
Abstract
RNA trans-splicing has become a versatile tool in the gene therapy of monogenetic diseases. This technique is especially valuable for the correction of mutations in large genes such as COL7A1, which underlie the dystrophic subtype of the skin blistering disease epidermolysis bullosa. Over 800 mutations spanning the entire length of the COL7A1 gene have been associated with defects in type VII collagen, leading to excessive fragility of epithelial tissues, the hallmark of dystrophic epidermolysis bullosa (DEB). In the present study, we designed an RNA trans-splicing molecule (RTM) that is capable of repairing any given mutation within a 4200 nucleotide region spanning the 3′ half of COL7A1. The selected RTM, RTM28, was able to induce accurate trans-splicing into endogenous COL7A1 pre-mRNA transcripts in a type VII collagen-deficient DEB patient-derived cell line. Correct trans-splicing was detected at the RNA level by semiquantitative RT-PCR and correction of full-length type VII collagen was confirmed at the protein level by immunofluorescence and western blot analyses. Our results demonstrate that RTM28, which covers >60% of all mutations reported in DEB and is thus the longest RTM described so far for the repair of COL7A1, represents a promising candidate for therapeutic applications.
Similar content being viewed by others


Introduction
Dystrophic epidermolysis bullosa (DEB) defines a severe variant of inherited epidermolysis bullosa (EB) characterized by cutaneous and mucosal fragility, resulting in blisters and superficial ulcerations that develop below the lamina densa of the cutaneous basement membrane associated with significant scarring and milia formation.1, 2 Approximately 25% of all EB patients have DEB, which can be inherited in an autosomal-dominant (DDEB) or -recessive (RDEB) manner. All variants of hereditary DEB are caused by mutations in the COL7A1 gene, coding for type VII collagen, the primary component of anchoring fibrils.3 These structures contribute to the firm adhesion of the dermis and epidermis. In DEB patients, anchoring fibrils may appear morphologically abnormal, reduced in number or altogether absent, depending on the nature of the mutations. To date, more than 800 reported and unpublished sequence variants in COL7A1 have been identified in DEB patients.4
DEB is associated with unique challenges for the development of promising therapies that can have a significant impact on survival and quality of patient’s life. Recent studies deal with allogeneic bone marrow transplantation,5 fibroblast injections,6 systemic application of allogeneic mesenchymal stem cells,7 gene therapy8, 9 or protein therapy10 and demonstrate progress towards clinical applications.11 Challenges of the current ex vivo gene therapy strategies include stable accommodation of the large COL7A1 cDNA in viral vectors, the risk of genetic rearrangements8 and the inability of cDNA gene therapy to correct autosomal-dominant variants of EB. The technical hurdles of supplementation of the mutated cDNA of the gene in target cells with its wild-type cDNA can be circumvented by using an alternative gene correction strategy such as spliceosome-mediated RNA trans-splicing.12 In principle, gene expression can be targeted and reprogrammed at the pre-mRNA level, which requires the coexistence of three distinct components: (i) the cellular splicing machinery, (ii) the endogenous target pre-mRNA of interest and (iii) exogenously delivered RNA trans-splicing molecules (RTMs). The RTM induces the trans-splicing event between the endogenous target pre-mRNA and the RTM, resulting in a chimeric gene product. RTMs are designed to carry a binding domain (BD), which defines the target specificity, splicing elements for efficient trans-splicing and a coding sequence that is desired to replace a part of the target pre-mRNA.13
The trans-splicing repair method provides several advantages over gene replacement strategies: (i) size reduction of the transgene vector to be delivered into the patient cells increasing the efficiency. Shorter stretches of homologous sequences within the vectors also reduce the possibility of rearrangements within the COL7A1 coding sequence.12 (ii) Expression of the transgene is regulated by endogenous control mechanisms, as the RTM is spliced into the endogenous pre-mRNA. As trans-splicing only occurs in cells endogenously expressing the pre-mRNA, this method minimizes the risk of unwanted ectopic expression of the recombinant protein spatially, temporally and quantitatively in all layers of the epidermis. (iii) Trans-splicing removes mutations on the transcript regardless of their mode of inheritance, and is thus suitable to decrease dominant-negative gene products.12, 14, 15 (iv) The use of non-viral vectors such as minicircle DNA, or stabilized mRNA molecules for RTM delivery into the skin in vivo, circumvent the necessity of transplantations in ‘classical’ ex vivo gene therapy.16, 17, 18
Recent studies demonstrated that the BD is crucial for success of the trans-splicing process. Marginal variations in sequence, length or target-binding position influence the outcome of mRNA repair. Rational design of RTMs has failed to reveal a consistent formula. Our group has therefore established a reporter-based screening system, which provides a tool for the identification of highly specific BDs for genes involved in EB.13, 19, 20, 21 In a fluorescence-based RTM screen, randomly generated BDs are cloned into reporter RTMs, which can be quantified for their trans-splicing efficiency by flow cytometry.
To improve the robustness of the fluorescence-based screening system for efficient RTM selection, a stable COL7A1-MG expressing human embryonic kidney 293 (HEK293) cell line was included in the experimental design. This is intended to mimic an endogenous target pre-mRNA expression at a constant level, thereby reducing RTM efficiency variations because of variable amounts of transiently transfected COL7A1-MG plasmids in the cells. The identification of the most efficient RTMs by flow cytometric analysis is currently a promising way to obtain high-quality RTMs for endogenous purposes. In this manner, it was demonstrated that 3′ trans-splicing is capable of fully reverting the RDEB phenotype in vitro.22 The study presented here describes the design and generation of the longest RTM so far available for DEB, capable of replacing over 4 kb of the 3′ COL7A1 coding sequence at the transcript level. As a consequence, around 2/3 of all known COL7A1 mutations can be corrected with one RTM.
Results
An improved fluorescence-based screening model facilitates RTM selection
As the BD is crucial for the efficiency and specificity of the trans-splicing process, the development of a BD screen can facilitate and accelerate the construction of highly efficient RTMs for subsequent endogenous applications.19, 20 The RTM screening system consists of an RTM library, in which each molecule contains the 3′ part of a green fluorescence protein (GFP), an internal ribosomal entry site (IRES), the reporter molecule dsRED, splicing elements for efficient splicing and a randomly designed BD hybridizing to the COL7A1 target region intron 46/exon 47 (Figure 1a). The target minigene (COL7A1-MG) was cloned into a screening vector carrying the 5′ GFP portion and COL7A1 intron 46 and exon 47. This fluorescence-based RTM screening system allows us to identify the most efficient RTMs from of a large pool of RTM variants containing complementary BDs for the target region (Figure 1a). Co-transfection of RTMs and COL7A1-MG into HEK293 cells induces the trans-splicing reaction, leading to the fusion of the GFP split portions, resulting in full-length GFP expression. The intensity of the fluorescence signal and the amount of GFP-expressing cells, analysed by flow cytometry, correlate with the trans-splicing efficiency of the given RTM.

A fluorescence-based screening system for RTM selection. (a) The screening system is built up by a COL7A1-MG, containing the 5′ portion of GFP and the selected COL7A1 pre-mRNA region of intron 46/exon 47, and an RTM, carrying a randomly cloned BD, splicing elements for efficient trans-splicing, the 3′ portion of GFP, an IRES and dsRED. Co-expression of both screening molecules in HEK293 cells leads to the restoration and expression of GFP upon accurate trans-splicing. (b – d) The transfection of the RTM library into HEK293 cells, stably expressing the COL7A1-MG (COL7A1-MG-CL) and the red fluorescence reporter molecule mRuby, results in a diverse expression of the red and green reporter molecules. This is dependent on the levels of RTM and COL7A1-MG expression and more importantly on the functionality of the introduced RTM. A high GFP expression indicates trans-splicing through highly efficient RTMs in the cell, detected by microscopic (b, c) and FACS analysis (d). Overlay of the different fluorochromes in trans-spliced cells results in a yellow to orange colour, depending on the strength of green GFP expression together with red target/RTM expression (c). The cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue fluorescence). Scale bars: 50 μm (b); 20 μm (c). PPT, polypyrimidine tract.
To increase the robust and straightforward nature of our screening system, we have constructed an HEK293 cell line stably expressing the COL7A1-MG (COL7A1-MG-CL) together with a puromycin/mRuby expression cassette under the control of an EF1 (elongation factor 1) promoter lacking in the original COL7A1-MG expression plasmid for co-transfection experiments (Figure 1a). The cell line was generated via co-transfection of a PiggyBac (pB) transposon vector, harbouring the COL7A1-MG and an mRuby-puromycin cassette, together with a PiggyBac transposase expression vector. An initial transfection of the generated RTM library, specific for the COL7A1 target region intron 46/exon 47, into the COL7A1-MG-CL, resulted in a diverse expression pattern of GFP detectable by fluorescence microscopy (Figures 1b and c) and flow cytometric analysis (Figure 1d).
The variable expression profile of GFP and mRuby in transfected HEK293 cells indicates a diverse functionality of the introduced RTMs determined by their binding ability to the target region. To investigate the trans-splicing behaviour of RTMs harbouring different BDs, 80 highly variable molecules from the RTM library were analysed by sequencing. Five individual RTMs with complementary sequences for intron 46/exon 47 of COL7A1 were selected for further experiments (Figure 2).

Investigation of RTM binding regions within the target intron 46/exon 47 of COL7A1 and their trans-splicing efficiencies analysed by flow cytometry. Eighty individual clones of the RTM library were analysed for the presence of a BD sequence by sequence analysis. (A) Five single RTMs, termed RTM02, RTM28, RTM37, RTM40 and RTM78, contained one or more specific BD(s) complementary to the selected COL7A1 target region intron 46/exon 47. These RTMs were included in initial experiments to investigate their potential to induce accurate trans-splicing into COL7A1. (B) Co-transfection of COL7A1-MG together with a representative low-efficiency (RTM02) (b) or high-efficiency RTM (RTM28) (c) into HEK293 cells resulted in the expression of GFP in ~40% or ~88% of all analysed cells, respectively, upon accurate 3′ RNA trans-splicing. Similar results were achieved after RTM02 (e) and RTM28 (f) transfection into the COL7A1-MG-CL-expressing cell line, in which ~40% and ~78% GFP expression, respectively, were detectable by flow cytometric analysis. GFP expression was undetectable in HEK293 cells exclusively transfected with RTM28 (a) and in the COL7A1-MG-CL-expressing cell line (d). Regardless of whether the COL7A1-MG was transiently or stably expressed in HEK293 cells, the RTM efficiencies were comparable. The most efficient RTMs, RTM28, RTM37 and RTM40, showed higher trans-splicing efficiencies than RTM02 and RTM78 in both screening settings, measured by flow cytometric analysis.
To test the robustness of our stably integrated COL7A1-MG cell line, we compared this screening system with our well-established co-transfection screening model.13, 19, 20, 21 After co-transfection of the COL7A1-MG and the selected individual RTMs into HEK293 cells, we noted a diverse expression ratio of GFP upon trans-splicing induction. The total amount of GFP-expressing cells varied between 40 and 88%, confirming the impact of the RTM binding site on the trans-splicing efficiency (Figure 2b). Transfection experiments of single RTMs into stable COL7A1-MG-expressing cells revealed results similar to those achieved in the co-transfection assay (Figures 2 and 3), and can thus be considered as a reliable screening model, facilitating the selection of highly potent RTMs. In the co-transfection system, RTM28 induced correct trans-splicing in ~88% of all analysed cells represented by GFP restoration (Figure 2). RTM28, RTM37 and RTM40 showed a promising trans-splicing efficiency (78% for RTM28) in stable COL7A1-MG-expressing cells, highly comparable to the co-transfection experiment. For the initial endogenous experiments in RDEB patient keratinocytes, we selected RTM28, which displayed a high trans-splicing capacity in both experimental settings, which was validated via fluorescence microscopy (Figure 3). RTM28 contains two complementary target-specific BDs (38 and 79 bp in length) for intron 46.

Validation of FACS analysis by fluorescence-based microscopy. Co-transfection of COL7A1-MG together with high-efficiency RTM (RTM28) (a) into HEK293 cells resulted in the expression of GFP upon accurate 3′ RNA trans-splicing. Similar results were achieved after RTM28 (b) transfection into the COL7A1-MG-CL-expressing cell line. GFP expression was undetectable in HEK293 cells exclusively transfected with RTM28 (a, upper panel) and the COL7A1-MG-CL-expressing cell line (b, upper panel). HEK293 cells transfected with RTM28 containing dsRED and the 3′ half of GFP (a, upper panel) and stably expressing COL7A1-MG cells (COL7A1-MG-CL) containing mRuby (b, upper panel) showed red fluorescence upon reporter molecule expression. Overlay of the different flurochromes in trans-spliced cells resulted in a yellow to orange colour, depending on the strength of the merged green GFP expression and the red target expression (a, b, lower panel). Scale bars: 50 μm.
Endogenous trans-splicing into the COL7A1 pre-mRNA
Trans-splicing detection at the RNA level
For endogenous studies, RTM28 was modified to create an RNA molecule capable of replacing the wild-type coding region of COL7A1 spanning from exons 47 to 118. A 3xFLAG tag and silent mutations were inserted into the coding domain to facilitate the detection of accurate trans-splicing into COL7A1 pre-mRNA transcripts at the RNA and protein level (Figure 4a). After cloning of the RTM into the retroviral vector, pMXs-IRES-Blasticidin, an RDEB keratinocyte cell line, harbouring a homozygous nonsense mutation in exon 105 was transduced. Eight days after antibiotic selection total RNA was isolated, and semiquantitative RT-PCR (sqRT-PCR) analysis revealed the expression of correctly trans-spliced COL7A1-RTM fusion mRNA (Figure 4b). As a negative control, total RNA isolated from RDEB keratinocytes treated with the retroviral vector pMXs-IRES-Blasticidin either without any insert (RDEB-mock) or with the RTM backbone lacking a BD (RTM w/o BD) for intron 46/exon 47 was used.

Detection of COL7A1-RTM fusion transcripts by sqRT-PCR. (a) Schematic depiction of the endogenous RTM28, containing a FLAG tag and silent mutations within the wild-type coding domain of COL7A1. (b) SqRT-PCR analysis, performed on total RNA isolated from RTM28-transduced RDEB keratinocytes using primers spanning exons 46–49, revealed the expression of COL7A1-RTM fusion transcripts at the RNA level. The respective PCR product with a size of 205 bp was verified by sequence analysis. (c) SqRT-PCR was performed to quantify total COL7A1 transcripts present in RTM-treated keratinocytes. As a result, a fivefold increase of COL7A1 expression was obtained after RTM28 treatment in comparison with RDEB patient cells transduced with an empty retroviral vector (RDEB-mock).
Using an exon 46-specific forward and an exon 49-specific reverse primer, binding exclusively to the silent mutations within the RTM backbone, it was possible to discriminate between uncorrected (cis-spliced) and corrected (trans-spliced) transcripts. Accurate trans-splicing was confirmed by sequencing of the 205 nucleotide PCR product, which represented the trans-spliced COL7A1-RTM fusion product (Figure 4b). In addition, sqRT-PCR analyses, using a primer pair spanning exons 17–19 of COL7A1, revealed a fivefold increase of total COL7A1 expression in RTM-transduced RDEB patient cells compared with the RDEB-mock control as a result of specific trans-splicing repair (Figure 4c).
Trans-splicing detection at the protein level
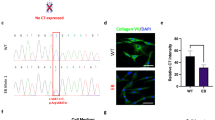
To verify the restoration of type VII collagen expression in RTM-transduced RDEB patient keratinocytes by trans-splicing, we performed immunofluorescence staining and western blot analysis using antibodies directed against either the NC1 domain of COL7A1 or the 3xFLAG tag in cultured RDEB keratinocytes. Neither the wild-type human keratinocyte cell line (Figure 5a4) nor RDEB keratinocytes (Figure 5a5) showed any specific FLAG staining, whereas RTM28-transduced cells (Figure 5a6) showed a strong staining, indicating the expression of FLAG-tagged COL7A1-RTM fusion protein. The specific FLAG staining indicated the correct restoration of collagen type VII by accurate 3′ trans-splicing. These data provide evidence that type VII collagen-deficient RDEB keratinocytes can recover the expression of type VII collagen protein as a consequence of accurate trans-splicing between the endogenous COL7A1 pre-mRNA and the introduced RTM. Additionally, the restoration of type VII collagen production in RTM-transduced RDEB patient keratinocytes was confirmed by an immunofluorescence staining specific for type VII collagen in cultured RDEB patient cells transduced with the RTM (Figure 5a3) and two control cell lines (Figures 5a1 and a2). The type VII collagen- and FLAG-stained samples, respectively, were merged with a DAPI (4′,6-diamidino-2-phenylindole) staining. RDEB patient cells showed a faint fluorescence signal, whereas wild-type human keratinocytes, serving as positive control for type VII collagen expression, showed a strong fluorescence signal. As a consequence, type VII collagen protein expression was increased in the RTM28-transduced RDEB patient keratinocytes compared with untreated RDEB cells. Second step controls did not show any specific staining (data not shown). These data could be confirmed by western blot analysis using cell lysates of the respective cell lines (Figure 5b). RTM28-transduced RDEB keratinocytes showed a weak type VII collagen band at the expected size of ~290 kDa, indicating accurate trans-splicing (Figure 5b3). Further, no band was detectable in wild-type keratinocytes (Figure 5b4; lower panel) and untransduced RDEB keratinocytes (Figure 5b5; lower panel) using a FLAG-specific antibody, whereas a specific ~293 kDa FLAG-tagged trans-spliced type VII collagen was visible in lysates of RTM28-transduced RDEB keratinocytes (Figure 5b6; lower panel).

Correction of type VII collagen in RTM28-transduced patient keratinocytes. (a) Immunofluorescene staining of a wild-type human keratinocyte line (1, 4), an RDEB keratinocyte line (2, 5) and an RTM28-transduced RDEB keratinocyte line (3, 6). The upper panel shows type VII collagen staining using an antibody against the NC1 domain of type VII collagen, whereas staining against the FLAG tag is shown in the lower panel. Staining with the type VII collagen-specific antibody resulted in a strong signal in wild-type human keratinocytes (1), while the staining was absent in the untransduced RDEB keratinocyes (2). RTM28-transduced RDEB keratinocytes displayed a specific staining with the anti-type VII collagen antibody, corresponding to corrected type VII collagen expression upon trans-splicing (3). As expected, wild-type keratinocytes (4) and RDEB keratinocytes (5) did not show a specific signal using an anti-FLAG tag antibody, whereas staining of RTM28-transduced RDEB keratinocytes (6) resulted in a specific signal (green) in the cytoplasm of the cells. The cell nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. (b) Western blot analysis of cell lysates using either an antibody against type VII collagen (upper panel) or the FLAG tag (lower panel). Wild-type human keratinocytes showed a strong type VII collagen band (1), whereas there was no band detectable in untransduced RDEB keratinocytes (2). RTM28-transduced RDEB keratinocytes (3) showed a weak band at the correct size of ~290 kDa, indicating accurate trans-splicing. Further, no band was detectable in wild-type keratinocytes (4) and untransduced RDEB keratinocytes (5) using a FLAG-specific antibody, whereas a specific ~293 kDa FLAG-tagged trans-spliced type VII collagen was visible in lysates of RTM28-transduced RDEB keratinocytes (6). Actinin staining served as loading control.
Discussion
Here we have described the stepwise design of the longest RNA trans-splicing construct designed for the correction of skin diseases up to now, RTM28, capable of correcting a homozygous nonsense mutation in COL7A1 exon 105 in keratinocytes from an RDEB patient by accurate trans-splicing. RTM28 is a functional 3′ RTM, which will also be able to correct any other pathogenic mutation (~450) occurring over a 3′ region of >4 kb of the COL7A1 transcript (http://www.col7.info).
RNA trans-splicing is an elegant tool for the repair of pathogenic mutations in inherited diseases such as DEB, especially when large transcript sizes complicate the application of a conventional gene replacement therapy approach. Our RNA-based therapy approach allows reduction of the size of the transgene, significantly increasing the choice of viral or non-viral vectors used to deliver the curative coding sequence into patient’s cells. RNA trans-splicing shows further advantages over gene replacement approaches such as increased efficiency of viral cell transductions due to reduced transgene size, the prevention of overexpression or ectopic expression of the transgene and the possibility to decrease dominant-negative gene products in dominantly inherited diseases. Furthermore, mRNA trans-splicing offers the opportunity for non-viral delivery into the skin using gene transfer methods such as the gene gun technology23 or microneedles.24
Although RNA trans-splicing-induced RNA repair strategies were successfully applied for the restoration of gene functions in preclinical models for various diseases such as cystic fibrosis,25, 26, 27 spinal muscular atrophy,28, 29, 30 haemophilia A,31 tauopathies,32, 33 EB simplex,34 X-linked immunodeficiency with hyper-IgM,35 severe combined immune deficiency,36 myotonic dystrophy type 1,37 frontotemporal dementia with parkinsonism linked to chromosome 17,32 Duchenne muscular dystrophy38 and dysferlinopathy,39 there is still scope for improvement of the trans-splicing efficiency to achieve a sustained and efficient correction of the mutated transcript. In recent studies, we have reported that the BD of the RTM is crucial for the efficiency of the trans-splicing process. Various experiments have shown that minor variations in length, sequence or localization of the target-binding position influence the splicing characteristics of a given RTM.20, 21, 22, 40 The rational design of RTMs is difficult since up to now only few guidelines for generating potential RTMs exist. Consequently, we have recently developed an RTM reporter-based screening system that accelerates and facilitates RTM construction for any target pre-mRNA region of choice in a co-transfection assay.15 In the present work, we extended the usefulness and robustness of this screening system by the generation of a stable target minigene expressing cell line, which allows for rapid screening of highly potent RTMs. Here we present the reliability of our improved reporter-based screening system that enabled the selection of an efficient RTM capable of inducing the restoration of type VII collagen in transduced RDEB patient keratinocytes by accurate trans-splicing in vitro.
We aimed to increase the size of the COL7A1 cDNA sequence to be replaced, to correct all possible mutations occurring on the 3′ half of the gene, covering a sequence of over 4200 nucleotides. Therefore, we adapted our BD screening procedure to the COL7A1 pre-mRNA region intron 46/exon 47. The inclusion of a stably expressed COL7A1-MG target in an easy to transfect cell line improved the RTM screening procedure as the target sequence expression is more uniform compared with the co-transfection system, where the RTM and MG were introduced separately.41 Therefore, this new system is comparable to the endogenous situation in patient cells. First attempts to create a stable COL7A1-MG-expressing cell line using a retroviral vector system for stable transduction of HEK293 cells was not successful, due to cryptic splicing events within the MG during viral packaging (data not shown). Therefore, we used a transposon-based delivery system for COL7A1-MG integration. Transposon-based systems are simple and efficient transfection tools, suitable for a variety of gene transfer applications. These plasmid-based gene delivery vehicles represent alternatives to popular integrating viral approaches to avoid the disadvantages of viral-based delivery systems. Advantages of the transposon-based systems over viral vectors include immune tolerance, decreased preference for integration into genes and increased cargo capacity.42, 43 Recently, the eukaryotic transposon pB, isolated from the moth Trichoplusia ni, has emerged as a highly efficient gene delivery vector for numerous in vitro and in vivo applications. To examine the feasibility of the pB transposon system, the COL7A1-MG was stably integrated into the genome of HEK293 cells with the aim that the cells mimic endogenous target pre-mRNA expression at a constant level. The COL7A1-MG cell line was transfected with RTMs carrying different BDs for the target region following functional analysis using flow cytometry. To prove the reliability of our screening systems with respect to the trans-splicing efficiency, we compared both systems by flow cytometric analysis.
In general, we found that the near proximity of the RTM and the COL7A1-MG pre-mRNA enhances the possibility that accurate RNA trans-splicing takes place, resulting in the fusion of split GFP parts, provided by both transcripts, and subsequently to the restoration of full-length GFP expression. The intensity of the GFP signal correlates with the efficiency of introduced RTMs varying in their binding behaviour to the target sequence. This is true for both the co-transfection system and the RTM transfection into the COL7A1-MG cell line, and we obtained comparable results for our most efficient RTM28 regardless of whether a transient or endogenous setting was applied. A high trans-splicing efficiency was detectable by flow cytometric analysis in the presence of transient (~88% GFP-expressing cells) or stable COL7A1-MG (78% GFP-expressing cells) expressing plasmids. The slightly reduced trans-splicing efficiency in the stable cell line compared with co-transfection studies can be explained by a lower expression level of the stably integrated MG compared with that of the plasmid provided by transient transfection. However, our aim is to correct mutations in the endogenously expressed pre-mRNA in patient cells, and therefore our MG cell line reflects a more comparable situation to a clinical setting. Most importantly, we have shown that our selected RTM28 was capable of type VII collagen correction in an RDEB patient keratinocyte line harbouring a homozygous nonsense mutation in exon 105. After RTM transduction into RDEB keratinocytes, the reprogrammed chimeric COL7A1-RTM mRNA was detectable by sqRT-PCR analysis, and a fivefold increase in the total COL7A1 mRNA level was measured. Additionally, immunofluorescence staining of type VII collagen and of the FLAG tag integrated in the trans-spliced protein revealed the presence of corrected type VII collagen in RTM-treated patient cells.
These results demonstrate that a robust RTM screening system provides a solid basis on which to select an efficient RTM before its application in a more endogenous setting. The stable COL7A1-MG-expressing cell line can be used for future investigations to analyse which factors influence the splicing characteristics of a designed RTM.40 Taken together, these results underscore the potential of RNA trans-splicing to induce correction of inherited monogenic disorders and we suggest trans-splicing as a promising technology paving the way towards an RNA therapy approach in RDEB patients, with the potential to be adapted to other monogenetic diseases.
Materials and methods
Construction of COL7A1-MG
The target molecule, termed COL7A1-MG, for the fluorescence-based screening procedure was designed and cloned according to Bauer et al.19 The target region, consisting of COL7A1 intron 46 and exon 47, was amplified from human genomic DNA using a forward primer including an EcoRV restriction site (5′-CTAGGATATCGTGAGTGACGGGAGGATG-3′) and a reverse primer including a NotI site (5′-CTAGGCGGCCGCCTTTTCTCCTTTGGGTCCAGC-3′). The PCR fragment was cloned into the target screening vector using the restriction sites for EcoRV and NotI (both Fermentas, Thermo Scientific, Waltham, MA, USA). All DNA plasmids and PCR products were verified by sequence analysis using a 3130 ABI Prism Automated Sequencer and ABI PRISM Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Waltham, MA, USA).
Generation of a stable COL7A1-MG-expressing cell line
The COL7A1-MG was stably integrated into the genome of HEK293AD (Stratagene, Santa Clara, CA, USA), a cell line that does not express COL7A1, using the PB transposase-based delivery system (System Biosciences (SBI), Mountain View, CA, USA) to mimic an endogenous target pre-mRNA expression at a constant level. The COL7A1-MG was transferred from the target screening vector to the PB transposon vector (PB514B-2 PB-CMV-MCS-EF1 vector; System Biosciences) using the restriction enzymes BamHI and NotI for cloning. HEK293 cells were seeded in 60 mm plates and were co-transfected with the PB transposon vector, harbouring the COL7A1-MG and an mRuby-puromycin cassette, together with a PB transposase expression vector using the jetPEI reagent (Polyplus-transfection SA, Illkirch, France) according to the manufacturer’s protocol. Antibiotic selection with puromycin was performed for 10 days at a concentration of 2 μg ml−1. Full-length integration of the COL7A1-MG into the cell’s genome was further confirmed by target-specific PCR amplification from genomic DNA, using a vector-specific forward primer (5′-TGGATAGCGGTTTGACTCAC-3′) and a COL7A1-MG-specific reverse primer (5′-CATTCAGCTCGATCAGGATGGGCACGATGCC-3′) (data not shown).
Construction of the RTM library
To construct an RTM library for the GFP screening system, containing randomly generated BDs, intron 46 and exon 47 of COL7A1 were PCR-amplified using Pfu Turbo polymerase (Stratagene, La Jolla, CA, USA) and human genomic wild-type DNA as PCR template. The resulting 420 bp fragment was purified after gel electrophoresis using the GFX Gel Purification Kit (GE Healthcare, Buckinghamshire, UK). The 3′ BD library was constructed by fragmentation of the PCR-amplified target sequence of COL7A1 by CviJI* digestion (Roboklon, Berlin, Germany). Partial digestion of the target region was performed for 10 s at 37 °C. The RTM vector backbone, carrying the reporter molecule dsRed and the 3′ portion of GFP,19 was digested with HpaI for 1 h at 37 °C and subsequently treated with calf intestine alkaline phosphatase (Thermo Fisher Scientific, Waltham, MA, USA) for further 15 min at 37 °C. The DNA fragments created from CviJI* digestion were ligated (T4 DNA ligase; New England Biolabs, Ipswich, MA, USA) into the linearized RTM vector for 3 h at room temperature and overnight at 4 °C. Chemical transformation of the ligation was performed into DH5α chemically competent cells (Invitrogen, Waltham, MA, USA) according to the manufacturer’s protocol. After cloning, the presence of RTMs with distinct BDs was analysed by colony PCR using Firepol polymerase I (Solis Biodyne, Tartu, Estonia) and vector-specific primers (forward: 5′-TAATACGACTCACTATAGGG-3′; reverse: 5′-CATTGTGGGCGTTGTAG-3′). Eighty single clones were analysed by PCR. Sequence analysis was performed to determine the exact BD sequence.
Constructs for retroviral delivery
The engineered RTM for endogenous experiments consisted of a 120 bp BD, complementary to two different regions (38 and 79 nucleotides in length, respectively, separated through 38 nucleotides) within intron 46 of COL7A1, a 31 bp spacer sequence, a branch point (BP) sequence and a polypyrimidine tract followed by a 3′ acceptor splice site and the 3′ wild-type coding sequence (4200 bp) of human COL7A1. The functional BD (BD28) was amplified from the RTM screening vector using GoTaq polymerase (Promega, Mannheim, Germany) according to the manufacturer’s protocol. The forward primer included a BamHI restriction site (5′-GATCGGATCCCTCAGTCCCCGCCCAGAAGTCACAG-3′), whereas the reverse primer inserted a single EcoRI site (5′-CTAGGAATTCCTCGTTCTCTCCTTCTGCTAGAAC-3′). The amplified PCR products were digested with BamHI and EcoRI (Fermentas) and cloned into the pMXs-IRES-Blasticidin retroviral vector (Cell Biolabs, San Diego, CA, USA). Correct insertion was confirmed by sequence analysis. For specific detection and distinction between the trans- and cis-spliced COL7A1 product at the protein level, a FLAG tag was included at the 3′ end of the COL7A1 coding sequence on the RTM. A start codon (ATG) within the BD sequence was modified (ATG→ATC) via in vitro mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) to reduce the possibility of direct RTM expression.
The 4.5 kb 3′ sequence of human COL7A1, spanning from exons 47 to 118 together with the FLAG sequence was amplified using the long-range DNA polymerase (Roche, Mannheim, Germany). The forward primer included an EcoRI restriction site, the spacer sequence, a BP sequence and a polypyrimidine tract sequence (5′-GATCGAATTCTCTTCTTTTTTTTCTGCAGGGACCTGCTGTTGCTGGACCC-3′). The reverse primer included the FLAG sequence and a NotI restriction site (5′-CTAGGCGGCCGCTCATTTATCATCATCATCTTTATAATCGTCCTGGGCAGTACCTGTC-3′). The resulting PCR product was cloned into the EcoRI/NotI-digested pMXs-IRES-Blasticidin retroviral vector, containing either BD28 (RTM28) or no BD (RTM w/o BD).
To enhance FLAG signals in immunofluorescence (IF) and western blot analysis a 3xFLAG tag instead of a 1xFLAG tag was introduced. The 4.5 kb 3′ sequence of human COL7A1, spanning from exons 47 to 118 together with the BD28 were cloned into the CMV-3xFLAG-14.1 vector (Sigma-Aldrich, St Louis, MO, USA). In the first cloning step, the 5′ part spanning from exons 47 to 94 was cut out of the pMXs-BD28-Ex47-Ex118-FLAG-IRES-Blasticidin retroviral vector using EcoRI/BglII (Fermentas) and cloned into the CMV-3xFLAG-14.1 vector. In a second step, the BD28 was PCR amplified using a forward (5′-GATCGAATTCCTCAGTCCCCGCCCAGAAGTCACAG-3′) and a reverse primer (5′-GATCGAATTCAGTTAGTACTCGAGCAACGCTATAATAATGTTC-3′), which both included an EcoRI restriction site. The amplified PCR products were digested with EcoRI (Fermentas) and cloned into the CMV-Ex47-Ex94-3xFLAG-14.1 vector. Subsequently, the 3′ part spanning from exons 86 to 118 was amplified. The forward primer bound to exon 86 (5′-GAAAAGATGGAGACAGAGGGAGCCC-3′), whereas the reverse primer inserted a single XbaI site (5′-GATCTCTAGAGTCCTGGGCAGTACCTGTCCCCTGG-3′). The amplified PCR products were digested with BglII and XbaI (Fermentas) and cloned into the CMV-BD28-Ex47-Ex94-3xFLAG-14.1 vector. For retroviral delivery the RTM, containing BD28, spacer sequence, a BP sequence and a polypyrimidine tract followed by a 3′ acceptor splice site, Ex47-Ex118 and the 3xFLAG tag, was amplified using a BD28 forward primer, which included an EcoRI restriction site (5′-GATCGAATTCCTCAGTCCCCGCCCAGAAGTCACAG-3′) and a reverse primer, which included a NotI restriction site (5′-GATCGCGGCCGCCTACTTGTCATCGTCATCCTTGTAGTCGATGTC-3′). The amplified PCR product was digested with EcoRI and NotI (Fermentas) and cloned into the pMXs-IRES-Blasticidin retroviral vector, containing BD28 (RTM28).
Cell culture and transfection
For all co-transfection experiments, the HEK293 cell line was used. HEK293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% foetal calf serum and 100 U ml−1 penicillin/streptomycin (Biochrom, Berlin, Germany) at 37 °C and 5% CO2 in a humidified incubator. RTM plasmids harbouring different BDs with a sequence complementary to the target sequence were transfected into COL7A1-MG-expressing HEK293 cells using the jetPEI reagent (Polyplus-transfection SA) according to the manufacturer’s protocol. The cells were analysed 48 h after transfection by fluorescence microscopy and flow cytometric analysis. The trans-splicing efficiency was measured by quantification of GFP-expressing cells.
Retroviral transduction of human RDEB patient keratinocytes
For the production of infectious retroviral particles, Phoenix amphotropic cells (ATCC LGC Standards GmbH, Wesel, Germany) were used. Cells were seeded in and grown until 60% confluence in Dulbecco's modified Eagle's medium (Hyclone, Perbio Science, Bezons, France) containing 10% FCII (Hyclone), 4 mm l-glutamine (Sigma-Aldrich) and 1 mm Na-pyruvate. The viral plasmid DNA was transfected into the amphotropic Phoenix packing cells using the jetPEI transfection reagent (Polyplus-transfection SA). At 24 h after transfection and incubation at 37 °C, the medium was changed and cells were further cultivated at 32 °C in Dulbecco's modified Eagle's medium (Hyclone, Perbio Science, Bezons, France) containing 10% FCII (Hyclone). Supernatant containing infectious viral particles was harvested after 48 h up to 96 h every 8 h and stored at 4 °C until keratinocyte transduction. An RDEB keratinocyte line derived from a patient harbouring a homozygous mutation in exon 105 (R2610X) was used, which was kindly provided by Prof Guerrino Meneguzzi (Nice, France). Keratinocytes were seeded into T-25 flasks (0.8 × 106 cells) and transduced at a confluence of 50% with the viral suspension in the presence of 5 μg ml−1 of polybrene (Sigma-Aldrich) at 32 °C in humid atmosphere (5% CO2). Flasks were centrifuged at 600 g for 90 min. After the centrifugation step, 2.5 ml of SFM keratinocyte medium (Keratinocyte-SFM Medium; Thermo Scientific) was added and transduced cells were incubated overnight at 32 °C. After washing the cells four times with Dulbecco's phosphate-buffered saline (PBS) (Biochrom), the keratinocytes were incubated for 2 days in cell-specific medium in the presence of 25 mg l−1 Primocin (Invivogen, San Diego, CA, USA). Cells were selected using blasticidin (Invitrogen) for 8 days at a concentration of 200 mg l−1.
RNA isolation and cDNA synthesis of cultured adherent cells
Cells were harvested at 100% confluence (1 × 107 cells) and RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Purified RNA (1–2 μg) was digested with DNase I (Sigma-Aldrich) for 20 min at room temperature, which was subsequently inactivated by incubation for 10 min at 70 °C, followed by cDNA synthesis using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA).
Semiquantitative RT-PCR
SqRT-PCR was performed to detect the trans-spliced fusion mRNA. To distinguish cis- from trans-splicing, six silent mutations were inserted within exon 49 of COL7A1 on the RTM vector. For amplification of the trans-spliced COL7A1 transcripts, the following primers were used: forward primer, 5′-CACCAGGACCCACTGGCCGCCA-3′ and reverse primer, 5′-GGATCGCCTTTCGGCCCTG-3′. To quantify the relative amounts of total COL7A1 mRNA, we used a COL7A1 exon 17-specific forward primer (5′-GTGAGGACTGCCCCTGAG-3′) and a COL7A1 exon 19-specific reverse primer (5′-GACTCCACCTTCGAGACCC-3′). PCR conditions were 3 min at 95 °C followed by 50 cycles of 30 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C. SqRT-PCR was performed using the Bio-Rad iCycler System and Bio-Rad iQ SYBR Green Supermix Kit (Bio-Rad), and all individual reactions were performed in duplicates and standard deviation was calculated from three independently performed experiments. Transcript levels were calculated after normalization to glyceraldehyde 3-phosphate dehydrogenase.
Flow cytometric analysis
GFP expression in treated HEK293 cells was quantified 48–96 h after transfection using a Beckman Coulter FC-500 FACS (Beckman Coulter, Brea, CA, USA) analyser. The CXP software (Beckman Coulter) was used for data analysis.
Immunofluorescence staining of type VII collagen in cultured cells
For immunofluorescence analysis of monolayer keratinocyte cultures, cells (5.5 × 104) were seeded into chamber slides (growth surface 1.7 cm2). After 24 h, the cells were fixed in cold 4% paraformaldehyde for 30 min and afterwards permeabilized in 1% BSA 0.5% Triton X-100 in PBS. As primary antibody against the FLAG tag a polyclonal anti-FLAG antibody (Sigma-Aldrich) (1:200 in PBS) was used. As secondary antibody Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) (1:400 in PBS) was used. The cells were additionally analysed for COL7A1 expression. The cells were incubated with a rabbit serum directed against type VII collagen (kindly provided by Dr Alexander Nyström, Freiburg, Germany) diluted at 1:1000 in PBS. The secondary antibody used was Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen). Cells were analysed using an epifluorescence Zeiss Axiophot microscope (Carl Zeiss, Oberkochen, Germany).
Western blot analysis of trans-spliced type VII collagen
RTM28-transduced RDEB keratinocytes were grown to confluence in conditioned medium. The medium was changed containing 50 μg ml−1 ascorbic acid, and the cultures were maintained for an additional 48 h. Cells were harvested and lysed in 50–100 μl RIPA lysis buffer (Santa Cruz Biotechnology, Dallas, TX, USA). After 20 min of centrifugation at 4 °C, 10–20 μg protein samples were mixed with 4x sodium dodecyl sulphate-polyacrylamide gel electrophoresis sample buffer (0.25 m Tris-HCl; 10% sodium dodecyl sulphate; 40% glycerol; 0.008% bromphenol blue; 2.86 m β-mercaptoethanol; pH 6.8), denatured for 5 min at 95 °C and loaded onto a Bolt 8% Bis-Tris Plus Gel (Novex, Life Technologies, Carlsbad, CA, USA). Western blot analysis was performed as described previously.23, 40 For detection of type VII collagen, the primary antibody rabbit anti-type VII collagen (kindly provided by Dr Alexander Nyström) was added to the blocking solution at a dilution of 1:3000 and incubated overnight at 4 °C, followed by incubation with the secondary antibody, anti-rabbit horse radish peroxidase (Dako, Glostrup, Denmark) 1:1000 in blocking buffer (Roche) for 1 h at room temperature.
For the detection of FLAG-tagged fusion proteins, blots were incubated with the anti-FLAG M2mAb (Sigma-Aldrich) diluted at 1:1000 in blocking buffer (Roche).
References
Fine JD, Hintner H (eds). Life with Epidermolysis Bullosa: Etiology, Diagnosis, and Multidisciplinary Care and Therapy. Springer: Wien, Germany and New York, NY, USA, 2009.
Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70: 1103–1126.
Lin AN, Carter DM . Epidermolysis bullosa. Annu Rev Med 1993; 44: 189–199.
Wertheim-Tysarowka K, Sobczyńska-Tomaszewska A, Kowalewski C, Skroński M, Swięćkowski G, Kutkowska-Kaźmierczak A et al. The COL7A1 mutation database. Hum Mutat 2012; 33: 327–331.
Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med 2010; 363: 629–639.
Wong T, Gammon L, Liu L, Mellerio JE, Dopping-Hepenstal PJ, Pacy J et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2008; 128: 2179–2189.
Petrof G, Lwin SM, Martinez-Queipo M, Abdul-Wahab A, Tso S, Mellerio JE et al. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2015; 135: 2319–2321.
Titeux M, Pendaries V, Zanta-Boussif MA, Décha A, Pironon N, Tonasso L et al. SIN retroviral vectors expressing COL7A1 under human promoters for ex vivo gene therapy of recessive dystrophic epidermolysis bullosa. Mol Ther 2010; 18: 1509–1518.
Siprashvili Z, Nguyen NT, Gorell E, Khuu P, Furukawa L, Lorenz HP et al. Phase I clinical trial of genetically corrected autologous epidermal keratinocytes for recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2014; 134: 75.
Woodley DT, Wang X, Amir M, Hwang B, Remington J, Hou Y et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol 2013; 133: 1910–1913.
Murauer EM, Koller U, Pellegrini G, De LM, Bauer JW . Advances in gene/cell therapy in epidermolysis bullosa. Keio J Med 2015; 64: 21–25.
Wally V, Murauer EM, Bauer JW . Spliceosome-mediated trans-splicing: the therapeutic cut and paste. J Invest Dermatol 2012; 132: 1959–1966.
Koller U, Wally V, Bauer JW, Murauer EM . Considerations for a successful RNA trans-splicing repair of genetic disorders. Mol Ther Nucleic Acids 2014; 3: e157.
Puttaraju M, Jamison SF, Mansfield SG, Garcia-Blanco MA, Mitchell LG . Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat Biotechnol 1999; 17: 246–252.
Wally V, Brunner M, Lettner T, Wagner M, Koller U, Trost A et al. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum Mol Genet 2010; 19: 4715–4725.
Peking P, Koller U, Hainzl S, Kocher T, Mayr E, Kitzmueller S et al. Non-viral RNA trans-splicing strategy for in situ treatment of dystrophic epidermolysis bullosa. Mol Ther Nucleic Acids 2016; 5: e287.
Sahin U, Kariko K, Tureci O . mRNA-based therapeutics—developing a new class of drugs. Nat Rev Drug Discov 2014; 13: 759–780.
Pardi N, Muramatsu H, Weissman D, Kariko K . In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol 2013; 969: 29–42.
Bauer JW, Murauer EM, Wally V, Koller U . RNA trans-splicing for genodermatoses. Methods Mol Biol 2013; 961: 441–455.
Murauer EM, Koller U, Hainzl S, Wally V, Bauer JW . A reporter-based screen to identify potent 3' trans-splicing molecules for endogenous RNA repair. Hum Gene Ther Methods 2013; 24: 19–27.
Koller U, Wally V, Mitchell LG, Klausegger A, Murauer EM, Mayr E et al. A novel screening system improves genetic correction by internal exon replacement. Nucleic Acids Res 2011; 39: e108.
Murauer EM, Gache Y, Gratz IK, Klausegger A, Muss W, Gruber C et al. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Invest Dermatol 2011; 131: 74–83.
Peking P, Koller U, Hainzl S, Kitzmueller S, Kocher T, Mayr E et al. A gene gun-mediated nonviral rna trans-splicing strategy for Col7a1 repair. Mol Ther Nucleic Acids 2016; 5: e287.
Pearton M, Saller V, Coulman SA, Gateley C, Anstey AV, Zarnitsyn V et al. Microneedle delivery of plasmid DNA to living human skin: formulation coating, skin insertion and gene expression. J Control Rel 2012; 160: 561–569.
Liu X, Jiang Q, Mansfield SG, Puttaraju M, Zhang Y, Zhou W et al. Partial correction of endogenous DeltaF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat Biotechnol 2002; 20: 47–52.
Mansfield SG, Kole J, Puttaraju M, Yang CC, Garcia-Blanco MA, Cohn JA et al. Repair of CFTR mRNA by spliceosome-mediated RNA trans-splicing. Gene Therapy 2000; 7: 1885–1895.
Liu X, Luo M, Zhang LN, Yan Z, Zak R, Ding W et al. Spliceosome-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells. Hum Gene Ther 2005; 16: 1116–1123.
Coady TH, Baughan TD, Shababi M, Passini MA, Lorson CL . Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS One 2008; 3: e3468.
Coady TH, Shababi M, Tullis GE, Lorson CL . Restoration of SMN function: delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol Ther 2007; 15: 1471–1478.
Coady TH, Lorson CL . Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J Neurosci 2010; 30: 126–130.
Chao H, Mansfield SG, Bartel RC, Hiriyanna S, Mitchell LG, Garcia-Blanco MA et al. Phenotype correction of hemophilia A mice by spliceosome-mediated RNA trans-splicing. Nat Med 2003; 9: 1015–1019.
Rodriguez-Martin T, Anthony K, Garcia-Blanco MA, Mansfield SG, Anderton BH, Gallo JM . Correction of tau mis-splicing caused by FTDP-17 MAPT mutations by spliceosome-mediated RNA trans-splicing. Hum Mol Genet 2009; 18: 3266–3273.
Rodriguez-Martin T, Garcia-Blanco MA, Mansfield SG, Grover AC, Hutton M, Yu Q et al. Reprogramming of tau alternative splicing by spliceosome-mediated RNA trans-splicing: implications for tauopathies. Proc Natl Acad Sci USA 2005; 102: 15659–15664.
Wally V, Brunner M, Lettner T, Wagner M, Koller U, Trost A et al. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum Mol Genet 2010; 19: 4715–4725.
Tahara M, Pergolizzi RG, Kobayashi H, Krause A, Luettich K, Lesser ML et al. Trans-splicing repair of CD40 ligand deficiency results in naturally regulated correction of a mouse model of hyper-IgM X-linked immunodeficiency. Nat Med 2004; 10: 835–841.
Zayed H, Xia L, Yerich A, Yant SR, Kay MA, Puttaraju M et al. Correction of DNA protein kinase deficiency by spliceosome-mediated RNA trans-splicing and sleeping beauty transposon delivery. Mol Ther 2007; 15: 1273–1279.
Chen HY, Kathirvel P, Yee WC, Lai PS . Correction of dystrophia myotonica type 1 pre-mRNA transcripts by artificial trans-splicing. Gene Therapy 2009; 16: 211–217.
Lorain S, Peccate C, Le HM, Griffith G, Philippi S, Precigout G et al. Dystrophin rescue by trans-splicing: a strategy for DMD genotypes not eligible for exon skipping approaches. Nucleic Acids Res 2013; 41: 8391–8402.
Monjaret F, Bourg N, Suel L, Roudaut C, Le RF, Richard I et al. Cis-splicing and translation of the pre-trans-splicing molecule combine with efficiency in spliceosome-mediated RNA trans-splicing. Mol Ther 2014; 22: 1176–1187.
Koller U, Hainzl S, Kocher T, Hüttner C, Klausegger A, Gruber C et al. Trans-splicing improvement by the combined application of antisense strategies. Int J Mol Sci 2015; 16: 1179–1191.
Wally V, Koller U, Bauer JW. High-throughput screening for highly functional RNA-trans-splicing molecules: correction of plectin in epidermolysis bullosa simplex. In: Plaseska-Karanfilska D (ed). Human Genetic Diseases. InTech Open Access Publisher: Rijeka, Croatia, 2011, pp 223–240.
Hackett PB, Largaespada DA, Cooper LJ . A transposon and transposase system for human application. Mol Ther 2010; 18: 674–683.
Owens JB, Mathews J, Davy P, Stoytchev I, Moisyadi S, Allsopp R . Effective targeted gene knockdown in mammalian cells using the piggyBac transposase-based delivery system. Mol Ther Nucleic Acids 2013; 2: e137.
Acknowledgements
We thank Prof Guerrino Meneguzzi from Nice for kindly providing the RDEB patient keratinocyte line, and Dr Alexander Nyström from the Department of Dermatology, University Medical Center, Freiburg, Germany who kindly provided the type VII collagen antibody. We also thank Dr Andrea Zurl from the University Clinic of Ophthalmology and Optometry Research Program for Experimental Ophthalmology and Glaucoma Research, SALK, Salzburg, Austria for assistance with immunofluorescence microscopy. Additionally, we thank Dr Thomas Lener from the University Clinic of Blood Group Serology and Transfusion Medicine, Core Facility for Microscopy, SCI-TRECS, Paracelsus Medical University, Salzburg, Austria for performing fluorescence microscopy. This work was supported by DEBRA Austria, and by grants from DEBRA International (Bauer 4) to JWB, INTERREG IV (Therapie für Schmetterlingskinder; project ID 4162) to JWB and the Austrian Science Fund (FWF) under the frame of Erare-2 (SpliceEB; Project I1175-B13) to EMM.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
JWB is an inventor on US (US8735366) and European (EP2320952) patent for ‘Improved pre-mRNA trans-splicing molecules (RTM) and their uses’. The other authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tockner, B., Kocher, T., Hainzl, S. et al. Construction and validation of an RNA trans-splicing molecule suitable to repair a large number of COL7A1 mutations. Gene Ther 23, 775–784 (2016). https://doi.org/10.1038/gt.2016.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2016.57
This article is cited by
-
Pathogenetic Therapy of Epidermolysis Bullosa: Current State and Prospects
Bulletin of Experimental Biology and Medicine (2021)
-
Molecular Therapeutics in Development for Epidermolysis Bullosa: Update 2020
Molecular Diagnosis & Therapy (2020)
-
Inside out: regenerative medicine for recessive dystrophic epidermolysis bullosa
Pediatric Research (2018)
