
Abstract
The Central American seasonally dry tropical (SDT) forest biome is one of the worlds’ most endangered ecosystems, yet little is known about the genetic consequences of its recent fragmentation. A prominent constituent of this biome is Calycophyllum candidissimum, an insect-pollinated and wind-dispersed canopy tree of high socio-economic importance, particularly in Nicaragua. Here, we surveyed amplified fragment length polymorphisms across 13 populations of this species in Nicaragua to elucidate the relative roles of contemporary vs historical factors in shaping its genetic variation. Genetic diversity was low in all investigated populations (mean HE=0.125), and negatively correlated with latitude. Overall population differentiation was moderate (ΦST=0.109, P<0.001), and Bayesian analysis of population structure revealed two major latitudinal clusters (I: ‘Pacific North’+’Central Highland’; II: ‘Pacific South’), along with a genetic cline between I and II. Population-based cluster analyses indicated a strong pattern of ‘isolation by distance’ as confirmed by Mantel’s test. Our results suggest that (1) the low genetic diversity of these populations reflects biogeographic/population history (colonisation from South America, Pleistocene range contractions) rather than recent human impact; whereas (2) the underlying process of their isolation by distance pattern, which is best explained by ‘isolation by dispersal limitation’, implies contemporary gene flow between neighbouring populations as likely facilitated by the species’ efficient seed dispersal capacity. Overall, these results underscore that even tree species from highly decimated forest regions may be genetically resilient to habitat fragmentation due to species-typical dispersal characteristics, the necessity of broad-scale measures for their conservation notwithstanding.
Similar content being viewed by others
Introduction
The seasonally dry tropical (SDT) forest of Central America ranks among the most threatened forest ecosystems in the tropical lowlands and is even more endangered than the tropical rainforests of Amazonia (Janzen, 1988; Pennington et al., 2006). Formerly extending over very large areas along the Pacific side of Central America, these SDT forest habitats have been so seriously impacted by human activities that nearly all of them have been lost. Historically, the first clearings of these forests probably date back to at least the Archaic Agricultural Period (ca. 6000–5550 BP) and destruction intensified after the 16th century Spanish Conquest (Janzen, 1988; Piperno, 2011). Nonetheless, up to the early-to-mid 20th century, there were still ca. 550 000 km2 of SDT forest left, extending in an almost continuous strip along the Pacific side of Central America, from south-western Mexico to north-western Costa Rica, and disjunctively to Panama’s Azuero Peninsula (Janzen, 1988; Olson et al., 2001; Griscom and Ashton, 2011). It is now estimated that only 2.0–1.7% of the original SDT forest in Central America escaped deforestation, degradation and conversion into agricultural land and/or pastures, especially between the 1930s and 1970s (Griscom and Ashton, 2011), and only <0.1% of this remaining forest is protected (Miles et al., 2006). This massive habitat loss and disturbance of Central American SDT forests is well recognised today as a major threat to the region’s high biodiversity and endemism (Janzen, 1988; Sánchez-Azofeifa et al., 2005; Linares-Palomino et al., 2011). However, relatively little is known about the impact of this forest degradation on regional-scale genetic structure of constituent tree species.
Especially for outcrossing tree species it has long been assumed that population isolation and decreases in population size associated with anthropogenic forest fragmentation can lead to genetic erosion and increased population differentiation through increased genetic drift, inbreeding and/or reduced gene flow. In turn, such genetic erosion may negatively affect the long-term viability and adaptive potential of remnant populations (Aguilar et al., 2008; Frankham et al., 2010). However, there is increasing evidence that a large number of forest tree species (from both temperate and tropical regions) are able to ameliorate the population genetic consequences of recent habitat fragmentation (reviewed in Lowe et al., 2005, 2015). Potential mechanisms underlying such ‘resilience’ include a flexible mating system (Ward et al., 2005), extensive gene flow via pollen and/or seed (Bacles et al., 2006) and/or longevity coupled with overlapping generations (Petit and Hampe, 2006; Davies et al., 2010; but for exceptions see, for example, Kettle et al., 2007; Dubreuil et al., 2010).
In Central America, genetic surveys of SDT forest trees are relatively scarce, highly heterogeneous (for example, in terms of geographical scale of sampling, time or severity of impact, species’ life history/dispersal characteristics) and mostly limited to heavily exploited timber trees (Lowe et al., 2005; Ratnam et al., 2014). Nonetheless, the few broad-scale studies available typically identified low-to-moderate spatial genetic structure (for example, Chase et al., 1995; Gillies et al., 1999; Novick et al., 2003; Poelchau and Hamrick, 2012; but see Cavers et al., 2003, 2013), whereas more fine-scale surveys usually found little or no significant impact of recent habitat loss on genetic diversity parameters (for example, Dawson et al., 1997; White et al., 2002; Fuchs et al., 2003; Lowe et al., 2005; but see Gillies et al., 1999). Overall, these assessments emphasise the need for more individual case studies, as the genetic impacts of fragmentation likely vary by species, geographical scale, current environmental conditions and/or historical context (Lowe et al., 2005; Poelchau and Hamrick, 2013). Moreover, there is a gap left for phylogeographic studies of SDT forest trees, especially in those countries that continued to experience particular high rates of deforestation up to the last few decades, namely Honduras, Guatemala and Nicaragua (Redo et al., 2012; Hansen et al., 2013). Such regional surveys are also important to guideline how remaining SDT forest populations may be effectively conserved in situ and optimally sampled for ex situ conservation, breeding and restoration programmes (Ceska et al., 1997; Frankham et al., 2010; Diniz-Filho et al., 2012).
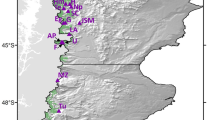
Here, we applied dominant amplified fragment length polymorphism (AFLP) markers to assess genetic variation in Nicaraguan populations of Calycophyllum candidissimum (Vahl) DC. (Rubiaceae, Ixoroideae, Condamineeae), one of the most common constituents of Central America’s degraded SDT forest (Faber-Langendoen et al., 2012). This insect-pollinated and wind-dispersed canopy tree is naturally distributed from southern Mexico through Panama to northern South America (Colombia, Venezuela, Ecuador) and also occurs in the Greater Antilles (Grandtner and Chevrette, 2013; Figure 1a; see Methods section for more details). In Nicaragua, where <1% of the area is covered with SDT forest (Stevens et al., 2001), C. candidissimum is valued for its showy inflorescences (for example, sold during religious festivities) and hard, fine-grained wood (used for, for example, house building, carpentry, cabinet work, tool handles, combs, firewood, charcoal; Standley and Williams, 1975; Cordero and Boshier, 2003). Since 1971, it also has received the distinction of being the country’s ‘National Tree’ (Salas, 1993). Apparently for those reasons, this ‘multi-purpose’ tree has often been left when forests were cleared for agricultural practices, timber, livestock grazing and cotton or other plantations (Barrance et al., 2009; Griscom and Ashton, 2011). Although this habitat degradation has occurred only recently (that is, ca.⩽85 years ago; see above), its presumably negative genetic impact (for example, diversity loss, reduced gene flow) might already be discernible in species with rather short generation times, such as C. candidissimum, which only requires ~3–5 years to first flowering under open-grown conditions (R Dolmus/UNAN-Léon; JB Quezada/Universidad Nacional Agraria (UNA), Managua; personal communications). On the other hand, in this wind-dispersed tree, such negative effects might be mitigated through sustained levels of seed flow, but this is something to be tested against molecular data.

(a) Simplified map of the geographical distribution of C. candidissimum in Central America, the Greater Antilles and northern South America based on geo-referenced specimen records (N=595) available at TROPICOS (http://www.tropicos.org/Name/27903157 (accessed 20 October 2016)) and the Global Biodiversity Facility (GBIF.org (6th September 2016) GBIF Occurrence Download http://doi.org/10.15468/dl.wc19nm). (b) Geographical locations of 13 populations of C. candidissimum in Nicaragua represented in this study for AFLP analysis (red circles), plus additional specimen records (N=90) according to TROPICOS/GBIF (small white circles). The light-brown areas represent the original distribution of seasonal dry forest derived from the World Wildlife Fund (WWF) ecoregion dataset (modified from Olson et al. (2001)). The dashed lines delimitate the physiographic regions of Nicaragua (Pacific, Central Highland, Atlantic) according to Taylor (1963). See Table 1 for identification of population codes.
Because of its massive habitat loss, C. candidissimum essentially lacks primary forest populations that could serve as ‘controls’ for pre-fragmentation conditions. However, a priori expectations about its genetic architecture might be derived from AFLP data available for a close relative from the western Amazonian rainforest, C. spruceanum (Benth.) Hook.f. ex K.Schum. (Russell et al., 1999). Based on nine populations from the Peruvian Amazon Basin, this study revealed relatively high genetic diversity within and moderate variation among populations, without any major influence of river structure (viz. hydrochory) or geographic distance among sites on genetic distance measures (for example, there was no evident pattern of ‘isolation by distance’ IBD; Wright, 1943). Overall, these data were indicative of high levels of gene flow among tree populations (mostly via insect-transferred pollen and/or wind-dispersed seed) in a non-fragmented rainforest setting (Russell et al., 1999). For C. candidissimum in Nicaragua, therefore, the null hypothesis would be that recent SDT forest fragmentation has resulted in much lower genetic diversity within populations (for example, owing to drift and/or inbreeding), and concomitantly higher genetic variation viz. ‘insularization’ among populations, in comparison with C. spruceanum.
We investigated geographical patterns of AFLP variation in 13 populations of C. candidissimum in Nicaragua, which cover much of the country’s original dry forest vegetation zone (Figure 1b), and also differ in their degree of disturbance and/or status of legal protection (Table 1). Our major aim was to gain first insights into the relative roles of contemporary vs historical factors in shaping the genetic variation of this emblematic tree species of Central America’s highly endangered SDT forest. Concomitantly, we anticipated that the results obtained might help assist future conservation measures that aim at preserving the genetic diversity of this species in Nicaragua. We addressed the following specific questions: (1) to what extent has human-driven landscape fragmentation affected levels of genetic diversity within populations and patterns of gene flow/dispersal (viz. the connectivity) between them? (2) Is there evidence that other factors such as past population dynamics and/or environmental dispersal barriers had a confounding effect on the genetic patterns observed? And (3) what is the conservation value of these populations (for example, are protected ones sufficiently representative of the species’ genetic diversity in Nicaragua, or is a broad-scale coverage of conservation measures advisable)?
Materials and methods
The study species
C. candidissimum, also called ‘Lemonwood’ or ‘Madroño’, is a diploid, light-requiring pioneer tree (with 2n=2 × =34 chromosomes; Kiehn, 1995), which grows as tall as 15–20 (30) m (Cordero and Boshier, 2003; Gonzáles-Rivas et al., 2009; Lorence, 2012). Above the glossy leaves, the small, white and bisexual flowers are clustered in large inflorescences (thyrses) in which some flowers bear a large, white-to-greenish calyx lobe or ‘calycophyll’ (Delprete, 1996). The sweet-scented, protogynous flowers are primarily visited by bees and butterflies (Villegas-Durán et al., 2003; Ramírez-Arriaga et al., 2011; A Dávila-Lara, personal observation) and develop into small, septicidal capsules releasing numerous small (3–5 mm), short-winged seeds that are wind-dispersed (Lorence, 2012; Hilje et al., 2015). Although Bawa (1974) stated that C. candidissimum is self-compatible, no concrete experimental evidence was provided due to ‘the lack of adequate numbers’. We are unaware of any report indicating that this species regularly reproduces clonally (for example, via root-sprouting).
In Nicaragua, C. candidissimum mainly occurs along shady hill sides and along water streams within fragments of secondary and/or riparian SDT forest in the ‘Pacific Region’ (sensu Taylor, 1963) and, more rarely, in isolated stands of semi-deciduous (for example, Quercus-Pinus) forest in the ‘Central Highland Region’ (Harvey et al., 2006; Lorence, 2012; see Figure 1b). The former region has high temperatures and moderate-to-high rainfall, but with strong seasonal drought, whereas the latter has a sub-tropical climate (cf. Taylor, 1963). Throughout its distribution range, C. candidissimum is still considered to be locally abundant (Stevens et al., 2001; Lorence, 2012; Kappelle, 2016) and neither listed by the CITES (Convention on Endangered Species; see http://www.cites.org) or the International Union for Conservation of Nature (IUCN, 2016). However, in Nicaragua, it is classified as ‘endangered’ (threat index ‘4’) according to the Food and Agriculture Organisation of the United Nations (FAO; Roa, 2003).
Plant material and sampling
Between April and August 2013 an extensive survey of the distribution of C. candidissimum in Nicaragua was carried out by one of us (AD-L) within the UNAN-León/Department of Biology project ‘Conservation and sustainable use of timber and non-timber tree species’ in cooperation with the MARENA (Ministry of Environment and Natural Resources)/Nicaragua (MARENA, 2002). Based on this, 13 natural populations were chosen as sampling sites to cover the full geographic range of the species in Nicaragua (see Figure 1b and Table 1): two populations (SO, QU) from the Central Highland Region (407–905 m above sea level), and 11 from the lowland and premontane areas of the Pacific Region (1–432 m above sea level), including one island population (OM) of Lake Nicaragua. Estimated pairwise geographical distances among sampling sites ranged between 25 km (MO–TE) and 287 km (SO–OS) with a mean (±s.d.) of 139 (±63.8) km (data not shown). These 13 sites differed in their degree of disturbance level and/or status of legal protection (Table 1). Five populations (QU, MO, CH, OM, EO) were located in relatively undisturbed, more or less-continuous forest (A Dávila-Lara, personal observation), of which three (MO, CH, OM) were protected; by contrast, all the remaining populations were from at least partially logged or highly degraded areas, and only two of those (TE, SC) were protected. At each site, young leaves were collected from, if possible, up to 20 mature trees that were sampled according to the line transect method of González (2006), with a minimum distance of 100 m between trees to avoid selecting closely related individuals (Gillies et al., 1999; Inza et al., 2012). To ensure sufficient individual sampling, the sizes of the areas surveyed (in total ca. 2638 ha) had to be adjusted accordingly, ranging from 61 ha (for the relatively dense QU population) to 453 ha (for the sparse LV and ES populations) (Table 1). Logistic constraints and the sampling scheme used did not allow for a detailed investigation of the tree density of C. candidissimum at any individual site, but it provided the basis for an estimation of the number of sampled trees per hectare at all sites (Table 1). This, together with visual assessments during sampling, allowed for approximating the disturbance level of populations, as indicated above. In total, leaf materials of 250 individuals were collected, dried with silica gel, and stored at room temperature.
DNA extraction and AFLP genotyping
Total genomic DNA was extracted from the dried leaf tissue using a modified cetyltrimethyl ammonium bromide protocol (Doyle, 1991). The AFLP analysis was performed according to Vos et al. (1995), with minor modifications as described by Jaros et al. (2016) for the use of fluorescent-dye-labelled primers. For a preliminary AFLP screen, we selected one individual each from four populations (LE, CH, OM EO), using 18 selective primer pair combinations with two or three selective nucleotides (data not shown). Based on this, the following primer combinations that produced the best results with respect to polymorphism and clarity of AFLP profiles were chosen for the complete survey: EcoRI-AAC/MseI-CA (NED), EcoRI-AC/MseI-CTC (VIC) and EcoRI-AT/MseI-CTG (6-FAM). Each PCR plate (48 slots) contained two positive controls, one negative control and one blind sample. The selective PCR products were purified using Sephadex G-50 Superfine (GE Healthcare BioSciences, Uppsala, Sweden) following the manufacturer’s instructions. AFLP fragments were separated on a MegaBACE 1000 (GE Healthcare Biosciences, Pittsburgh, PA, USA), using the ET400-R-Rox-MegaBACE sizing standard, and manually scored in DAX version 8.0 (Van Mierlo Software Consultancy, Eindhoven, The Netherlands) as described in Bendiksby et al. (2011). AFLP products were scored as presence (1) or absence (0), whereby a fragment was considered polymorphic if at least one individual showed a variant pattern. Separate test runs with 16 individuals (representing all 13 populations) were used to obtain an AFLP error rate, calculated as the percentage of differently scored loci over all replicates and the three primer pair combinations (Bonin et al., 2004). In total, 220 individuals were finally available for data analysis because 30 individuals failed to amplify in at least one of the three primer combinations. Accordingly, the mean (±s.d.) sample size was 17.0 (±2.6) individuals per population (see Table 2).
Data analysis
Using the program GENALEX version 6.5 (Peakall and Smouse, 2012), three different measures of genetic diversity were calculated for each locus and then averaged across all loci to represent a single estimate for each population: the mean number of observed alleles per locus (NA), the percentage of polymorphic fragments (PPF), and Nei’s (1987) unbiased gene diversity (HE) for small sample sizes. In addition, we used the AFLPDAT R package (Ehrich, 2006) to estimate frequency down-weighted marker (DW) values (or ‘rarity-indices’), with 95% confidence intervals obtained by bootstrapping over all loci (9999 replicates), and significance tested by a permutation procedure (A Tribsch and D Ehrich, unpublished data) implemented in AFLPDAT (1000 replicates). Significantly high vs low values of DW may indicate long-term population isolation vs genetic drift and/or founder effects (Skrede et al., 2009). Nonparametric Mann–Whitney U-tests were performed to determine whether there was a significant difference in genetic diversity and rarity estimates between populations differing in disturbance level or protection status. Likewise, Spearman’s rank correlations were employed to test for a potential relationship between these genetic parameters and various extrinsic factors (latitude, longitude, precipitation, temperature; see Table 1). All these statistical tests were made using the ‘Social Science Statistics’ online calculators (http://www.socscistatistics.com/).
Genetic structure analysis was performed in STRUCTURE version 2.2 with a Bayesian clustering approach based on a recessive allele model, as applicable for dominant markers (Pritchard et al., 2000; Falush et al., 2007). We run this analysis after pruning markers that proved uninformative (that is, monomorphic) across the entire AFLP dataset (ca. 35%; see Results) and assumed both recent ancestry and some current gene flow among populations (that is, admixture and correlated allele frequencies). The number of clusters (K) was set to vary from 1 to 15. For each value of K, we performed 10 runs with a burn-in length of 50 000 and a run length of 100 000 Markov chain Monte Carlo replications. To identify the most probable value of K, post processing STRUCTURE runs were performed in STRUCTURE HARVESTER version 0.6.94 (Earl and vonHoldt, 2012) to produce graphical files representing the final posterior probability [lnP(D)] (Pritchard et al., 2000) and ΔK (Evanno et al., 2005) per K and repeated run. The CLUMPP program version 1.1.2 (Jakobsson and Rosenberg, 2007) was used to align the membership coefficients from the 10 replicates for each K based on the LargeK Greedy algorithm.
Analyses of molecular variance (AMOVAs) were performed in ARLEQUIN version 3.5 (Excoffier and Lischer, 2010) to quantify the partitioning of AFLP variance (1) among populations (ΦST) and within populations; and (2) between regional groups of populations (ΦCT) and among populations within such groups (ΦSC). Significance levels of variance components were evaluated by a permutation approach (1023 replicates). We also used GENALEX to calculate values of ΦPT (an analogue of ΦST) for all pairs of populations and to infer their significance with 9999 random permutations. Genetic relationships among populations were visualised by subjecting their pairwise ΦPT distances to a principal coordinates analysis (PCoA) in GENALEX. In addition, we constructed a Neighbour-Joining (NJ) phenogram in PHYLIP version 3.69 (Felsenstein, 2005) based on Nei’s (1987) unbiased genetic distances calculated using the fragment-frequency approach in AFLPSURV version 1.0 (Vekemans, 2002). The NJ phenogram was midpoint-rooted, with BS (bootstrap support) obtained by resampling with replacement over loci (9999 replicates). Isolation by distance (IBD) was assessed by regressing values of ΦPT/(1−ΦPT) against the logarithm (log10) of geographical distance (in km) for all pairs of populations (Rousset, 1997), and a Mantel test was employed to evaluate significance with 9999 permutations using IBDWS version 3.23 (Jensen et al., 2005).
Results
Population genetic diversity
For the 220 individuals (13 populations) of C. candidissimum, the three AFLP primer combinations resolved a total of 226 unambiguous fragments, ranging from 87 to 411 base pairs (bp), of which 147 markers were polymorphic (PPF=65%; Table 2). Different primer pairs generated a variable number of fragments (‘NED’: 88; ‘VIC’: 49; ‘6-FAM’: 89), with an average of 75.3 (±22.8 s.d.) fragments per primer combination. The overall error rate calculated on the basis of 16 replicates (1.16%) is at the lower end of the range reported for other AFLP studies in plant species (Bonin et al., 2004; Poelchau and Hamrick, 2012).
Estimates of genetic diversity (in terms of NA, PPF and HE) for each population (Table 2) were positively correlated to each other (all P<0.001), but not with the number of individuals (N) sampled per population (r=0.29–0.36, P=0.140–0.675). Values of these diversity parameters were similar across populations and generally low (Table 2). For example, within-population gene diversity (HE) averaged 0.125 (±0.015 s.d.), with the lowest value (0.111) observed in a Central Highland population (OU) and the highest (0.166) in the southernmost population from the Pacific Region (EO). Although populations in disturbed forest displayed on average slightly lower gene diversity (mean HE=0.121) than those in undisturbed forest (0.132), this difference was not significant (U-test: Z=−0.732, P=0.465), and the same was true for NA and PPF (P=0.418 and 0.509, respectively). Similarly, there were no significant differences between populations from unprotected vs protected sites in any of the three diversity parameters (all P⩾0.660). Levels of allelic rarity (in terms of DW) varied among populations but were generally low, averaging 0.669 (±0.113 s.d.) across all populations (Table 2). Based on permutation tests, one Central Highland (SO) and two Pacific North/Central populations (TE/SL) had significantly low DW values, whereas the southernmost Pacific population (EO) had a significantly high value (Table 2). Notably, the proportion of rare markers was on average significantly lower in populations from disturbed (0.617) vs undisturbed forest (0.754; U-test, two-tailed significance: Z=−2.269, P=0.023). However, no such difference was found between unprotected vs protected populations (0.689 vs 0.638; P=0.826). Correlation analyses revealed a negative and significant relationship between latitude and both NA (r=−0.72, P=0.006) and PPF (r=−0.65, P=0.016), whereas the same trend was marginally significant for HE (r=−0.53, P=0.061). None of the other factors examined (that is, longitude, precipitation, temperature) showed a significant effect (P⩽0.05) on genetic diversity or allelic rarity within populations (data not shown).
Population genetic structure and relationships
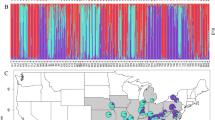
In the STRUCTURE analysis, log probabilities of the data [lnP(D)] showed some tendency to plateau at K≈4, whereas the ad hoc statistic ΔK clearly showed the highest likelihood at K=2 (see Supplementary Appendix Figure S1). Based on the latter model, all populations from the Central Highland (SO, QU) and Pacific North Region (LV, ES, TE, MO, LE) were fixed for cluster I (‘black’ in Figure 2), while cluster II (‘grey’) was predominant in the three Pacific South populations (CH, OM, EO). By contrast, populations from the Pacific Central Region were differently admixed with clusters I vs II, whereby SL and SC showed higher membership proportions to the northern cluster I and ST to the southern cluster II. At K=4, the Pacific Far South population (EO) became clearly distinguishable (cluster III), whereas few samples (mostly from QU and LV) were erratically assigned to a fourth ‘gene pool’, without reflecting a geographical pattern (see Supplementary Figure S2). We therefore retained K=2 as the likely more robust model, albeit without dismissing the genetic distinctiveness of EO (see also below).

Distribution of the two major AFLP gene pool clusters (I and II) within and among 13 populations (220 individuals) of C. candidissimum from Nicaragua as identified by STRUCTURE based on the ad hoc statistic ΔK (see also Supplementary Figure S1). The bar-plot displays the assignment of individuals to the two clusters, with the smallest vertical bar representing one individual. The y axis presents the estimated membership coefficient (Q) for each individual in the two clusters. The x axis corresponds to population codes as identified in Table 1. The pie charts at the top represent the average proportion of cluster membership computed across individuals per site. Populations are ordered from left to right according to decreasing latitude (see also Table 1 and Figure 1b).
The non-hierarchical AMOVA (Table 3) indicated that most of the total genetic variance resided within populations (89.08%) even though the among-population component was highly significant (ΦST=0.109, P<0.001). Pairwise ΦPT values (as measures of connectivity between pairs of sites) ranged from 0.005 to 0.229, whereby 93.6% (73/78) were significant (P<0.05) based on permutation tests (see Supplementary Appendix Table S1 and Supplementary Figure S3). The PCoA plot of the first two coordinates (Figure 3a) revealed a more or less continuous north-south alignment of most populations along the first coordinate (44.3% of the total variance), which nonetheless allowed their grouping into: (1) a well-delimited ‘North’ group (SO, QU, LV, ES, TE, MO, LE); (2) the genetically heterogeneous ‘Pacific Central’ group (SL/SC and ST); and (3) the ‘Pacific South’ group (CH, OM). In addition, the second coordinate (15.6%) separated the Pacific Far South population (EO) from the latter group. Hierarchical AMOVA (Table 3) attributed a relatively high percentage of the total variance (9%) to differences among these four population groups, compared with 4.5% among populations, and 86.5% within populations. The PCoA for the first and third coordinates (Figure 3b) suggested a further subdivision of the ‘North’ group into ‘Central Highland’ and ‘Pacific North’ populations. However, the corresponding AMOVA based on five groups resulted in the same amount of among-group partitioning (9%; Table 3). Consistent with the PCoA, the midpoint-rooted NJ phenogram (Figure 4) placed the three populations from the Pacific Central Region (SL, SC, ST) at intermediate branches between the strongly supported ‘North’ group (BS 95%) and the genetically more distinct southern populations (CH, OM, EO; BS 87%), as would be expected for admixed groups (Verdu et al., 2014). Not unexpectedly, therefore, the Mantel regression of pairwise ΦPT/(1−ΦPT) values against the geographical distances among all 13 populations (Figure 5) revealed a positive and highly significant relationship (r=0.652, R2=0.425, P<0.001), indicating a strong pattern of IBD.

Principal coordinates analysis (PCoA) for 13 populations of C. candidissimum from Nicaragua based on pairwise ΦPT values of AFLP data. Plots of (a) the first and second, and (b) the first and third coordinates. Percentages of total variance explained by each coordinate are noted in parentheses. Population symbols and colours indicate, respectively, regional affiliation (Table 1; Figure 1b) and gene pool (I–III) structure (black, I; grey, II; grey/black, I and II; see Figure 2; and white, III; see Supplementary Figure S2). Note in (b) the subdivision between ‘Central Highland’ and ‘Pacific North’ populations. Population codes are identified in Table 1.

Midpoint-rooted Neighbour-Joining (NJ) phenogram depicting AFLP-derived Nei’s (1987) unbiased genetic distances between 13 populations of C. candidissimum from Nicaragua. Numbers above branches indicate bootstrap values (⩾70%) based on 9999 replicates. Populations are labelled according to their geographic origin. Population codes are identified in Table 1.

Regression of AFLP-derived ΦPT/(1−ΦPT) values against the logarithm (log10) of geographic distance (km) for 78 pairwise comparisons of 13 Nicaraguan populations of C. candidissimum (r=0.651, R2=0.425; Mantel probability (9999 permutations) P<0.001).
Discussion
Our analysis of AFLP variation within and among 13 populations of C. candidissimum from fragmented SDT forest in Nicaragua revealed three major findings. (1) Genetic diversity is low in all investigated populations and negatively correlated with latitude. (2) Genetic variation is partitioned into two major regional (latitudinal) clusters and a cline, resulting in an overall moderate level of population differentiation (ΦST=0.109). (3) The IBD pattern found may indicate spatially restricted gene flow, but this interpretation requires careful examination. In the following sections, we discuss these results and comment on their implications for conservation.
Low levels of genetic diversity in populations of C. candidissimum in Nicaragua
Tropical trees in general, and especially those that are outcrossing, and pollinated and/or dispersed by wind, tend to harbour high levels of genetic diversity at local and regional spatial scales (for example, Hamrick et al., 1993; Chase et al., 1995; Alvarez-Buylla et al., 1996; Collevatti et al., 2014). However, comparisons across such studies have to be taken with caution owing to discordances in sampling scale, genetic markers and/or data analysis. Nonetheless, our data suggest that populations of C. candidissimum in Nicaragua exhibit unexpectedly low diversity (in terms of NA, PPF, HE; Table 2). For example, if we focus on values for gene diversity (mean HE=0.125, range: 0.111–0.166), those are much lower than for populations of the likewise insect-pollinated and wind-dispersed C. spruceanum (mean: 0.284, range: 0.249–0.349) surveyed for the same marker system in the Peruvian Amazon Basin (Russell et al., 1999). The same discrepancy emerges if we consider the averages of within-population diversity (H) reported by Kremer et al. (2005) for 10 Neotropical outcrossing tree species using AFLPs under the assumption of Hardy–Weinberg equilibrium (mean: 0.23, range: 0.14–0.32) or full selfing (0.24, 0.17–0.38). Although realising that dominant AFLPs cannot provide estimates of inbreeding, we do not consider a highly selfing mating system to be a compelling explanation for the reduced genetic diversity of C. candidissimum. First, there is no firm experimental evidence to suggest that this species is truly self-compatible, as surmised by Bawa (1974; see also Methods section). And second, our global estimate of population differentiation (ΦST=0.109; Table 3) is much lower when compared to selfers (ΦST=0.65; Nybom, 2004) yet more characteristic of a highly outcrossing species (see below). This is in further agreement with field observations and melissopalynogical research indicating that C. candidissimum is a highly melliferous tree and important nectar/pollen resource, especially for butterflies (Villegas-Durán et al., 2003; Ramírez-Arriaga et al., 2011).
In the following, therefore, we discuss three alternative factors that may have acted additively and/or synergistically in causing this reduced diversity over time: biogeographic history, Pleistocene range dynamics, and recent anthropogenic deforestation.
First, this reduced diversity may reflect a signature of the species’ colonisation of Central America from South America. This scenario gains support from two congruent lines of evidence. (1) Considering Calycophyllum DC. in its traditional circumscription (ca. 10–15 spp.; Delprete, 1996; Lorence, 2012), C. candidissimum is the only Central American representative of an otherwise South American genus. (2) Although the most recent molecular phylogeny of tribe Condamineeae renders the genus polyphyletic (Kainulainen et al., 2010), it still supports a clade comprised of C. candidissimum (the type species) as sister to the South American C. multiflorum Griseb. and C. spruceanum, which together are likely sister to a clade consisting of genera from mainly South America (that is, the monotypic Bothriospora Hook.f., Dialypetalanthus Kuhlm., Wittmackanthus Kuntz (also found in eastern Panama) and Alseis Schott. (ca. 18 spp., three present in Central America)). Although the phylogeny and taxonomy of the entire group requires more work (Delprete and Jardim, 2012), all current evidence seems to indicate that C. candidissimum arrived in Central America from South America, perhaps facilitated by the most recent uplift of the Panama land bridge (ca. 3 million years ago), as proposed for many other constituents of the Central American lowland flora (for example, Burnham and Graham, 1999; Novick et al., 2003; Antonelli et al., 2009; Poelchau and Hamrick, 2013; but see Cavers et al., 2013). Thus, the overall depletion of genetic diversity in Nicaraguan C. candidissimum could, at least in part, reflect a historical legacy of the species’ colonisation from South America (see also Novick et al., 2003). This also concurs with a significant south-to-north trend of declining diversity (in terms of NA and PPF), consistent with successive (‘serial’) founder events in the direction of spread (Hewitt, 2004) and the nested (that is, potentially derived) position of northern populations in the NJ phenogram (Figure 4; see also below).
Second, other historical events that may have further contributed to the loss of genetic diversity in Nicaraguan C. candidissimum are repeated range shifts in response to the climatic fluctuations of the Quaternary (⩽2.5 million years ago), which likely had a major impact on the current genetic structure of many Neotropical plant species (reviewed in Ramírez-Barahona and Eguiarte, 2013). Some authors have hypothesised that the ranges of SDT forest plant species in Central America were not restricted during the cooler and drier periods of the Pleistocene (Piperno, 2006) but rather attained a more continuous distribution (for example, Prado and Gibbs, 1993; Pennington et al., 2000; Novick et al., 2003). This hypothesis seems intuitively obvious as such plants are tolerant to drought resulting from seasonal rainfall or drought spells during rainy seasons (Pineda-García et al., 2013). In support of this, a recent palaeodistribution modelling of multiple Central American tree species (Poelchau and Hamrick, 2013) found little evidence to suggest that, during the LGM (Last Glacial Maximum; ca. 21 000 years ago; and possibly earlier cold periods), constituents of SDT forest (including C. candidissimum but also those of wet-topical forest) retreated into isolated humid refugia (for example, extending from southern Nicaragua to northern Panama), as postulated previously (Brown, 1987); instead, this modelling approach inferred a decrease of potential range size since the LGM for at least some of the SDT forest species studied, including C. candidissimum. Hence, following its initial colonisation of Central America, this species may have experienced subsequent reductions in historical population sizes owing to inter-/postglacial range contractions, accentuating the loss of diversity through drift.
Finally, recent anthropogenic forest fragmentation may have also contributed to the genetic erosion of Nicaraguan C. candidissimum. In general, however, molecular studies to date have found only little evidence for such an effect on levels of genetic diversity in forest trees from the Neotropics and elsewhere, which is usually ascribed to their long generation times relative to the time since deforestation (Lowe et al., 2005; Kramer et al., 2008; Ratnam et al., 2014; but for exceptions see, for example, Gillies et al., 1999; André et al., 2008; Inza et al., 2012). Yet another problem is that measures of genetic diversity differ in their sensitivity to detect such recent impact. For example, while gene diversity (HE) should be appropriate for assessing long-term (for example, historical) loss of diversity (including common alleles), it often fails to register reductions in diversity in recently bottlenecked populations owing to the loss of rare alleles (Luikart et al., 1998; Aguilar et al., 2008). This is in line with our findings in C. candidissimum populations from disturbed vs undisturbed forest in Nicaragua (Table 2): although average levels of HE (as well as NA and PPF) were reduced in the former, this difference was not significant (for example, HE=0.121 vs 0.132; P=0.248). By contrast, populations from disturbed forest exhibited on average lower levels of rare makers compared with those from undisturbed sites (DW=0.617 vs 0.754; P=0.026). We therefore conclude that the record of intensified deforestation in Nicaragua (since about the 1930’s; Griscom and Ashton, 2011) has not yet impacted overall levels of diversity (NA, PPF, HE), but has been sufficiently long (compared to the relatively short generation time of our study species) for genetic drift having caused a significant loss of rare alleles in disturbed forest fragments.
Population genetic differentiation, spatial structure and IBD
Despite the highly fragmented landscape occupied by C. candidissimum in Nicaragua, we found little evidence for strong genetic structuring across the 13 sampled populations. Although the great majority (93.6%) of estimated pairwise ΦPT values (ranging from 0.005 to 0.229) were significant (Supplementary Table S1), AMOVA revealed an overall ΦST value of 0.109 (Table 3), which is generally considered to indicate ‘moderate’ genetic differentiation (Hollingsworth and Ennos, 2004; Freeland et al., 2011). While this value is much lower than the corresponding averages reported for outcrossing (0.27) or wind-dispersed (0.25) species (Nybom, 2004), it is comparable to those of some other insect-pollinated Neotropical trees (for example, Swietenia macrophylla, ΦST=0.126, Gillies et al., 1999; Cedrela lilloi, ΦPT=0.130, Inza et al., 2012; Ficus insipida, ΦPT=0.140, Poelchau and Hamrick, 2012) and almost identical to that of Amazonian C. spruceanum (ΦST=0.094; Russell et al., 1999). Furthermore, although the STRUCTURE analysis (Figure 2) inferred that individuals do cluster into two major population groups that correspond to geographic regions (‘North’ vs ‘Pacific South’), several geographically intermediate ‘Pacific Central’ populations (SL, SC, ST) had individuals with membership to either the ‘North’ or ‘Pacific South’ group. These populations most likely reflect a continuous genetic gradation or admixture of these neighbouring groups (Rosenberg et al., 2005). This interpretation also accords with the among-population relationships detected in the PCoA and NJ analyses (Figures 3 and 4). When coupled with the strong pattern of IBD (Figure 5), these results seem to suggest that population structure in this highly fragmented landscape is still mediated through an equilibrium between genetic drift and gene flow structured by space (Wright, 1943; Slatkin, 1993).
In turn, this would imply ongoing gene flow between presently isolated populations, yet more commonly between neighbouring ones. There are three arguments in support of such a process, recently called 'IBDL' (isolation by dispersal limitation; Orsini et al., 2013). First, our estimates of population differentiation (ΦST, ΦPT) are probably not strongly biased towards pre-fragmentation conditions (Lowe et al., 2005). Although exact measurements of tree DBH (diameter at breast height) were not undertaken during this study, most sampled individuals had a DBH of ca. 23–80 cm, with only few (mostly from population OM) approaching 110 cm (A Dávila-Lara, personal observation). Assuming a DBH growth rate (mean±s.d.) of ca. 1.9±1.0 cm per year for C. candidissimum (Caughlin et al., 2016), this may indicate that at least a sizable portion of trees we have genotyped were probably not present prior to the onset of intensified deforestation around the 1930’s. Second, ensuing loss of SDT forest in Nicaragua must have greatly reduced the effective population size (Ne) of C. candidissimum there, leaving migration rate (m) viz. gene flow as the predominant, if not sole factor responsible for counteracting drift to reach, and probably maintain current equilibrium (see also Galbusera et al., 2004). And finally, as typical for a SDT forest tree, the small, winged seeds of this species are released during the dry season (January to April, when mean wind-gust speeds are at their highest in Nicaragua; Stevens et al., 2001), and thus may be dispersed by wind several kilometres, facilitating gene flow beyond the local neighbourhood (Clark et al., 2002). By comparison, pollen flow via bees or butterflies (as main pollinators) might be expected to decay more rapidly with distance from paternal trees, particularly in disturbed tropical forest canopies (for example, Hamrick and Apsit, 2004; Bonebrake et al., 2010; Duminil et al., 2016). In sum, this would suggest that the studied populations still exchange genes (mostly via seed) at a rate dependent upon the geographic distance between habitat fragments.
However, there are two potential caveats to this classical interpretation of IBD. First, estimators of population genetic differentiation (for example, FST, ΦST) only represent an integrated indirect measure of the number of migrants across generations (Nem), and therefore must be treated cautiously when used to infer levels of contemporary gene flow (Whitlock and McCauley, 1999). And second, there is increasing awareness that IBD can also be generated through a particular history of ‘serial’ colonisation and drift (reviewed in Orsini et al., 2013), unless being confounded by environmental conditions and/or local adaptation (Wang and Bradburd, 2014). At first glance, this variant scenario of ‘isolation by colonisation’ (IBC; sensu Orsini et al., 2013) seems to fit the notion that C. candidissimum is a Central American colonist (see above). However, under any persisting signature of IBC, one would expect highest levels of genetic differentiation among the most recently colonised populations (Boileau et al., 1992; Slatkin, 1993; Orsini et al., 2013; Spurgin et al., 2014), but this is not observed. Instead, the potentially derived populations of Nicaraguan C. candidissimum from the ‘Pacific North’ and Central Highland are genetically more similar to each other than to the remainder (see Figures 2,–4; Supplementary Table S1), and in spite of their dissimilar elevation ranges and (macro-)climatic conditions (Table 1; Taylor, 1963). Hence, the current data provide no conclusive evidence for IBC or particular environmental (for example, physiographic, climatic) factors influencing spatial population divergence, but also cannot exclude the possibility that other processes than IBDL (for example, historic-demographic, adaptive) underlie the observed pattern of IBD. Disentangling those phenomena, even though difficult, may be achievable through comparisons of correlations between FST and geographic and ecological distances for, respectively, neutral loci, non-neutral loci and genotypic trait values (see Orsini et al., 2013 for details) and/or coalescent-based approaches to model colonisation (including bottleneck) scenarios (for example, Henn et al., 2012; Spurgin et al., 2014; Dewoody et al., 2015).
Implications for conservation
Measures to conserve the genetic resources of C. candidissimum in Nicaragua have been called upon repeatedly (Roa, 2003; Weaver et al., 2003; Noguera-Talavera et al., 2011), mainly because of its socio-economic importance. However, apart from serving as important pollen/nectar resource for insects, this SDT forest tree also functions as foraging, nesting, shelter, and/or resting site (and so on) for a diversity of other animals (for example, howler monkeys; Garber et al., 1999; birds, bats: Harvey et al., 2006). The present data, therefore, have implications for devising conservation plans for Nicaragua’s ‘National Tree’ in the context of a community-based conservation policy also to preserve the integrity of plant-animal interactions (Ruiz-Mallén and Corbera, 2013). Currently, all populations of C. candidissimum in this country, regardless of their protection status, are facing risks of losing even more of their already low genetic diversity through reductions in population size, whether due to increased urbanisation/agricultural intensification, especially in the Pacific Region (for example, Redo et al., 2012), or stochastic factors (for example, natural or man-made fires; de Dixmude, 2001), with likely detrimental consequences for their long-term fitness and adaptation (Frankham et al., 2010). In fact, when compared with their protected counterparts (TE, MO, SC, CH, OM), the eight populations analysed here from outside nature reserves do not harbour significantly lower genetic diversity (Table 2), thus emphasising the need to extend close monitoring, preventive action, and in situ conservation to them as well. In view of limited resources, and based on our ΦST estimate of 0.109, one might argue to focus ex situ sampling efforts (for example, for provenance trials) on just two populations, representing >95% of the total genetic variation (that is, 1−(ΦST)2=0.99; see Ceska et al. (1997)). However, our data show that Nicaraguan C. candidissimum is comprised of two major regional genetic clusters (I and II), and a remarkable cline (between I and II; Figure 2), calling for a broad coverage of both in situ and ex situ conservation measures. Hence, ‘North’ (I) vs ‘Pacific South’ (II) populations should be valued separately under conservation programmes (for example, by avoiding human assisted gene exchange between them), whereas those from the Pacific Central Region (SL, SC, ST) likewise warrant special attention, as they are likely stepping-stones for natural gene flow between these groups. The same should apply to the southernmost EO population, given its unusually high diversity/rarity (Table 2), divergent gene pool (detectable at K=4; Supplementary Figure S2), and location in a transition zone between SDT and seasonal moist forest (Taylor, 1963; Olson et al., 2001). Notably, too, this divergence occurs in a region (southern Isthmus of Rivas/north-western Costa Rica) where previous large-scale phylogeographic studies have registered genetic discontinuities in dry-forest trees (Cedrela odorata: Cavers et al., 2003, 2013; Brosimum alicastrum: Poelchau and Hamrick, 2012) and various animals (for example, ants, bats, birds, reptiles; reviewed in Bagley and Johnson, 2014). Hence, only a wider sampling could clarify whether EO represents a genetically unique population or just delimitates the northernmost edge of a perhaps more widespread southern lineage.
Conclusions and research gaps
This study has assessed for the first time genetic variation for a SDT forest tree in Nicaragua. When compared with C. spruceanum from continuous Amazonian rainforest (Russell et al., 1999), Nicaraguan C. candidissimum exhibits lower genetic diversity and, despite similarly moderate overall population differentiation (ΦST), stronger geographical structure (IBD). We propose this reduced diversity more likely reflects population history (that is, the species’ colonisation from South America; Pleistocene (inter-/postglacial) range contractions) rather than recent human impact, whereas the strong pattern of IBD is probably best explained by ‘dispersal limitation’ rather than ‘isolation by serial colonisation’ (sensu Orsini et al., 2013). The spatial genetic patterns observed thus conform to expectations of a subdivided meta-population (Wright, 1943) in which drift–migration equilibrium is still maintained by ongoing gene flow, possibly owing to the species’ efficient seed dispersal capacity. Overall, these results underscore that even tree species from one of the world’s most decimated forest regions may be genetically resilient to habitat fragmentation due to species-typical dispersal characteristics that ensure nearest-neighbouring population connectivity and viability (Bacles et al., 2006; Kramer et al., 2008; Lowe et al., 2015). However, future research is required to arrive at more conclusive evidence about the recurrent and historical factors shaping genetic variation in C. candidissimum.
First, our inference of ongoing gene flow should be re-examined using approaches that can provide more direct estimates of pollen and seed movement at both local and regional scales, such as parentage and/or paternity analyses based on nuclear microsatellite loci (for example, Gerber et al., 2014). Second, such co-dominant markers are also critical to examine whether inbreeding (due to selfing and/or sib mating) is increased by the small size and isolation of these populations (that is, in terms of FIS), and whether levels of genetic diversity differ between adult trees and seedlings (for example, Davies et al., 2010; Duminil et al., 2016). Third, experimental and field data are required to assess whether the genetic processes leading to the loss of rare alleles, as found in some of the populations studied here, also contribute to decreased survival, reproductive output and/or progeny fitness of individual trees, as indicated by a recent monitoring of C. candidissimum in Southwest Nicaragua (Noguera-Talavera et al., 2011). Fourth, more integrated approaches using both neutral and non-neutral genetic markers in relation to spatial and environmental factors would be useful to test for local adaptation in C. candidissimum across its entire range in Central/northern South America (Orsini et al., 2013; Wang and Bradburd, 2014). And finally, this Neotropical tree and its allies should provide a promising study model for phylogenetic and phylogeographic research as indicated by the biogeographic and population demographic scenarios we have invoked here.
Data archiving
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.3274n.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J . (2008). Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17: 5177–5188.
Alvarez-Buylla ER, Garcia-Barrios R, Lara-Moreno C, Martinez-Ramos M . (1996). Demographic and genetic models in conservation biology: applications and perspectives for tropical rain forest tree species. Ann Rev Ecol Syst 27: 387–421.
André T, Lemes MR, Grogan J, Gribel R . (2008). Post-logging loss of genetic diversity in a mahogany (Swietenia macrophylla King, Meliaceae) population in Brazilian Amazonia. For Ecol Manage 255: 340–345.
Antonelli A, Nylander JAA, Persson C, Sanmartín I . (2009). Tracing the impact of the Andean uplift on Neotropical plant evolution. Proc Natl Acad Sci USA 106: 9749–97544.
Bacles CFE, Lowe AJ, Ennos RA . (2006). Effective seed dispersal across a fragmented landscape. Science 311: 628.
Bagley JC, Johnson JB . (2014). Phylogeography and biogeography of the lower Central American Neotropics: diversification between two continents and between two seas. Biol Rev 89: 767–790.
Barrance A, Schreckenberg K, Gordon J . (2009) Conservation Through Use: Lessons from the Mesoamerican Dry Forest. Overseas Development Institute: London, UK.
Bawa KS . (1974). Breeding systems of tree species of a lowland tropical community. Evolution 28: 85–92.
Bendiksby M, Tribsch A, Borgen L, Trávnícěk P, Brysting AK . (2011). Allopolyploid origins of the Galeopsis tetraploids – revisiting Muüntzing’s classical textbook example using molecular tools. New Phytol 191: 1150–1167.
Boileau MG, Hebert PDN, Schwartz SS . (1992). Non-equilibrium gene frequency divergence: persistent founder effects in natural populations. J Evol Biol 5: 25–39.
Bonebrake TC, Ponisio LC, Boggs CL, Ehrlich PR . (2010). More than just indicators: a review of tropical butterfly ecology and conservations. Biol Conserv 143: 1831–1841.
Bonin A, Bellemain E, Eidesen PB, Pompanon F, Brochmann C, Taberlet P . (2004). How to track and assess genotyping errors in population genetics studies. Mol Ecol 13: 3261–3273.
Brown KS . (1987). Areas where humid tropical forest probably persisted. In: Whitmore TC, Prance GT (eds). Biogeography and Quaternary History in Tropical America. Clarendon Press: Oxford, UK. pp 44–45.
Burnham RJ, Graham A . (1999). The history of Neotropical vegetation: new developments and status. Ann Missouri Bot Gard 86: 546–589.
Caughlin TT, Graves SJ, Asner GP, van Breugel M, Hall JS, Martin RE et al. (2016). A hyperspectral image can predict tropical tree growth rates in single-species stands. Ecol Appl 26: 2367–2373.
Cavers S, Navarro C, Lowe AJ . (2003). A combination of molecular markers identifies evolutionarily significant units in Cedrela odorata L. (Meliaceae) in Costa Rica. Conserv Genet 4: 571–580.
Cavers S, Telford A, Arenal Cruz F, Perez Castaneda AJ, Valencia R, Navarro C et al. (2013). Cryptic species and phylogeographical structure in the tree Cedrela odorata L. throughout the Neotropics. J Biogeogr 40: 732–746.
Ceska JF, Affolter JM, Hamrick JC . (1997). Developing a sampling strategy for Baptisia arachnifera based on allozyme diversity. Conserv Biol 11: 1133–1139.
Chase MR, Boshier DH, Bawa KS . (1995). Population genetics of Cordia alliodora (Boraginaceae), a Neotropical tree. 1. Genetic variation in natural populations. Am J Bot 82: 468–475.
Clark JS, Beckage B, HilleRisLambers J, Ibanez I, LaDeau S, MacLachlan J et al. (2002). Dispersal and plant migration. In: Mooney H, Canadell J (eds). Encyclopedia of Global Environmental Change Vol 3, Wiley: Chichester, UK. pp 81–93.
Collevatti RG, Estolano R, Ribeiro ML, Rabelo SG, Lima EJ, Munhoz CBR . (2014). High genetic diversity and contrasting fine-scale spatial genetic structure in four seasonally dry tropical forest species. Plant Syst Evol 300: 1671–1681.
Cordero J, Boshier DH . (2003) Árboles de Centroamérica: un manual para extensionistas. Oxford Forestry Institute (OFI), Centro Agronómico Tropical de Investigación y Enseñanza (CATIE): Oxford, UK, Costa Rica.
Davies SJ, Cavers S, Finegan B, Navarro C, Lowe AJ . (2010). Genetic consequences of multigenerational and landscape colonisation bottlenecks for a neotropical forest pioneer tree, Vochysia ferruginea. Trop Plant Biol 3: 14–27.
Dawson IK, Waugh R, Simons AJ, Powell W . (1997). Simple sequence repeats provide a direct estimate of pollen-mediated gene dispersal in the tropical tree Gliricidia sepium. Mol Ecol 6: 179–183.
de Dixmude AJ . (2001). Fire situation in Nicaragua. In: Goldammer JG, Mutch RW, Pugliese P (eds). ‘Global Forest Fire Assessment 1990–2000’, Forest Resources Assessment WP 55. FAO: Rome, Italy. pp 427–431.
Delprete PG . (1996). Notes on calyphyllous Rubiaceae. Part I. Morphological comparison of the genera Chimarrhis, Bathysa, Calycophyllum, with new combinations and a new species Chimarrhis gentryana. Brittonia 48: 35–44.
Delprete PG, Jardim JG . (2012). Systematics, taxonomy and floristics of Brazilian Rubiaceae: an overview of the current status and future challenges. Rodriguésia 63: 101–128.
Dewoody J, Trewin H, Taylor G . (2015). Genetic and morphological differentiation in Populus nigra L.: isolation by colonization or isolation by adaptation? Mol Ecol 24: 2641–2655.
Diniz-Filho JAF, Melo DB, de Oliveira G, Collevatti RG, Soares TN, Nabout JC et al. (2012). Planing for optimal conservation of geographical genetic variability within species. Conserv Genetics 13: 1085–1093.
Doyle JJ . (1991) DNA protocols for plants-CTAB total DNA isolation. In: Hewitt GM, Johnston A (eds). Molecular Techniques in Taxonomy. Springer-Verlag: Berlin, Germany. pp 283–293.
Dubreuil M, Riba M, González-Martínez SC, Vendramin GG, Sebastiani F, Mayol M . (2010). Genetic effects of chronic fragmentation revisited: strong genetic differentiation in a temperate tree, Taxus baccata (Taxaceae), with great dispersal capability. Am J Bot 97: 303–310.
Duminil J, Mendene Abessolo DT, Ndiade Bourobou D, Douect JL, Loo J, Hardy OJ . (2016). High selfing rate, limited pollen dispersal and inbreeding depression in the emblematic African rain forest tree Baillonella toxisperma – Management implications. For Ecol Manage 379: 20–29.
Earl DA, vonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4: 359–361.
Ehrich D . (2006). Aflpdat: a collection of R functions for convenient handling of AFLP data. Mol Ecol Notes 6: 603–604.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Lischer HE . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10: 564–567.
Faber-Langendoen D, Keeler-Wolf T, Meidinger D, Josse C, Weakley A, Tart D et al. (2012) Classification and Description of World Formation Types. Part I (Introduction) and Part II (Description of Formation Types). Hierarchy Revisions Working Group, Federal Geographic Data Committee, FGDC Secretariat, U.S. Geological Survey: Reston, VA, and NatureServe: Arlington, VA, USA.
Falush D, Stephens M, Pritchard JK . (2007). Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578.
Felsenstein J . (2005) PHYLIP (Phylogeny Inference Package) Version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington: Seattle.
Frankham R, Ballou JD, Briscoe DA, McInnes KH . (2010) Introduction to Conservation Genetics.2nd edn. Cambridge University Press: Cambridge, UK.
Freeland JR, Kirk H, Petersen SD . (2011) Molecular Ecology..2nd edn. Wiley-Blackwell: Chichester, UK.
Fuchs EJ, Lobo JA, Quesada M . (2003). Effects of forest fragmentation and flowering phenology on the reproductive success and mating patterns of the tropical dry forest tree Pachira quinata. Conserv Biol 17: 149–157.
Galbusera P, Githiru M, Lens L, Matthysen E . (2004). Genetic equilibrium despite habitat fragmentation in an Afrotropical bird. Mol Ecol 13: 1409–1421.
Garber PA, Pruetz JD, Lavallee AC, Lavallee SG . (1999). A preliminary study of mantled howling monkey (Alouatta palliata ecology and conservation on isla de Ometepe, Nicaragua. Neotrop Primates 7: 113–117.
Gerber S, Chadœuf J, Gugerli F, Lascoux M, Buiteveld J, Cottrell J et al. (2014). High Rates of gene flow by pollen and seed in oak populations across Europe. PLoS One 9: e85130.
Gillies ACM, Navarro C, Lowe AJ, Newton AC, Hernández M, Wilson J et al. (1999). Genetic diversity in Mesoamerican populations of mahogany (Swietenia macrophylla, assessed using RAPDs. Heredity 83: 722–732.
González AR . (2006) Ecología: Métodos de muestreo y análisis de poblaciones y comunidades. Pontificia Universidad Javeriana: Bogotá, Colombia.
Gonzáles-Rivas B, Tigabu M, Castro-Marín, Odén PC . (2009). Seed germination and seedling establishment of Neotropical dry forest species in response to temperature and light conditions. J For Res 20: 99–104.
Grandtner MM, Chevrette J . (2013) Dictionary of Trees. Volume 2: South America. Nomenclature, Taxonomy and Ecology. Academic Press (Elsevier): Amsterdam, The Netherlands.
Griscom HP, Ashton MS . (2011). Restoration of dry tropical forest in Central America: a review of pattern and process. For Ecol Manage 261: 1564–1579.
Hamrick JL, Murawski DA, Nason JD . (1993). The influence of seed dispersal mechanisms on the genetic structure of tropical tree populations. Vegetatio 107: 281–297.
Hamrick JL, Apsit VJ . (2004). Breeding structure of Neotropical dry-forest tree species in fragmented landscapes. In: Frankie GW, Mata A, Vinson SB (eds). Biodiversity Conservation in Costa Rica: Learning the Lessons in a Seasonal Dry Forest. University of California Press: Berkeley, CA, USA. pp 30–37.
Hansen MC, Potapov PV, Moore R, Hancher M, Turubanova SA, Tyukavina A et al. (2013). High-resolution global maps of 21st-century forest cover change. Science 342: 850–853.
Harvey CA, Medina A, Merlo Sánchez D, Vílchez S, Hernández B, Saenz J et al. (2006). Patterns of animal diversity in different forms of tree cover in agricultural landscapes. Ecol Appl 16: 1986–1999.
Henn BM, Cavalli-Sforza LL, Feldman MW . (2012). The great human expansion. Proc Natl Acad Sci USA 109: 17758–17764.
Hewitt GM . (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc B 359: 183–195.
Hilje B, Calvo-Alvarado J, Jiménez-Rodríguez C, Sánchez-Azofeifa A . (2015). Tree species composition, breeding systems, and pollination and dispersal syndromes in three forest successional stages in a tropical dry forest in Mesoamerica. Trop Conserv Sci 8: 76–94.
Hollingsworth PM, Ennos RA . (2004). Neighbour joining trees, dominant markers and population genetic structure. Heredity 92: 490–498.
Inza MV, Zelener N, Fornes L, Gallo LA . (2012). Effect of latitudinal gradient and impact of logging on genetic diversity of Cedrela lilloi along the Argentine Yungas Rainforest. Ecol Evol 2: 2722–2736.
IUCN. (2016). The IUCN Red List of Threatened Species. Annual Release. Available at www.iucnredlist.org (last accessed August 2016).
IUCN and UNEP-WCMC. (2016) The World Database on Protected Areas (WDPA). Annual Release. UNEP-WCMC: Cambridge, UK. Available at. www.protectedplanet.net (last accessed August 2016).
Jakobsson M, Rosenberg NA . (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Janzen DH . (1988). Tropical dry forests. The most endangered major tropical ecosystem. In: Wilson EO (ed). Biodiversity. National Academy Press: Washington, DC, USA. pp 130–137.
Jaros U, Fischer GA, Pallier T, Comes HP . (2016). Spatial patterns of AFLP diversity in Bulbophyllum occultum (Orchidaceae) indicate long-term refugial isolation in Madagascar and long-distance colonization effects in La Réunion. Heredity 116: 434–446.
Jensen JL, Bohonak AJ, Kelley ST . (2005). Isolation by distance, web service. BMC Genetics 6: 13 Available at http://ibdws.sdsu.edu/ (last accessed October 2016).
Kainulainen K, Persson C, Eriksson T, Bremer B . (2010). Molecular systematics and morphological character evolution of the Condamineeae (Rubiaceae). Am J Bot 97: 1961–1981.
Kappelle M . (2016) Costa Rican Ecosystems. University of Chicago Press: Chicago, USA.
Kettle CJ, Hollingsworth PM, Jaffré T, Moran B, Ennos RA . (2007). Identifying the early genetic consequences of habitat degradation in a highly threatened tropical conifer, Araucaria nemorosa de Laub. Mol Ecol 16: 3581–3591.
Kiehn M . (1995). Chromosome survey of the Rubiaceae. Ann Missouri Bot Gard 82: 398–408.
Kramer AT, Ison JL, Ashley MV, Howe HF . (2008). The paradox of forest fragmentation genetics. Conserv Biol 22: 878–885.
Kremer A, Caron H, Cavers S, Colpaert N, Gheysen L, Gribel R et al. (2005). Monitoring genetic diversity in tropical trees with multilocus dominant markers. Heredity 95: 274–280.
Linares-Palomino R, Oliveira-Filho AT, Pennington RT . (2011) Neotropical seasonally dry forests: diversity, endemism and biogeography of woody plants. In: Dirzo R, Young HS, Mooney HA, Ceballos G (eds). Latin American Seasonally Dry Tropical Forests: Ecology and Conservation. Island Press: Washington, DC, USA. pp 3–21.
Lorence DH . (2012) Calycophyllum DC. In: Davidse G, Sousa Sánchez M, Knapp S, Chiang Cabrera F (eds). Flora Mesoamericana Vol. 4 Rubiaceae a VerbenaceaeMissouri Botanical Garden Press: St. Louis, MO, USA. pp 45.
Lowe AJ, Boshier D, Ward M, Bacles CFE, Navarro C . (2005). Genetic resource impacts of habitat loss and degradation; reconciling evidence and predicated theory for neotropical trees. Heredity 95: 255–273.
Lowe AJ, Cavers S, Boshier D, Breed MF, Hollingsworth PM . (2015). The resilience of forest fragmentation genetics – no longer a paradox – we were just looking in the wrong place. Heredity 115: 97–99.
Luikart G, Sherwin WB, Steele B, Allendorf FW . (1998). Usefulness of molecular markers for detecting population bottlenecks and monitoring genetic change. Mol Ecol 7: 963–974.
MARENA. (2002). Estrategia Nacional de Biodiversidad y su plan de acción. La Gaceta–Diario Oficial de Nicaragua 156: 5471–5476.
Miles L, Newton AC, Defries RS, Ravilious C, May I, Blyth S et al. (2006). A global overview of the conservation status of tropical dry forests. J Biogeogr 33: 491–505.
Nei M . (1987) Molecular Evolutionary Genetics. Columbia University Press: New York, USA.
Noguera-Talavera A, González-Rivas B, Castro-Marín G . (2011). Monitoring applied to the preparation of a conservation plan of Calycophyllum candidissimum in fragments of a dry forest in the Southwest of Nicaragua. La Calera 11: 48–54.
Novick RR, Dick C, Lemes MR, Navarro C, Caccone A, Bermingham E . (2003). Genetic structure of Mesoamerican populations of Big-leaf mahogany (Swietenia macrophylla inferred from microsatellite analysis. Mol Ecol 12: 2885–2893.
Nybom H . (2004). Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13: 1143–1155.
Olson DM, Dinerstein E, Wikramanayake ED, Burgess ND, Powell GVN, Underwood EC et al. (2001). Terrestrial ecoregions of the world: a new map of life on Earth. BioScience 51: 933–938.
Orsini L, Vanoverbeke J, Swillen I, Mergeay J, De Meester L . (2013). Drivers of population genetic differentiation in the wild: isolation by dispersal limitation, isolation by adaptation and isolation by colonization. Mol Ecol 22: 5983–5999.
Peakall R, Smouse PE . (2012). GENALEX 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28: 2537–2539.
Pennington RT, Lewis GP, Ratter JA . (2006) Neo-tropical Savannas and Seasonally Dry Forests: Plant Diversity, Biogeography, and Conservation. CRC/Taylor and Francis: Boca Raton, FL, USA.
Pennington RT, Prado DE, Pendry CA . (2000). Neotropical seasonally dry forests and Quaternary vegetation changes. J Biogeogr 27: 261–273.
Petit R, Hampe A . (2006). Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst 37: 187–214.
Pineda-García F, Paz H, Meinzer FC . (2013). Drought resistance in early and late secondary successional species from a tropical dry forest: the interplay between xylem resistance to embolism, sapwood water storage, and leaf shedding. Plant Cell Environ 36: 405–418.
Piperno DR . (2006). Quaternary environmental history and agricultural impact on vegetation in Central America. Ann Missouri Bot Gard 93: 274–296.
Piperno DR . (2011). The origins of plant cultivation in the New World Tropics. Curr Anthropol 52: S453–S470.
Poelchau MF, Hamrick JL . (2012). Differential effects of landscape-level environmental features on genetic structure in three codistributed tree species in Central America. Mol Ecol 21: 4970–4982.
Poelchau MF, Hamrick JL . (2013). Palaeodistribution modelling does not support disjunct Pleistocene refugia in several Central American plant taxa. J Biogeogr 40: 662–675.
Prado DE, Gibbs PE . (1993). Patterns of species distributions in the dry seasonal forests of South America. Ann Missouri Bot Gard 80: 902–927.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Ramírez-Arriaga E, Navarro-Calvo LA, Díaz-Carbajal E . (2011). Botanical characterisation of Mexican honeys from a subtropical region (Oaxaca) based on pollen analysis. Grana 50: 40–54.
Ramírez-Barahona S, Eguiarte LE . (2013). The role of glacial cycles in promoting genetic diversity in the Neotropics: the case of cloud forests during the Last Glacial Maximum. Ecol Evol 3: 725–738.
Ratnam W, Rajora OP, Finkeldey R, Aravanopoulos F, Bouvet JM, Vaillancourt RE et al. (2014). Genetic effects of forest management practices: global synthesis and perspectives. For Ecol Manage 333: 52–65.
Redo DJ, Grau HR, Aide TM, Clark ML . (2012). Asymmetric forest transition driven by the interaction of socioeconomic development and environmental heterogeneity in Central America. Proc Natl Acad Sci USA 109: 8839–8844.
Roa MG . (2003) Estado de la diversidad biológica de los árboles y bosques en Nicaragua. Documentos de Trabajo: Recursos Genéticos Forestales. RGF/49S Servicio de Desarrollo de Recursos Forestales, Dirección de Recursos Forestales, FAO: Roma, Italy. Available at ftp://ftp.fao.org/docrep/fao/007/j0602s/j0602s00.pdf (last accessed August 2016).
Rosenberg NA, Mahajan S, Ramachandran S, Zhao C, Pritchard JK, Feldman MW . (2005). Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet 1: e70.
Rousset F . (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Ruiz-Mallén I, Corbera E . (2013). Community-based conservation and traditional ecological knowledge: implications for socio-ecological resilience. Ecol Soc 18: 12.
Russell JR, Weber JC, Booth A, Powell W, Sotelo-Montes C, Dawson IK . (1999). Genetic variation of Calycophyllum spruceanum in the Peruvian Amazon Basin, revealed by amplified fragment length polymorphism (AFLP) analysis. Mol Ecol 8: 199–204.
Salas JB . (1993) Arboles de Nicaragua. Instituto Nicaragüense de Recursos Naturales y del Ambiente: Managua, Nicaragua.
Sánchez-Azofeifa GA, Quesada M, Calvo J, Rodriguez JP, Nassar J, Garvin T et al. (2005). Research priorities for tropical secondary dry forests. Biotropica 37: 477–485.
Skrede I, Borgen L, Brochmann C . (2009). Genetic structuring in three closely related circumpolar plant species: AFLP versus microsatellite markers and high-arctic versus arctic-alpine distributions. Heredity 102: 293–302.
Slatkin M . (1993). Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47: 264–279.
Spurgin LG, Illera JC, Jorgensen TH, Dawson DA, Richardson DS . (2014). Genetic and phenotypic divergence in an island bird: isolation by distance, by colonization or by adaptation? Mol Ecol 23: 1028–1039.
Standley PC, Williams LO . (1975). Flora of Guatemala – Part XI. Fieldiana. Botany 24: 1–274.
Stevens WD, Ulloa Ulloa C, Pool A, Montiel OM . (2001). Flora de Nicaragua. Monogr Syst Bot Mo Bot Gard 85: 1–2666.
Taylor BW . (1963). An outline of the vegetation of Nicaragua. J Ecol 51: 27–54.
Vekemans X . (2002) AFLP-SURV Version 1.0. Distributed by the author. Laboratoire de Génétique et Ecologie Végétale, Université Libre de Bruxelles: Belgium.
Verdu P, Pemberton TJ, Laurent R, Kemp BM, Gonzalez-Oliver A, Gorodezky C et al. (2014). Patterns of admixture and population structure in native populations of Northwest North America. PLoS Genet 10: e1004530.
Villegas-Durán G, Bolaños-Medina A, Miranda-Sánchez, Sandoval-Hernández R . (2003) Flora nectarífera y polinífera en el Estado de Veracruz. Secretaría de Agricultura, Ganadería, Desarrollo Rural, Pesca y Alimentacíon. Fundación Produce de Veracruz: Xalapa Enríquez, México.
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23: 4407–4414.
Wang IJ, Bradburd GS . (2014). Isolation by environment. Mol Ecol 23: 5649–5662.
Ward M, Dick CW, Gribel R, Lowe AJ . (2005). To self, or not to self… a review of outcrossing and pollen-mediated gene flow in neotropical trees. Heredity 95: 246–254.
Weaver PL, Lombardo DM, Martínez Sánchez JC . (2003) Biodiversity and tropical forest conservation, protection and management in Nicaragua: assessment and recommendations US Department of Agriculture. Foreign Agricultural Service: Washington DC. Available at http://pdf.usaid.gov/pdf_docs/Pnadf173.pdf (last accessed August 2016).
White GM, Boshier DH, Powell W . (2002). Increased pollen flow counteracts fragmentation in a tropical dry forest: an example from Swietenia humilis Zuccarini. Proc Nat Acad Sci USA 99: 2038–2042.
Whitlock MC, McCauley DE . (1999). Indirect measures of gene flow and migration: FST≠1/(4Nm+1). Heredity 82: 117–125.
WNBR. (2016). The World Network of Biosphere Reserves (WNBR) of UNESCO’s ‘Man and the Biosphere Programme’ (MAB). Annual release. Available at http://www.unesco.org/new/en/natural-sciences/environment/ecological-sciences/man-and-biosphere-programme/networks/ (last accessed August 2016).
Wright S . (1943). Isolation by distance. Genetics 28: 114–138.
Wright S . (1978) Evolution and the Genetics of Populations. Volume 4: Variability Within and Among Natural Populations. University of Chicago Press: Chicago.
Acknowledgements
We are thankful to Carlos Mejia (MARENA) for processing of collection/export permits and Ursula Jaros (Salzburg University) for help with the data analyses. For scientific advice and information we are indebted to Indiana Coronado and Charlotte Taylor (Missouri Botanical Garden), Rolando Dolmus (UNAN-Léon), Daniel H Janzen (University of Pennsylvania), and José Benito Quezada (Universidad Nacional Agraria, Managua). Alberto Dávila-Lara’s research stay at Salzburg University, as part of his ‘Licenciatura’ work, was supported by a PLUS studentship. Special thanks are extended to Waltraud Kofer (Pontifical Catholic University of Peru; German Academic Exchange Service/DAAD), Hermann Suida and Walter Gruber (Salzburg University) for their encouragement and support throughout this project. We also gratefully acknowledge the insightful comments from three anonymous reviewers and the handling editor on earlier versions of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Dávila-Lara, A., Affenzeller, M., Tribsch, A. et al. AFLP diversity and spatial structure of Calycophyllum candidissimum (Rubiaceae), a dominant tree species of Nicaragua’s critically endangered seasonally dry forest. Heredity 119, 275–286 (2017). https://doi.org/10.1038/hdy.2017.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2017.45
