iTregs.125 As direct contact between hepatocytes and T cells located in the sinusoids is possible due to the fenestration of LSECs,126 Notch-dependent conversion of iTreg by hepatocytes could occur. More speculative, hepatocyte expressed MBP could be handed around via trogocytosis to other parenchymal cells, including LSECs that are potent inducers of iTregs via membrane-bound latent TGF-β. The source for hepatic RA are HSCs, which due to their vitamin A storage function, can generate RA via expression of RALDH1.105 Although HSCs do not present antigens to CD4+ T cells, they were critically involved in iTreg generation by DCs in the presence of TGF-β,106 which depended on RA production. As HSCs are an abundant source of vitamin A, other APCs take advantage of its availability. For instance, LSECs express functional retinal dehydrogenases and can convert vitamin A to RA, which was essential for the induction of gut homing molecules CCR9 and α4β7.36 As the induction of gut-homing receptors on Tregs by RA markedly improves their suppressive function,127 the presence of RA during induction of iTregs by latent TGF-β on LSECs105 could support induction of potent suppressive Tregs. Other anti-inflammatory soluble factors, like IL-10 or PGE2, that are pivotal to the tolerogenic immune status in the liver, can influence Treg function. IL-10, for instance, that is produced by KCs, is required in the Treg-promoting microenvironment to maintain their suppressive phenotype by stabilizing Foxp3 expression.128 In tolerance induced after ConA-mediated hepatitis, both KCs and CD4+CD25+ Tregs are major producers of IL-10, which is responsible for the tolerogenic effect upon repeated injections of ConA,9 suggesting that an autocrine IL-10 loop is present in Tregs to maintain Foxp3 and suppressive function. Tregs are not only generated in situ, but can also be recruited into the liver. CD4+CD25+Tregs are reported to express the chemokine receptor CXCR3.129 In immune-mediated liver inflammation induced by ConA, induction of hepatic CXCR3-ligand expression (CXCL9, CXCL10 and CXCL11) is accompanied by increased hepatic infiltration of CXCR3+ Tregs.130 These Tregs are important regulators in ConA hepatitis, as in Cxcr3−/− mice, Treg accumulation following ConA administration was compromised and exacerbated liver damage due to induction of an extensive Th1/Th17 response.130 As a reaction to this inflammation, Tregs increase in number, and this expansion exerts hepato-protective effects in liver injury of different etiologies and upkeep of tolerance toward liver and secondary organ allografts. But in strong contrast, it also supports perpetuation of viral hepatitis and establishment and progression of hepatocellular carcinomas. In animal models for liver transplantation, ablation of recipient Treg by CD25 antibodies, for example, abrogates allograft tolerance, and induces acute graft rejection.131
Controversial discussion arose, however, about immune suppressive competence of Tregs in patients with AIH. Peiseler et al.132 reported that they did not observe differences in the number of Tregs in peripheral blood of AIH patients or healthy subjects, together with comparable suppressive capacity. In addition, in adult AIH patients, numbers of intrahepatic Tregs were increased when compared with liver biopsies from NASH patients. This was argued by Vergani’s group,133 and references therein, as they found reduction in Tregs suppressive capacity from pediatric AIH patients. However, both groups appear to use alternate methodology to test Tregs suppressive function, and to purify Tregs and their observations may be due to methodological variances /discrepancies. In addition, it cannot be ruled out that early onset of AIH in pediatric patients yields alterations in Treg phenotype compared with adults.132, 133
Innate lymphocytes in the liver and their function in tolerance
NKT cells in liver tolerance
The liver supports an unusually high frequency of NKT cells. Type 1 invariant (i)NKT cells express a distinct TCRα chain (Vα24-Jα18) and are thought to recognize a broad spectrum of self and microbial lipids. Type 2 NKT cells express a variable TCR-type pattern and recognize mammalian and pathogen-related phospholipids.134, 135 In the mouse, up to 30–40% of lymphocytes in the liver constitute NK1.1+CD3+ NKT cells3 (opposed to 0.5–2% in peripheral blood) of which 80% express the invariant TCR,136 whereas in humans, NKT cells predominantly belong to type 2 NKT cells.137 In the human liver, there are ~5–10% CD56+CD3+ NKT cells among the hepatic lymphocyte population.3 Furthermore, ~10–30% of hepatic NKT cells express the (phospho)lipid binding MHC-like molecule CD1d, a population that is largely absent in human livers and only constitutes less than 1% of the hepatic lymphocytes.3 Therefore, it is of note that in murine models for immune-mediated liver injury, NKT cell-mediated damage and immune regulation prevail, in contrast to human liver diseases. Consequently, mouse models addressing immune-mediated liver damage must be discussed carefully when it comes to interpretations of the relevance for human disease.
Both type I and II NKT recognize bacterial (phospho)lipids in the context of CD1d. In vitro studies show that LSECs, KCs and DCs can present lipid antigens to NKT cells; however, in vivo upon bacterial infection, NKT activation is predominantly mediated by KCs.138 NKT cells can be activated by both pathogen-derived or self-lipids and pro-inflammatory cytokines like IL-12 and IL-18, predominantly produced by DCs and KCs upon infection. As they secrete Th1, Th2 and Th17-signature cytokines upon activation, depending on the nature of the APC stimulus and glycolipid ligand,139 NKT cells are believed to have pro-inflammatory properties. They exert their hepatocytotoxic effects via secretion of pro-inflammmatory cytokines, or killing of hepatocytes by release of FasL.140 This discriminates them from killing mechanisms of NK cells that use secretion of TNF-related apoptosis-inducing ligand and granzyme B. However, as described below, NKT cells can also act as regulators of tolerogenic Tregs and suppress autoimmunity.
In the liver, NKT cell activation seems to contribute to both the initiation or inhibition of liver inflammation and fibrosis, depending on the etiology of liver disease. The crucial involvement of NKT cells in inflammation-related liver injury after ConA challenge, in ischemia/reperfusion liver injury, and high-fat diet78, 141, 142, 143 was demonstrated, as mice that lack iNKT cells (in J 18−/− or Cd1−/− mice) are resistant to the aforementioned insults. In experimental models for NASH, primary biliary cirrhosis (PBC) and HBV infection, iNKT cell activation aggravates disease, whereas after chronic toxic liver injury, NKT cells are protective.144 The dichotomous role may in part be explained by the reverse roles of NKT cell-produced IL-4 and IFN-γ, their relative concentrations, and consequently, on IL-4 and STAT-6-controlled infiltration of neutrophils into the liver.145 Similarly, in hepatic ischemia and reperfusion injury, for example, sulfatide-mediated activation of type II NKT cells leads to the reduction of type I NKT-related IFN-γ secretion, which, in turn, diminishes hepatic recruitment of myeloid cells (CD11b+Gr.1int/CD11b+Gr.1−) and NK cells and yields liver protection.141 Even more so, type II NKT cells can prevent inflammatory liver disease by anergy induction in type I NKT cells:146 activation of sulfatide-reactive type II NKT cells and pDCs can recruit type I NKT cells to the liver; however, these iNKTs were anergic, indicating that hepatic CD11c+ DCs were rendered tolerogenic after activation of type II NKTs.146 Interestingly, proliferation of iNKT cells was impaired after challenge with α-GalCer in mice that had received DCs from sulfatide-injected animals after challenge with α-GalCer.146 In analogy, ConA-mediated liver injury is prevented if iNKTs are rendered anergic following NKT type II-restricted recognition of sulfated glycolipids or self-lysophospholipids.146148
Also, α-GalCer-induced immune hepatitis is ameliorated if iNKT cells are rendered hyporesponsive to α-GalCer restimulation after α-GalCer pre-treatment, even though α-GalCer also induces NKT cell-dependent NK cell activation.149, 150 Biburger and Tiegs report that α-GalCer-induced protection from liver injury is not a result of actively tolerizing factors but of activation-induced hyporesposiveness of hepatic NKT cells;149 they state that the function of KCs and Tregs in this context is not relevant for the development of hepatoprotection.149 Contradicting these observations, however, Swain’s group states that NKT cells induce recruitment of CXCR3-expressing Tregs (see above), and that ~50% of the hepatic Tregs express CXCR3;151 after α-GalCer challenge, they found that hepatic CXCL10 levels were significantly increased in WT but not in NKT cell-deficient mice, accompanied by increased numbers of TGF-β and IL-10 secreting Tregs. Hence, activated NKT cells induce a “cytokine-to-chemokine pathway” that controls hepatic inflammatory responses.151
In addition to self-antigens, recognition of different bacterial antigens derived from the gut further impacts NKT cell function. Pathogenic bacteria in the gut aggravate ConA-mediated liver damage by enhancing NKT cell cytotoxicity toward hepatocytes, and ConA treatment itself aids bacterial translocation into the systemic circulation.152 Contrary, depletion of gram-negative bacteria alleviated ConA-induced hepatitis, which concurred with suppressed NKT cell activation.152 Another level of CD4+ T cell and NKT cell-mediated liver injury is the secretion of IL-17, that acts upstream of KC activation.153 Neutralization of IL-17 release dampens ConA-mediated liver injury by reduction of IL-6 and TNF-α-levels, but Il-17A−/− mice displayed liver injury after ConA not different from WT controls. This phenotype was feasible, as hepatoprotective IL-22 is also produced by Th17 cells, and is not affected by IL-17 blockage.153 Importantly, blocking of the IL-17R does not ameliorate ConA-mediated liver injury, as negative feedback of IL-17A and IL-17F production is outruled—instead, in a TGF-β-rich environment, the impairment of IL-17A/-F-mediated IL-6 induction aids generation of Treg and thus favors induction of tolerance.154, 155 In ConA hepatitis, IL-17 is also produced by γδ T cells, which exert hepato-protective functions after ConA application (Vγ4 γδ T cells). This protective effect seems to rely on the negative regulation of NKT cells in an IL-17A-dependent manner.156 Interestingly, non-committed iNKT cells can be induced to produce IL-17 when activated in the presence of TGF-β and IL-1β, which emphasizes the critical function of the micromilieu in immune cell activation.157 Furthermore, hepatic iNKT cells specifically activated with α-GalCer rapidly produce IL-17, which inhibits the development of hepatitis.158 Altogether, (i)NKT cells have been discussed quite controversially, as mentioned above, as they are quite potent in the induction of inflammation and hepatotoxicity (“friend or foe?”159), but on the other hand, they are designated as “regulators regulating regulators”:160 With regards to the specific immunotolerogenic phenotype of NKT cells, iNKT (Vα14i) cells share properties with human iNKT (Vα24i) cells and are reduced in frequency in diverse animal models and human patients with autoimmune diseases.161 This accounts for findings reported for systemic lupus, and models/patients for type I diabetes or ob/ob mice, or EAE mouse models and patients with multiple sclerosis (refer to Hammond and Kronenberg, and Wilson and Delovitch, and references therein).161, 162 As iNKT cells are capable of producing a large spectrum of Th1, Th2 or Th17 cytokines, it is of note that in autoimmune disease, secretion of Th2 cell-associated cytokines correlates with NKT cell regulation. Importantly, NKT cells, upon their activation and IFN-γ secretion, can induce hepatic Treg recruitment.151 Furthermore, in humans, CD4+NKT cell-derived IL-2 production enhances Treg survival and proliferation in the presence of allogenic DCs,160 with the NKT cells acting as helper cells to facilitate Treg expansion.163
NK cells
NK cells develop from an Id2+ precursor, and belong to the group 1 innate lymphoid cells and depend on IL-15 for their development and maintenance.164, 165 NK cells reside in the sinusoids and, besides NKT cells and KCs, are among the first cells to encounter circulating tumor cells and virus-infected cells; their depletion facilitates hepatic metastatic seeding or fulminant courses of hepatitis.166 In infection, NK cell numbers significantly increase in the liver.147, 167 In mice, 5–10% of the hepatic lymphocytes constitute NK1.1+/CD3−/DX5+ NK cells, and in humans, 30–50% of the hepatic lymphocyte population are NK cells, characterized by expression of CD56, but not CD3.3 Their activation and inhibition is mediated by the balance of stimulatory and inhibitory receptors, such as the inhibitory receptor NKG2A, that is highly expressed on hepatic NK cells; simultaneously, hepatic NK cells are devoid of the MHC class I-binding Ly49 receptor and exhibit a dampened INFγ response upon challenge with IL-12/IL-18.168, 169 By the immuno-suppressive milieu of the liver, they are kept in a hyporesponsive state.170, 171
Hepatic NK cells remain liver-resident, and like NKT cells, are activated by IL-12/IL-18 to become cytotoxic and produce IFN-γ. NK cells directly and indirectly interact with hepatic APCs, such as KCs and DCs, to control liver immune regulation:170 KCs are critical in the maintenance of NK-mediated tolerance in the liver, as KCs are the main producers of IL-10, and TLR-induced release of IL-18. On the one hand, IL-10 suppresses NK cell activation and supports maintenance of their hypo-reactive state, whereas IL-18 potentially stimulates NK cell activity, especially when IL-10 levels are low.171,172,173 TLR2/4 engagement and subsequent activation of the MyD88 pathway, in turn, result in enhanced IL-10 secretion by KCs, and thus support the intrahepatic immune-suppressive milieu. Also, in vitro stimulation of human NK cells with apoptotic cells induces tolerogenic, TGF-β-secreting NK cells that suppresses their autocrine IFN-γ production.174 Hence, NK cells could contribute to liver protection by preventing exacerbation of liver damage.
In contrast, TLR3 engagement in response to dsRNA exposure after, for instance, viral infection, which is independent of MyD88 signaling, triggers activity of the TRIF–IRF3 pathway.172 The latter impairs KC-related IL-10 secretion, and thus enhances NK cell activity. In this way, tolerance toward a homeostatic endotoxin load is maintained with simultaneous upkeep of potent anti-viral defense. Still, in mice with transgenic expression of HBsAg, poly I:C-induced liver injury is predominantly mediated via secretion of IFN-γ by intrahepatic NK cells, and independent of KCs.175 In Pseudomonas aeruginosa-induced NK cell-mediated hepatotoxicity, enhanced recruitment of NK cells relies on the presence of KCs and secretion of TNF-α.176 However, the role of NK cells in liver injury is strictly context-dependent, as treatment of mice with poly I:C before ConA injection yielded protection toward ConA-induced damage.177 Contrary, mice with liver-specific expression of the HBsAg were over-sensitive toward ConA-hepatitis.175, 178 The differences in this sensitivity lie in the enhanced vulnerability of the HBsAg transgenic mice to IFN-γ, as IFN-γ-receptor levels were upregulated on their hepatocytes; also, IFN-γ and IL-4 derived from iNKT cells enhance the NK-ligand expression on hepatocytes, and therefore exacerbate hepatocellular cytoxicity.169 Furthermore, KC-derived IL-12 was critical in WT mice for NK cell activation, whereas NK cell activation was KC-independent in HBsAg-transgenic animals.175, 178
Importantly, besides the regulation of KC-NK cell crosstalk, NK cells are actively involved in the tolerogenic diversion of T cells into Tregs via bi-directional cross-talk with DCs:179 in co-culture experiments, IL-2-primed NK cells down-modulated DC-activity in a NKG2A-dependent fashion,180 and NK-cell and DC-derived cytokines counteract each other’s action. Furthermore, NK cell-DC contact via NKG2A induces DC-mediated activation of CD4+CD25+ Tregs.179 However, contrary to freshly isolated CD4+CD25+ Tregs, NK cell-induced Tregs exert their suppressive activity via PD-1, and independent of TGF-β, glucocorticoid-induced TNFR family related gene (GITR) or IL-10.179
THE RELEVANCE OF THE INTESTINAL BARRIER FOR LIVER TOLERANCE
Inflammatory bowel diseases are known to affect hepatic immune responses and the induction of systemic disease or tolerance.181 Shifts in the gut microbiome were also identified as key factors in the development of the metabolic syndrome and various immunopathologies.182 Importantly, the intestine–blood–barrier prevents excessive leakage of TLR ligands and bacterial debris in the healthy individual, and thus maintains liver-resident immune cells tolerant. As a consequence, intrahepatic immune responses are prevented and liver tolerance is preserved. Contrary, in acute and chronic liver inflammation or failure, concomitant endotoxemia provokes a breach of liver tolerance. Furthermore, patients with chronic or acute liver failure are prone to the development of sepsis and SIRS (see above). Endotoxemia and shift in the gut microbiome cause hepatic TLR/PRR activation, that leads to chronic liver inflammation and disease progression in viral hepatitis, ASH, NAFLD progression to NASH, cirrhosis and fibrosis, as well as PSC and PBC,182,183, 184, 185, 186 and references therein. Hence, integrity of the intestinal barrier is a critical factor in the modulation of gut-liver cross talk. Also, in AIH, concurrence with inflammatory bowel diseases is observed, albeit at a much lower frequency compared with PSC.187, 188
An important link between the microbiota and liver fibrosis was discovered, as fibrosis was alleviated by antibiotic treatment, and endotoxin-mediated TLR signaling via TLR4 enhances fibrosis.189 Hepatic TLR and inflammasome signaling mediates liver injury and tolerance: TLRs on hepatic cells are activated upon intestinal dysbiosis and microbial translocation, a critical step in the development of autoimmune and chronic inflammatory liver diseases; TLRs and inflammasomes are expressed by liver cells: hepatocytes (TLR2-4; NLRP3), HSCs (TLR1-9), KCs (TLR2-4,9, NLRP1,3, AIM2) and LSECs (TLR2, NLRP1,3, AIM2). Activation of these TLRs induces inappropriate release of TNF-α, IL-6 as well as inflammasome activation and IL-1β secretion.182, 190 In intestinal and systemic inflammation, aberrant homing of intestinal mucosal cells to the liver significantly contributes to the development of AIH and metabolic and chronic liver diseases.191 To date, tissue-specific homing receptors in the liver endothelium have not been identified. So, in autoimmune disease, the leukocyte addressin repertoire becomes diverted from a gut-homing to a less-restricted pattern ectopic expression of formerly gut endothelial-restricted antigens (an overview on hepatic leukocyte recruitment can be found in Oo and Adams192): aberrant expression of gut-homing ligand-receptor pairs like CCR9, and CCL25 or α4β7 integrin and MAdCAM1, are important to induce hepatic tolerance (see above), but on the other hand, diversion of inflammatory lymphocytes from the gut to the liver is a critical determinant in the development and progression of autoimmune liver disease.191, 192 Pro-inflammatory Th17 cells, for example, are involved in the development and progression of AIH, PBC and PSC, as well as in inflammatory bowel diseases; at the same time, they contribute to the immune response against pathogenic microorganisms.193, 194, 195, 196 IL-23 and TNF-α that are produced by pathogen-activated DCs induce Th17 cells and activate RORγt+ cells in the colon that secrete IL-17A.197 Disruption of the mucosal barrier in the gut facilitates translocation of bacteria and endotoxins into the portal circulation and initiate a hepatic immune response. In the liver, the local cytokine milieu determines the outcome between tolerance or inflammation and autoimmunity by balancing the levels of TGF-β, IL-6, IL-17, IL-23 or IL-1β, and thus Th17 cell or Treg polarization.194, 198 Activated Th17 cells that secrete IL-17, TNF-α and IL-23, promote hepatic leukocyte recruitment and therefore enhance liver inflammation in AIH, PBC and PSC. Contrary to Th17 cells, Tregs dampen liver inflammation in AIH, PBC and PSC, while these reports controversially describe the role of Tregs, or alterations in their number and function in these pathologies.199
RESUME
Hepatic tolerance resides in its unique composition and abundance of conventional and non-conventional antigen-presenting cells and their creating an immunological micromilieu that allows maintenance of tolerance on the one hand but renders activation of potent immune responses on the other. The increasing prevalence of AIH and metabolic diseases, like NAFLD, NASH, but also ASH, cirrhosis, viral hepatitis and hepatocellular carcinoma poses a significant challenge to develop satisfactory therapies that allows manipulation of immune responses without eliminating anti-infectious immune responses. Also, the pathobiological mechanisms of PBC, PSC and AIH are only understood to a very limited extent, and recently, pieces of evidence accumulated that connect immunological tolerance with alterations of the intestinal microflora. The breech of tolerance in acute viral or autoimmune hepatitis, or after organ transplantation, pose challenges that demand novel therapeutic approaches to overcome lack of vaccines, and immune-suppressive therapeutics that still leave sufficient capacity to combat life-threatening infections. The discovery of the significant impact of the intestinal microflora, or novel regulatory pathways utilized by innate lymphoid cells, may hold the potential to further develop novel insights into therapeutic applications.
References
Tiegs G, Lohse AW . Immune tolerance: what is unique about the liver. J Autoimmun 2010; 34: 1–6.
Thomson AW, Knolle PA . Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol 2010; 10: 753–766.
Gao B, Jeong WI, Tian Z . Liver: an organ with predominant innate immunity. Hepatology 2008; 47: 729–736.
Crispe IN . Immune tolerance in liver disease. Hepatology 2014; 60: 2109–2117.
Crispe IN . The liver as a lymphoid organ. Annu Rev Immunol 2009; 27: 147–163.
Parker GA, Picut CA . Immune functioning in non lymphoid organs: the liver. Toxicol Pathol 2012; 40: 237–247.
Racanelli V, Rehermann B . The liver as an immunological organ. Hepatology 2006; 43: S54–S62.
Di Marco R, Xiang M, Zaccone P, Leonardi C, Franco S, Meroni P et al. Concanavalin A-induced hepatitis in mice is prevented by interleukin (IL)-10 and exacerbated by endogenous IL-10 deficiency. Autoimmunity 1999; 31: 75–83.
Erhardt A, Biburger M, Papadopoulos T, Tiegs G . regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology 2007; 45: 475–485.
Heymann F, Hamesch K, Weiskirchen R, Tacke F . The concanavalin A model of acute hepatitis in mice. Lab Anim 2015; 49: 12–20.
Calne RY, Sells RA, Pena JR, Davis DR, Millard PR, Herbertson BM et al. Induction of immunological tolerance by porcine liver allografts. Nature 1969; 223: 472–476.
Jenne CN, Kubes P . Immune surveillance by the liver. Nat Immunol 2013; 14: 996–1006.
Callery MP, Kamei T, Flye MW . The effect of portacaval shunt on delayed-hypersensitivity responses following antigen feeding. J Surg Res 1989; 46: 391–394.
Yang R, Liu Q, Grosfeld JL, Pescovitz MD . Intestinal venous drainage through the liver is a prerequisite for oral tolerance induction. J Pediatr Surg 1994; 29: 1145–1148.
Gorczynski RM, Chan Z, Fau-Chung S, Chung S, Fau-Cohen Z, Cohen Z et al. Prolongation of rat small bowel or renal allograft survival by pretransplant transfusion and/or by varying the route of allograft venous drainage. Transplantation 1994; 58: 816–820.
Knolle PA, Limmer A . Control of immune responses by scavenger liver endothelial cells. Swiss Med Wkly 2003; 133: 501–506.
Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest 1997; 99: 2782–2790.
Menezes GB, Lee WY, Zhou H, Waterhouse CC, Cara DC, Kubes P . Selective down-regulation of neutrophil Mac-1 in endotoxemic hepatic microcirculation via IL-10. J Immunol 2009; 183: 7557–7568.
McDonald B, McAvoy EF, Lam F, Gill V, de la Motte C, Savani RC et al. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med 2008; 205: 915–927.
McDonald B, Jenne CN, Zhuo L, Kimata K, Kubes P . Kupffer cells and activation of endothelial TLR4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am J Physiol Gastrointest Liver Physiol 2013; 305: G797–G806.
Wolf D, Schumann J, Koerber K, Kiemer AK, Vollmar AM, Sass G et al. Low-molecular-weight hyaluronic acid induces nuclear factor-kappaB-dependent resistance against tumor necrosis factor alpha-mediated liver injury in mice. Hepatology 2001; 34: 535–547.
John B, Crispe IN . TLR-4 regulates CD8+ T cell trapping in the liver. J Immunol 2005; 175: 1643–1650.
John B, Crispe IN . Passive and active mechanisms trap activated CD8+ T cells in the liver. J Immunol 2004; 172: 5222–5229.
Schurich A, Berg M, Stabenow D, Bottcher J, Kern M, Schild HJ et al. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J Immunol 2010; 184: 4107–4114.
Smedsrod B, Le Couteur D, Ikejima K, Jaeschke H, Kawada N, Naito M et al. Hepatic sinusoidal cells in health and disease: update from the 14th International Symposium. Liver Int 2009; 29: 490–501.
Tang L, Yang J, Liu W, Tang X, Chen J, Zhao D et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic e5-cell immune response. Gastroenterology 2009; 137: 1498–1508.e5.
Knolle PA, Uhrig A, Hegenbarth S, Löser E, Schmitt E, Gerken G et al. IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin Exp Immunol 1998; 114: 427–433.
Knolle PA, Germann T, Treichel U, Uhrig A, Schmitt E, HEgenbarth S et al. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J Immunol 1999; 162: 1401–1407.
Kruse N, Neumann K, Schrage A, Derkow K, Schott E, Erben U et al. Priming of CD4+ T cells by liver sinusoidal endothelial cells induces CD25low forkhead box protein 3- regulatory T cells suppressing autoimmune hepatitis. Hepatology 2009; 50: 1904–1913.
Carambia A, Frenzel C, Bruns OT, Schwinge D, Reimer R, Hohenberg H et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J Hepatol 2013; 58: 112–118.
Klugewitz K, Blumenthal-Barby F, Schrage A, Knolle PA, Hamann A, Crispe IN . Immunomodulatory effects of the liver: deletion of activated CD4+ effector cells and suppression of IFN-gamma-producing cells after intravenous protein immunization. J Immunol 2002; 169: 2407–2413.
Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol 2015; 62: 1349–1356.
Neumann K, Rudolph C, Neumann C, Janke M, Amsen D, Scheffold A . Liver sinusoidal endothelial cells induce immunosuppressive IL-10-producing Th1 cells via the Notch pathway. Eur J Immunol 2015; 45: 2008–2016.
Morell CM, Strazzabosco M . Notch signaling and new therapeutic options in liver disease. J Hepatol 2014; 60: 885–890.
Geisler F, Strazzabosco M . Emerging roles of Notch signaling in liver disease. Hepatology 2015; 61: 382–392.
Neumann K, Kruse N, Szilagyi B, Erben U, Rudolph C, Flach A et al. Connecting liver and gut: murine liver sinusoidal endothelium induces gut tropism of CD4+ T cells via retinoic acid. Hepatology 2012; 55: 1976–1984.
Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol 2008; 181: 2277–2284.
Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hubscher SG et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med 2004; 200: 1511–1517.
Kunkel EJ, Campbell JJ, Haraldsen G, Pan J, Boisvert J, Roberts AI et al. Lymphocyte Cc chemokine receptor 9 and epithelial thymus-expressed chemokine (Teck) expression distinguish the small intestinal immune compartment: epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J Exp Med 2000; 192: 761–768.
Bottcher JP, Schanz O, Wohlleber D, Abdullah Z, Debey-Pascher S, Staratschek-Jox A et al. Liver-primed memory T cells generated under noninflammatory conditions provide anti-infectious immunity. Cell Rep 2013; 3: 779–795.
Heydtmann M, Hardie D, Shields PL, Faint J, Buckley CD, Campbell JJ et al. Detailed analysis of intrahepatic CD8 T cells in the normal and hepatitis C-infected liver reveals differences in specific populations of memory cells with distinct homing phenotypes. J Immunol 2006; 177: 729–738.
Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA . Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 2008; 47: 296–305.
Limmer A, Ohl J, Wingender G, Berg M, Jungerkes F, Schumak B et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur J Immunol 2005; 35: 2970–2981.
Limmer A, Ohl J, Kurts C, Ljunggren H-G, Reiss Y, Groetterup M et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results. Nat Med 2000; 6: 1348–1354.
Kaczmarek J, Homsi Y, van Uum J, Metzger C, Knolle PA, Kolanus W et al. Liver sinusoidal endothelial cell-mediated CD8 T cell priming depends on co-inhibitory signal integration over time. PLoS One 2014; 9: e99574.
Bottcher JP, Schanz O, Garbers C, Zaremba A, Hegenbarth S, Kurts C et al. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep 2014; 8: 1318–1327.
Wong YC, Tay SS, McCaughan GW, Bowen DG, Bertolino P . Immune outcomes in the liver: Is CD8 T cell fate determined by the environment? J Hepatol 2015; 63: 1005–1014.
Schildberg FA, Hegenbarth SI, Schumak B, Scholz K, Limmer A, Knolle PA . Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur J Immunol 2008; 38: 957–967.
Schildberg FA, Wojtalla A, Siegmund SV, Endl E, Diehl L, Abdullah Z et al. Murine hepatic stellate cells veto CD8 T cell activation by a CD54-dependent mechanism. Hepatology 2011; 54: 262–272.
Hutchins NA, Wang F, Wang Y, Chung CS, Ayala A . Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J Leukoc Biol 2013; 94: 963–970.
Eckert C, Klein N, Kornek M, Lukacs-Kornek V . The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front Immunol 2015; 6: 179.
Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M et al. The role of Kupffer cells and regulation of neutrophil migration into the liver by macrophage inflammatory protein-2 in primary listeriosis in mice. Pathol Int 1999; 49: 519–532.
Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol 2010; 11: 295–302.
Helmy KY, Katschke Jr KJ, Gorgani NN, Kljavin NM, Elliott JM, Diehl L et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006; 124: 915–927.
Boermeester MA, Houdijk APJ, Myer S, Cuesta MA, Appelmelk BJ, Wesdorp RIC et al. Liver failure inudes a systemic inflammatory response. Am J Pathol 1995; 147: 1428–1440.
Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R . The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32: 734–739.
Chensue SW, Terebuh PD, Rernick DG, Scales WE, Kunkel SL . In vivo biologic and immunohistochemical analysis of interleukin-1 alpha, beta and tumor necrosis factor during experimental endotoxemia. Am J Pathol 1991; 138: 395–402.
Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Buschenfelde KH, Gerken G . Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol 1995; 22: 226–229.
You Q, Cheng L, Kedl RM, Ju C . Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 2008; 48: 978–990.
Bingisser RM, Tilbrook PA, Holt PG, Kees UR . Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol 1998; 160: 5729–5734.
Biswas SK, Mantovani A . Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010; 11: 889–896.
Li X, Wang Z, Zou Y, Lu E, Duan J, Yang H et al. Pretreatment with lipopolysaccharide attenuates diethylnitrosamine-caused liver injury in mice via TLR4-dependent induction of Kupffer cell M2 polarization. Immunol Res 2015; 62: 137–145.
Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014; 59: 130–142.
Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015; 62: 279–291.
Maemura K, Zheng Q, Wada T, Ozaki M, Takao S, Aikou T et al. Reactive oxygen species are essential mediators in antigen presentation by Kupffer cells. Immunol Cell Biol 2005; 83: 336–343.
Wu K, Kryczek I, Chen L, Zou W, Welling TH . Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res 2009; 69: 8067–8075.
Oikawa T, Takahashi H, Ishikawa T, Hokari A, Otsuki N, Azuma M et al. Intrahepatic expression of the co-stimulatory molecules programmed death-1, and its ligands in autoimmune liver disease. Pathol Int 2007; 57: 485–492.
Xie Z, Chen Y, Zhao S, Yang Z, Yao X, Guo S et al. Intrahepatic PD-1/PD-L1 up-regulation closely correlates with inflammation and virus replication in patients with chronic HBV infection. Immunol Invest 2009; 38: 624–638.
Chen J, Wang XM, Wu XJ, Wang Y, Zhao H, Shen B et al. Intrahepatic levels of PD-1/PD-L correlate with liver inflammation in chronic hepatitis B. Inflamm Res 2011; 60: 47–53.
Dolina JS, Sung SS, Novobrantseva TI, Nguyen TM, Hahn YS . Lipidoid nanoparticles containing PD-L1 siRNA delivered in vivo enter Kupffer cells and enhance NK and CD8(+) T cell-mediated hepatic antiviral immunity. Mol Ther Nucleic Acids 2013; 2: e72.
Uchikura K, Wada T, Hoshino S, Nagakawa Y, Aiko T, Bulkley GB et al. Lipopolysaccharides induced increases in Fas ligand expression by Kupffer cells via mechanisms dependent on reactive oxygen species. Am J Physiol Gastrointest Liver Physiol 2004; 287: G620–G626.
Müschen M, Warskulat U, Peters-Regehr T, Bode JG, Kubitz R, Häussinger D . Involvement of CD95 (Apo-1/Fas) ligand expressed by rat Kupffer cells in hepatic immunoregulation. Gastroenterology 1999; 116: 666–677.
Sun ZL, Wada T, Uchikura K, Ceppa E, Klein AS . Role of Fas/Fasl in Kupffer cell-dependent deletion of alloantigen activated T cells following liver transplantation. Transplant Proc 2001; 33: 279–282.
Sun Z, Wada T, Maemura K, Uchikura K, Hoshino S, Diehl AM et al. Hepatic allograft-derived Kupffer cells regulate T cell response in rats. Liver Transpl 2003; 9: 489–497.
Tiegs G, Hentschel J, Wendel AA . T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest 1992; 90: 196–203.
Kunstle G, Hentze H, Germann PG, Tiegs G, Meergans T, Wendel A . Concanavalin A hepatotoxicity in mice: tumor necrosis factor-mediated organ failure independent of caspase-3-like protease activation. Hepatology 1999; 30: 1241–1251.
Kaneko Y, Harada M, Kawano T, Yamashita M, Shibata Y, Geiyo F et al. Augmentation of Va14 NKT cell–mediated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in the development of concanavalin A–induced hepatitis. J Exp Med 2000; 191: 105–114.
Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K . Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci USA 2000; 97: 5498–5503.
Schümann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N et al. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol 2000; 157: 1671–1683.
Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G . Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology 1995; 21: 190–198.
Kusters S, Gantner F, Kunstle G, Tiegs G . Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996; 111: 462–471.
Nicoletti F, Di Marco R, Zaccone P, Salvaggio A, Magro G, Bendtzen K et al. Murine concanavalin A-induced hepatitis is prevented by interleukin 12 (IL-12) antibody and exacerbated by exogenous IL-12 through an interferon-gamma-dependent mechanism. Hepatology 2000; 32: 728–733.
Matta BM, Castellaneta A, Thomson AW . Tolerogenic plasmacytoid DC. Eur J Immunol 2010; 40: 2667–2676.
Raich-Regue D, Glancy M, Thomson AW . Regulatory dendritic cell therapy: from rodents to clinical application. Immunol Lett 2014; 161: 216–221.
Lukacs-Kornek V, Schuppan D . Dendritic cells in liver injury and fibrosis: shortcomings and promises. J Hepatol 2013; 59: 1124–1126.
Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol 2012; 13: 888–899.
Tokita D, Mazariegos GV, Zahorchak AF, Chien N, Abe M, Raimondi G et al. High PD-L1/CD86 ratio on plasmacytoid dendritic cells correlates with elevated T-regulatory cells in liver transplant tolerance. Transplantation 2008; 85: 369–377.
Tokita D, Sumpter TL, Raimondi G, Zahorchak AF, Wang Z, Nakao A et al. Poor allostimulatory function of liver plasmacytoid DC is associated with pro-apoptotic activity, dependent on regulatory T cells. J Hepatol 2008; 49: 1008–1018.
Castellaneta A, Sumpter TL, Chen L, Tokita D, Thomson AW . NOD2 ligation subverts IFN-alpha production by liver plasmacytoid dendritic cells and inhibits their T cell allostimulatory activity via B7-H1 up-regulation. J Immunol 2009; 183: 6922–6932.
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE . Nod-like proteins in immunity, inflammation and disease. Nat Immunol 2006; 7: 1250–1257.
Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S . Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA 2008; 105: 10113–10118.
Chen X, Du Y, Huang Z . CD4+CD25+ Treg derived from hepatocellular carcinoma mice inhibits tumor immunity. Immunol Lett 2012; 148: 83–89.
Mikami Y, Mizuno S, Nakamoto N, Hayashi A, Sujino T, Sato T et al. Macrophages and dendritic cells emerge in the liver during intestinal inflammation and predispose the liver to inflammation. PLoS One 2014; 9: e84619.
Nakamoto N, Ebinuma H, Kanai T, Chu PS, Ono Y, Mikami Y et al. CCR9+ macrophages are required for acute liver inflammation in mouse models of hepatitis. Gastroenterology 2012; 142: 366–376.
Hadeiba H, Sato T, Habtezion A, Oderup C, Pan J, Butcher EC . CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nat Immunol 2008; 9: 1253–1260.
Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 2012; 143: 1061–1072.
Reynaert H, Thompson MG, Thomas T, Geerts A . Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut 2002; 50: 571–581.
Baba S, Fujii H, Hirose T, Yasuchika K, Azuma H, Hoppo T et al. Commitment of bone marrow cells to hepatic stellate cells in mouse. J Hepatol 2004; 40: 255–260.
Weiskirchen R, Tacke F . Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr 2014; 3: 344–363.
Friedman SL . Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008; 88: 125–172.
Trautwein C, Friedman SL, Schuppan D, Pinzani M . Hepatic fibrosis: concept to treatment. J Hepatol 2015; 62: S15–S24.
Lee YA, Wallace MC, Friedman SL . Pathobiology of liver fibrosis: a translational success story. Gut 2015; 64: 830–841.
Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 2007; 26: 117–129.
Winau F, Quack C, Darmoise A, Kaufmann SH . Starring stellate cells in liver immunology. Curr Opin Immunol 2008; 20: 68–74.
Ichikawa S, Mucida D, Tyznik AJ, Kronenberg M, Cheroutre H . Hepatic stellate cells function as regulatory bystanders. J Immunol 2011; 186: 5549–5555.
Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol 2013; 190: 2009–2016.
Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ et al. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology 2004; 40: 1312–1321.
Scholzel K, Schildberg FA, Welz M, Borner C, Geiger S, Kurts C et al. Transfer of MHC-class-I molecules among liver sinusoidal cells facilitates hepatic immune surveillance. J Hepatol 2014; 61: 600–608.
Davis DM . Intercellular transfer of cell-surface proteins is common and can affect many stages of an immune response. Nat Rev Immunol 2007; 7: 238–243.
Dhainaut M, Moser M . Regulation of immune reactivity by intercellular transfer. Front Immunol 2014; 5: 112.
Carambia A, Herkel J . Liver sinusoidal cells collecting MHC-I molecules: you can't get enough of a good thing. J Hepatol 2014; 61: 464–465.
Arnold B . Parenchymal cells in immune and tolerance induction. Immunol Lett 2003; 89: 225–228.
Herkel J, Jagemann B, Wiegard C, Lazaro JF, Lueth S, Kanzler S et al. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocyutes. Hepatology 2003; 37: 1079–1085.
Bertolino P, Trescol-Biémont M-C, Rabourdin-Combe C . Hepatocytes induce functional activation of naiveCD8+T lymphocytes but fail to promote survival. Eur J Immunol 1998; 28: 221–236.
Tay SS, Wong YC, McDonald DM, Wood NA, Roediger B, Sierro F et al. Antigen expression level threshold tunes the fate of CD8 T cells during primary hepatic immune responses. Proc Natl Acad Sci USA 2014; 111: E2540–E2549.
Benseler V, Warren A, Vo M, Holz LE, Tay SS, Le Couteur DG et al. Hepatocyte entry leads to degradation of autoreactive CD8 T cells. Proc Natl Acad Sci USA 2011; 108: 16735–16740.
Burghardt S, Erhardt A, Claass B, Huber S, Adler G, Jacobs T et al. Hepatocytes contribute to immune regulation in the liver by activation of the Notch signaling pathway in T cells. J Immunol 2013; 191: 5574–5582.
Burghardt S, Claass B, Erhardt A, Karimi K, Tiegs G . Hepatocytes induce Foxp3+ regulatory T cells by Notch signaling. J Leukoc Biol 2014; 96: 571–577.
Franco A, Barnaba V, Natali P, Balsano C, Musca A, Balsano F . Expression of class I and class II major histocompatibility complex antigens on human hepatocytes. Hepatology 1988; 8: 449–454.
Stross L, Gunther J, Gasteiger G, Asen T, Graf S, Aichler M et al. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology 2012; 56: 873–883.
Kido M, Watanabe N, Okazaki T, Akamatsu T, Tanaka J, Saga K et al. Fatal autoimmune hepatitis induced by concurrent loss of naturally arising regulatory T cells and PD-1-mediated signaling. Gastroenterology 2008; 135: 1333–1343.
Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 2007; 204: 1775–1785.
Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 2007; 204: 1757–1764.
Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S et al. TGF-beta-dependent induction of CD4(+)CD25(+)Foxp3(+) Tregs by liver sinusoidal endothelial cells. J Hepatol 2014; 61: 594–599.
Luth S, Huber S, Schramm C, Buch T, Zander S, Stadelmann C et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest 2008; 118: 3403–3410.
Warren A, Le Couteur DG, Fraser R, Bowen DG, McCaughan GW, Bertolino P . T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology 2006; 44: 1182–1190.
Menning A, Loddenkemper C, Westendorf AM, Szilagyi B, Buer J, Siewert C et al. Retinoic acid-induced gut tropism improves the protective capacity of Treg in acute but not in chronic gut inflammation. Eur J Immunol 2010; 40: 2539–2548.
Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol 2009; 10: 1178–1184.
Chen D, Bromberg JS . T regulatory cells and migration. Am J Transplant 2006; 6: 1518–1523.
Erhardt A, Wegscheid C, Claass B, Carambia A, Herkel J, Mittrucker HW et al. CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J Immunol 2011; 186: 5284–5293.
Li W, Kuhr CS, Zheng XX, Carper K, Thomson AW, Reyes JD et al. New insights into mechanisms of spontaneous liver transplant tolerance: the role of Foxp3-expressing CD25+CD4+ regulatory T cells. Am J Transplant 2008; 8: 1639–1651.
Peiseler M, Sebode M, Franke B, Wortmann F, Schwinge D, Quaas A et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol 2012; 57: 125–132.
Longhi MS, Ma Y, Mieli-Vergani G, Vergani D . Regulatory T cells in autoimmune hepatitis. J Hepatol 2012; 57: 932–933.
Kumar V . NKT-cell subsets: promoters and protectors in inflammatory liver disease. J Hepatol 2013; 59: 618–620.
Berzins SP, Smyth MJ, Baxter AG . Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol 2011; 11: 131–142.
Park S-H, Weiss A, Benlagha K, Kyin T, Teyton L, Bendelac A . The mouse Cd1d-restricted repertoire is dominated by a few autoreactive T cell receptor families. J Exp Med 2001; 193: 893–904.
Bendelac A, Savage PB, Teyton L . The biology of NKT cells. Annu Rev Immunol 2007; 25: 297–336.
Hickey MJ, Kubes P . Intravascular immunity: the host-pathogen encounter in blood vessels. Nat Rev Immunol 2009; 9: 364–375.
Zajonc DM, Girardi E . Recognition of microbial glycolipids by natural killer T cells. Front Immunol 2015; 6: 400.
Shimamura K, Kawamura H, Nagura T, Kato T, Naito T, Kameyama H et al. Association of NKT cells and granulocytes with liver injury after reperfusion of the portal vein. Cell Immunol 2005; 234: 31–38.
Arrenberg P, Maricic I, Kumar V . Sulfatide-mediated activation of type II natural killer T cells prevents hepatic ischemic reperfusion injury in mice. Gastroenterology 2011; 140: 646–655.
Deng ZB, Liu Y, Liu C, Xiang X, Wang J, Cheng Z et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 2009; 50: 1412–1420.
Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012; 61: 1323–1329.
Mattner J . Natural killer T (NKT) cells in autoimmune hepatitis. Curr Opin Immunol 2013; 25: 697–703.
Wang H, Feng D, Park O, Yin S, Gao B . Invariant NKT cell activation induces neutrophil accumulation and hepatitis: opposite regulation by IL-4 and IFN-gamma. Hepatology 2013; 58: 1474–1485.
Halder RC, Aguilera C, Maricic I, Kumar V . Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest 2007; 117: 2302–2312.
Gao B, Radaeva S, Park O . Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol 2009; 86: 513–528.
Maricic I, Girardi E, Zajonc DM, Kumar V . Recognition of lysophosphatidylcholine by type II NKT cells and protection from an inflammatory liver disease. J Immunol 2014; 193: 4580–4589.
Biburger M, Tiegs G . Activation-induced NKT cell hyporesponsiveness protects from alpha-galactosylceramide hepatitis and is independent of active transregulatory factors. J Leukoc Biol 2008; 84: 264–279.
Eberl G, MacDonald HR . Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol 2000; 30: 985–992.
Santodomingo-Garzon T, Han J, Le T, Yang Y, Swain MG . Natural killer T cells regulate the homing of chemokine CXC receptor 3-positive regulatory T cells to the liver in mice. Hepatology 2009; 49: 1267–1276.
Chen J, Wei Y, He J, Cui G, Zhu Y, Lu C et al. Natural killer T cells play a necessary role in modulating of immune-mediated liver injury by gut microbiota. Sci Rep 2014; 4: 7259.
Yan S, Wang L, Liu N, Wang Y, Chu Y . Critical role of interleukin-17/interleukin-17 receptor axis in mediating Con A-induced hepatitis. Immunol Cell Biol 2012; 90: 421–428.
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA . Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007; 27: 647–659.
Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci USA 2008; 105: 18460–18465.
Zhao N, Hao J, Ni Y, Luo W, Liang R, Cao G et al. Vgamma4 gammadelta T cell-derived IL-17A negatively regulates NKT cell function in Con A-induced fulminant hepatitis. J Immunol 2011; 187: 5007–5014.
Monteiro M, Almeida CF, Agua-Doce A, Graca L . Induced IL-17-producing invariant NKT cells require activation in presence of TGF-beta and IL-1beta. J Immunol 2013; 190: 805–811.
Wondimu Z, Santodomingo-Garzon T, Le T, Swain MG . Protective role of interleukin-17 in murine NKT cell-driven acute experimental hepatitis. Am J Pathol 2010; 177: 2334–2346.
Swain MG . Hepatic NKT cells: friend or foe? Clin Sci 2008; 114: 457–466.
La Cava A, Van Kaer L, Fu Dong S . CD4+CD25+ Tregs and NKT cells: regulators regulating regulators. Trends Immunol 2006; 27: 322–327.
Wilson SB, Delovitch TL . Janus-like role of regulatory iNKT cells in autoimmune disease and tumour immunity. Nat Rev Immunol 2003; 3: 211–222.
Hammond KJ, Kronenberg M . Natural killer T cells: natural or unnatural regulators of autoimmunity? Curr Opin Immunol 2003; 15: 683–689.
Jiang S, Game DS, Davies D, Lombardi G, Lechler RI . Activated CD1d-restricted natural killer T cells secrete IL-2: innate help for CD4+CD25+ regulatory T cells? Eur J Immunol 2005; 35: 1193–1200.
Walker JA, Barlow JL, McKenzie AN . Innate lymphoid cells—how did we miss them? Nat Rev Immunol 2013; 13: 75–87.
Hazenberg MD, Spits H . Human innate lymphoid cells. Blood 2014; 124: 700–709.
Subleski JJ, Hall VL, Back TC, Ortaldo JR, Wiltrout RH . Enhanced antitumor response by divergent modulation of natural killer and natural killer T cells in the liver. Cancer Res 2006; 66: 11005–11012.
Tian Z, Chen Y, Gao B . Natural killer cells in liver disease. Hepatology 2013; 57: 1654–1662.
Doherty DG . Immunity, tolerance and autoimmunity in the liver: a comprehensive review. J Autoimmun 2015; 66: 60–75; pii: S0896-8411(15)30038-X.
Sun H, Sun C, Tian Z, Xiao W . NK cells in immunotolerant organs. Cell Mol Immunol 2013; 10: 202–212.
Krueger PD, Lassen MG, Qiao H, Hahn YS . Regulation of NK cell repertoire and function in the liver. Crit Rev Immunol 2011; 31: 43–52.
Lassen MG, Lukens JR, Dolina JS, Brown MG, Hahn YS . Intrahepatic IL-10 maintains NKG2A+Ly49- liver NK cells in a functionally hyporesponsive state. J Immunol 2010; 184: 2693–2701.
Tu Z, Bozorgzadeh A, Pierce RH, Kurtis J, Crispe IN, Orloff MS . TLR-dependent cross talk between human Kupffer cells and NK cells. J Exp Med 2008; 205: 233–244.
Notas G, Kisseleva T, Brenner D . NK and NKT cells in liver injury and fibrosis. Clin Immunol 2009; 130: 16–26.
Chong WP, Zhou J, Law HK, Tu W, Lau YL . Natural killer cells become tolerogenic after interaction with apoptotic cells. Eur J Immunol 2010; 40: 1718–1727.
Chen Y, Sun R, Jiang W, Wei H, Tian Z . Liver-specific HBsAg transgenic mice are over-sensitive to Poly(I:C)-induced liver injury in NK cell- and IFN-gamma-dependent manner. J Hepatol 2007; 47: 183–190.
Muhlen KA, Schumann J, Wittke F, Stenger S, van Rooijen N, van Kaer L et al. NK cells, but not NKT cells, are involved in Pseudomonas aeruginosa exotoxin A-induced hepatotoxicity in mice. J Immunol 2004; 172: 3034–3041.
Wang J, Sun R, Wei H, Dong Z, Gao B, Tian Z . Poly I:C prevents T cell-mediated hepatitis via an NK-dependent mechanism. J Hepatol 2006; 44: 446–454.
Chen Y, Wei H, Sun R, Dong Z, Zhang J, Tian Z . Increased susceptibility to liver injury in hepatitis B virus transgenic mice involves NKG2D-ligand interaction and natural killer cells. Hepatology 2007; 46: 706–715.
Jinushi M, Takehara T, Tatsumi T, Yamaguchi S, Sakamori R, Hiramatsu N et al. Natural killer cell and hepatic cell interaction via NKG2A leads to dendritic cell-mediated induction of CD4+CD25+T cells with PD-1-dependent regulatory activities. Immunology 2007; 120: 73–82.
Jinushi M, Takehara T, Tatsumi T, Kanto T, Miyagi T, Suzuki T et al. Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell-induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J Immunol 2004; 173: 6072–6081.
Adams DH, Eksteen B . Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol 2006; 6: 244–251.
Henao-Mejia J, Elinav E, Thaiss CA, Licona-Limon P, Flavell RA . Role of the intestinal microbiome in liver disease. J Autoimmun 2013; 46: 66–73.
Schnabl B, Brenner DA . Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014; 146: 1513–1524.
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012; 482: 179–185.
in C, Henao-Mejia J, Flavell RA . Innate immune receptors: key regulators of metabolic disease progression. Cell Metab 2013; 17: 873–882.
Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA . Inflammasomes and metabolic disease. Annu Rev Physiol 2014; 76: 57–78.
Boberg KM, Chapman RW, Hirschfield GM, Lohse AW, Manns MP, Schrumpf E et al. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol 2011; 54: 374–385.
Trivedi PJ, Chapman RW . PSC, AIH and overlap syndrome in inflammatory bowel disease. Clin Res Hepatol Gastroenterol 2012; 36: 420–436.
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 2007; 13: 1324–1332.
Wang B, Trippler M, Pei R, Lu M, Broering R, Gerken G et al. Toll-like receptor activated human and murine hepatic stellate cells are potent regulators of hepatitis C virus replication. J Hepatol 2009; 51: 1037–1045.
Trivedi PJ, Adams DH . Mucosal immunity in liver autoimmunity: a comprehensive review. J Autoimmun 2013; 46: 97–111.
Oo YH, Adams DH . The role of chemokines in the recruitment of lymphocytes to the liver. J Autoimmun 2010; 34: 45–54.
Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y . Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol 2009; 157: 261–270.
Zhao L, Qiu de K, Ma X . Th17 cells: the emerging reciprocal partner of regulatory T cells in the liver. J Dig Dis 2010; 11: 126–133.
Katt J, Schwinge D, Schoknecht T, Quaas A, Sobottka I, Burandt E et al. Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology 2013; 58: 1084–1093.
Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One 2011; 6: e18909.
Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 2012; 37: 674–684.
Huber S, Gagliani N, Esplugues E, O'Connor Jr W, Huber FJ, Chaudhry A et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(-) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011; 34: 554–565.
Longhi MS, Hussain MJ, Mitry RR, Arora SK, Mieli-Vergani G, Vergani D et al. Functional study of CD4+CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol 2006; 176: 4484–4491.
Czabotar PE, Lessene G, Strasser A, Adams JM . Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15: 49–63.
Bertolino P, Trescol-Biémont M-C, Thomas J, de St Groth BF, Pihlgren M, Marvel J et al. Death by neglect as a deletional mechanism of peripheral tolerance. Int Immunol 1999; 11: 1225–1237.
Acknowledgements
This work was supported by the German Research Foundation (DFG), SFB841: ‘Liver inflammation: Infection, immune regulation and consequences’.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Horst, A., Neumann, K., Diehl, L. et al. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol Immunol 13, 277–292 (2016). https://doi.org/10.1038/cmi.2015.112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2015.112
Keywords
This article is cited by
-
Hepatic T-cell senescence and exhaustion are implicated in the progression of fatty liver disease in patients with type 2 diabetes and mouse model with nonalcoholic steatohepatitis
Cell Death & Disease (2023)
-
Novel antigens for targeted radioimmunotherapy in hepatocellular carcinoma
Molecular and Cellular Biochemistry (2023)
-
Synthetically glycosylated antigens for the antigen-specific suppression of established immune responses
Nature Biomedical Engineering (2023)
-
The immunological mechanisms and therapeutic potential in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity
Cell & Bioscience (2022)
-
The immunological function of CXCR2 in the liver during sepsis
Journal of Inflammation (2022)
