
Abstract
We used multilocus sequence analysis (MLSA) on a worldwide collection of the plant pathogenic Ralstonia solanacearum (Betaproteobacteria) to retrace its complex evolutionary history. Using genetic imprints left during R. solanacearum evolution, we were able to delineate distinct evolutionary complex displaying contrasting dynamics. Among the phylotypes already described (I, IIA, IIB, III, IV), eight groups of strains with distinct evolutionary patterns, named clades, were identified. From our recombination analysis, we identified 21 recombination events that occurred within and across these lineages. Although appearing the most divergent and ancestral phylotype, phylotype IV was inferred as a gene donor for the majority of the recombination events that we detected. Whereas this phylotype apparently fuelled the species diversity, ongoing diversification was mainly detected within phylotype I, IIA and III. These three groups presented a recent expanding population structure, a high level of homologous recombination and evidences of long-distance migrations. Factors such as adaptation to a specific host or intense trading of infected crops may have promoted this diversification. Whether R. solanacearum lineages will eventually evolve in distinct species remains an open question. The intensification of cropping and increase of geographical dispersion may favour situations of phylotype sympatry and promote higher exchange of key factors for host adaptation from their common genetic pool.
Similar content being viewed by others


Introduction
Despite the increasing availability of large amounts of sequence data, an understanding of the evolutionary processes that explain the extent diversity and distribution of pathogenic microorganisms still remains a challenging task. From the single genome scale to the microbial ecology scale, many imbricate and interdependent mechanisms determine the fate of a given pathogen, this possibly ranging from simple spillover to disease emergence and establishment.
In bacteria, polymorphism created by mutation is redistributed among strains by recombination and horizontal gene transfer. At the bacterial population scale, demographic processes such as selection, drift and migration also shape the diversity of bacterial taxa, so that from a single common ancestor different taxa displaying different host ranges or strong phylogeographic patterns can be derived. Although these processes frequently result in speciation for many eukaryotes, the species delineation remains blurred within bacteria because of their propensity to exchange DNA even between distantly related species (Kunin et al., 2011). Among the recombination processes, the evolutionary importance of horizontal gene transfer has been highlighted by the tremendously increasing number of whole-genome sequences available (Martin and Beiko, 2010), and has mainly been studied on animal or human pathogenic bacteria (Achtman, 2008; Didelot and Maiden, 2010). Although some recent studies address this question on Pseudomonas syringae (Yan et al., 2008) or Xanthomonas campestris (Fargier et al., 2011), data are far less numerous on plant-associated bacteria, and more specifically plant pathogenic bacteria (Vos and Didelot, 2008).
Recombination has been recognized as one of the main evolutionary forces driving bacterial adaptation to the environment and to hosts they may colonize (Ochman et al., 2000; Cohan, 2002). DNA exchanges most likely occur only between taxa sharing at least the same ecological niche or geographical location. Consequently, adaptations to host or ecological specificities are expected to drive the dynamics of recombination.
In this regard, and in order to understand the micro- and macro-evolution patterns leading to emergence, Ralstonia solanacearum appears to be one of the best models. R. solanacearum is a soilborne Betaproteobacteria able to infect more than 250 plant species in 54 botanical families both within monocots and dicots (Hayward, 1994). This bacterium encompasses a great diversity of ecotypes (defined as a group of bacteria ecologically similar to one another (Cohan, 2006), in that case sharing similar host, symptomatology, and inducing disease in similar climatic conditions), among which some are specialized to one host or family (Musaceae, ginger, mulberry tree). R. solanacearum induces bacterial wilt (BW) on Solanaceae and many other crops, Moko disease on banana and ornamental Musaceae and brown rot on potato (Hayward, 1991, 1994). R. solanacearum is regarded as a species complex (Gillings and Fahy, 1994), composed of four phylotypes of probable different geographical origins (I: Asian, II: American, III: African, and IV: Indonesian) as previously defined based on hrpB, egl and mutS sequence analysis (Poussier et al., 2000a, 2000b; Fegan and Prior, 2005; Prior and Fegan, 2005), whereas previous classifications were based on host range variations (races; Buddenhagen et al., 1962) and metabolic properties (biovars; Hayward, 1964). The four phylotypes, subdivided into sequevars based on egl sequence similarities (Fegan and Prior, 2005), were further validated with comparative genomic hybridization (Guidot et al., 2007) and complete genome comparisons (Remenant et al., 2010).
The first global population genetics study addressing these questions by multilocus sequence analysis (MLSA) and multilocus sequence typing concluded that R. solanacearum is an essentially clonal organism following estimations of linkage disequilibrium between loci (Castillo and Greenberg, 2007). However, some lines of evidence in this study, and several other studies (Bertolla et al., 1999; Fall et al., 2007; Guidot et al., 2009; Coupat-Goutaland et al., 2011), suggest an important contribution of recombination and horizontal gene transfer in the evolutionary dynamics of R. solanacearum.
Although it is clearly apparent that estimating recombination frequency within bacterial populations is essential to assess their evolutionary potential, recombination also hampered classical phylogenetic reconstruction, an approach often used when attempting to reconstruct the evolutionary history of an organism (Feil et al., 2001; Pérez-Losada et al., 2006). To cope with this problem, the use of the coalescent theory (Schierup and Wiuf, 2010) and gene genealogies has been increasing in bacteria (Moodley et al., 2009; Wilson et al., 2009; Doroghazi and Buckley, 2010).
Here we performed MLSA to assess and estimate the contributions of the different evolutionary mechanisms shaping the R. solanacearum diversity. Using both phylogenetic and coalescent analyses, we inferred and compared the population evolutionary story of the different phylotypes. We particularly aimed to assess (1) whether recombination was a significant driving force for R. solanacearum evolution, (2) whether recombination was evenly distributed within phylotypes, (3) whether ecotypes were distinct evolutionary lines, (4) the demographic and biogeographic patterns of phylotypes and (5) their most probable pattern of descent.
Materials and methods
Strains
A collection of 89 R. solanacearum strains was provided by the authors and obtained from different collections (Supplementary Table S1). It contained the R. solanacearum type strain K60, and at least one representative strain of each of the 51 egl-based sequevars (Fegan and Prior, 2005), and representatives of the different ecotypes described so far within R. solanacearum (‘Moko’, ‘bugtok’, ‘Banana Blood Disease’, ‘brown rot’, mulberry and ‘emerging’ strains, see Supplementary Information for details). Two closely related organisms included in the phylotype IV of the species complex (that is, the Blood Disease Bacterium (BDB), and the causing agent of the Sumatra Disease of Clove Ralstonia syzygii) and three outgroups were also used. Genomic DNA was obtained from each strain, as described in Supplementary Information.
MLSA of R. solanacearum
Nine single-copy genes were chosen for our MLSA scheme, as detailed in Supplementary Information, and shown in Figure 1. Seven were chromosome housekeeping genes (gdhA, mutS, ppsA, adk, leuS, rplB and gyrB) whereas two were megaplasmid virulence-associated genes (egl and fliC). Sets of primers used to amplify fragments of these nine genes (Supplementary Table S2) were either described previously (Prior and Fegan, 2005; Castillo and Greenberg, 2007; Ait Tayeb et al., 2008) or newly designed. PCR protocols, sequencing primers and alignment processes are detailed in Supplementary Information. All gene sequences were deposited in GenBank (see Supplementary Table S7 for accession numbers).

Schematic representations of the nine locus positions within the genomes of CMR15 (phylotype III), CFBP2957 (phylotype IIA), PSI07 (phylotype IV) and GMI1000 (phylotype I). Detailed positions are summarized in Supplementary Table S2.
Data analysis
Molecular diversity indices and haplotypes were determined using DnaSP 5.0 (Librado and Rozas, 2009). Tests of mutation/drift equilibrium (Tajima's D and Fu and Li's D* and F*) were calculated for each gene separately and for concatenated gene set, on all sequences as well as on each phylotype subset separately.
To detect the presence of recombination, we first performed the pairwise homoplasy index test, Φw (Bruen et al., 2006), implemented in Splitstree4 (Huson and Bryant, 2006). The standardized index of association was calculated with LIAN v3.5 (Haubold and Hudson, 2000) on allelic data. The LDhat module (McVean et al., 2002) available within RDP3.44 (Martin et al., 2010) was used to estimate recombination and mutation rates ρ and θ for both single and concatenated gene sequences, as detailed in the Supplementary Information.
Detection of potential recombinant sequences, identification of likely parental sequences and localization of possible recombination breakpoints were carried out on each gene separately and on concatenated sequences using the seven recombination detection methods as implemented in RDP v3.44 (Martin et al., 2010), as detailed in Supplementary Information. To bring a direct view of gene exchange patterns in the R. solanacearum species complex, recombination events were summarized at the phylotype level and represented in a recombination network (see Supplementary Information for details).
Phylogeny and gene genealogy reconstruction
After assessment of the best fit nucleotide substitution model, phylogenetic trees for each gene and for recombination-free concatenated data (that is, with recombinant section removed), were reconstructed using PhyML v3.0 (Guindon and Gascuel, 2003). Along with the phylogenetic reconstruction, we performed a network reconstruction on total concatenated data using the Neighbor-net algorithm available in Splitstree4.
To reconstruct the genealogy of the whole R. solanacearum species complex and each phylotype, we used ClonalFrame vs1.1 (Didelot and Falush, 2007) as described in Supplementary Information.
Phylogeography
Using sampling coordinates and a freely available hierarchical clustering method (the ‘hclust’ function) implemented in R (R Development Core Team, 2010), we were able to optimally define eight groups of sequences displaying definite geographical clustering. R. solanacearum sequences were then assigned with their defined geographical discrete states (that is, one of the eight groups) and the ancestral geographical states were reconstructed using the BayesTraits programs (Pagel et al., 2004) with bootstrap replicates to take into account possible sampling bias (see Supplementary Information for details).
Results
Genetic diversity within the R. solanacearum species complex
Sequence data of the nine genes studied were obtained for most of the 93 Ralstonia strains (Supplementary Table S6). Missing genes (see blank tracks on Figure 2; Supplementary Table S7) were considered as deletions in further analyses.

Maximum-likelihood tree of 90 Ralstonia solanacearum strains constructed with PhyML 3.0, using the non-recombinant concatenated sequences of nine loci (gdhA, mutS, ppsA, adk, leuS, rplB, gyrB, fliC and egl), with an overview of the occurrence and positions of the 21 recombination events, identified with RDP3.44 (Martin et al., 2010)—see details in Supplementary Table S3. The tree was rooted on the outgroups Ralstonia insidiosa LMG21421T, R. mannitolylitica LMG6866T and R. pickettii LMG5942T, not shown in the figure. Circles on the nodes indicate the bootstrap support values (closed circles: >70%; open circles: >50%). Horizontal bars represent concatenated sequences of each strain, coloured according to their phylotype assignation (I, IIa and IIb, III, IV; UNK, unknown donor). Blank bars within concatenates indicate genes that were absent, or could not be amplified, from the corresponding strain. Recombination events are identified by ID numbers (Supplementary Table S3), and coloured according to the phylogenetic position of the donor (minor parent). The events 1, 10 and 19 are absent from the figure, as the recombining sequence is a Ralstonia outgroup (R. insidiosa and R. pickettii, respectively).
Synonymous substitutions (Ks) were more frequent than nonsynonymous substitutions (Ka) at each locus (from 3 to 4 times more frequent within egl and fliC to 30 times more frequent within rplB; Table 1), indicating that the nine genes were suitable for population studies (Maiden, 2006). Nucleotide diversity (π in Tables 1 and 4) was 3.0% considering the entire R. solanacearum collection and were similar across genes (between 2.0% and 3.6%, Table 4), except for egl and ppsA that were more variable (π=5.5% and 3.9%, respectively, Table 1). Among phylotypes, highest π values were observed for phylotype IV (2.1%), and then II (1.3%) and III (1.1%); phylotype I was the least diverse (0.5%). Haplotype numbers for each of the nine genes ranged from 39 (fliC) to 65 (gyrB, Supplementary Table S4). Phylotypes III and IV had the maximal haplotype diversities (mean Hd=0.95 and 0.92, respectively) and phylotype I the minimal ones (mean Hd=0.77; Supplementary Table S4). Within the phylotype II, phylotype IIA (0.90) was more diverse than phylotype IIB (0.84).
Phylogenetic reconstruction and recombination
Phylogeny was reconstructed on each gene separately and on concatenated sequences. Interestingly, individual maximum likelihood phylogenies (Supplementary Figure S1A–I) displayed incongruent topologies. Along with the reticulate structure of the neighbor-net reconstruction inferred from the concatenated sequence set (Supplementary Figure S4), these inconsistencies suggested occurrence of recombination within or across genes. This assumption was confirmed with the rejection of the hypothesis of no recombination with the pairwise homoplasy index test (Φw) (Table 2). We therefore analysed our sequence data sets using RDP to detect individual recombination events within the aligned concatenated genes.
A total of 21 individual recombination events were detected in the concatenated data set (Figure 2 and Supplementary Table S3). Two genes appeared as recombination free (mutS and leuS), whereas the remaining seven genes displayed from one to four recombination events. The distribution of recombination events within and across the different phylotypes and clades is summarized in Figure 2. Recombination was detected in each of the phylotypes, with phylotype I being the most recombinagenic (nine events detected). Recombinant sequences originated primarily from phylotype IV and IIB strains. Phylotype IV also displayed two recombinant events, with one being an intraphylotype recombination. Within phylotype II, most of the strains displayed a phylotype III-originating or outgroup-related fliC recombinant sequences.
Two recombination events (17 and 21) were particularly interesting in that they were detected within several phylotypes, suggesting an ancient evolutionary origin, predating the phylotype divergence. Recombination event 17 was detected in strains from phylotypes I and III, with the strain ACH732 as possible donor. Recombination event 21 was detected in both phylotypes IIA and IIB with a donor most likely from phylotype III.
When summarizing this information at the phylotype level (Figure 3), patterns of genetic exchange became apparent. Within the recombination network, phylotype IV appeared to be a gene donor to all the other phylotypes, particularly phylotypes III and I, more secondarily to phylotype IIA. Phylotype IV was recipient only once from phylotype I. Phylotypes IIA and IIB were donors to phylotype I with three events being inferred; IIA was recipient from phylotypes IV and III, whereas IIB was recipient only from IV. No gene exchange was identified between phylotypes IIA and IIB or between phylotypes I and III.

Network of gene exchanges between phylotypes of Ralstonia solanacearum, established according to RDP results. Gene exchanges are represented with arrows and orientated from the donor (minor parent) to the recombinant. Arrow thickness is proportional to the numbers of recombination event inferred between genes. When a recombination event was inferred from or in sequences belonging to more than one phylotype, multiple arrows with a thickness proportional to the number of sequence involved were drawn.
Genealogies and demographic parameters of phylotypes inferred from coalescent analysis
According to coalescent genealogy reconstruction (Figure 4 and Supplementary Figure S2), ACH732 may be the most ancestral strain of this collection. Within the remaining strains, it is interesting to note that the phylotype structure is again clearly apparent over the genealogy. Phylotype IV was the first group to diverge, followed by phylotype II on one side and phylotypes I–III cluster on the other side, at similar times (coalescence time distributions of these two nodes widely overlap, data not shown). These two clusters split later into phylotypes IIA and IIB, and phylotypes I and III, respectively (Figure 4b).

Gene genealogies of the phylotypes and clades of Ralstonia solanacearum. (a) GraphViz network generated by ClonalFrame. Ancestral nodes inferred are marked in black, and the location of isolates in red, with each red line representing a single isolate. The ancestral node of each network component is indicated by a thicker circle (Didelot and Falush, 2007). Strains are identified by their RUN ID (see Supplementary Table S1). Yellow shades indicate banana-affecting ecotypes: Moko disease-inducing strains (IIA/sequevars 6 and 24, IIB/sequevars 3 and 4) and BDB (IV/sequevar 10); green shade indicates the ‘Brown Rot’ strains (IIB/sequevar 1, formerly race 3/biovar 2), which are preferentially pathogenic to potato and tomato, and are specifically able of inducing disease at cool temperatures (Milling et al., 2009). Red shade indicates ‘emerging’ strains that are pathogenic to Araceae, Solanaceae, Cucurbitaceae while not pathogenic to dessert banana (Wicker et al., 2007). Blue shade indicates mulberry-affecting strains (race 5/biovar 5). S., egl-based sequevars (Fegan and Prior, 2005). Outliers (1359 and 71 in phylotype IV; 43, 81, 299, 482 and 40 in phylotype II; 76 and 39 in phylotypes I and III) are identified by their RUN number, with their host, country and year of isolation. Pel., Pelargonium sp. (b) Schematic representation of the majority-rule tree based on the posterior distribution of genealogies inferred by ClonalFrame. D1, divergence of phylotypes IV and I/II/III; D2, divergence of phylotypes II and I/III; D3, divergence of IIA and IIB; D4, divergence of phylotypes I and III.
Based on the genealogy structure, known ecotypes and the inferred demographic parameters, we defined eight clades among the four phylotypes (Figure 4). Phylotype IV included two clades: (1) clade 7 encompassing both the ‘Blood Disease of Banana’ (BDB)- and BW-inducing strains and (2) clade 8 encompassing strains of the species R. syzygii. The strain 1359 (isolated from clove in West Java) appeared as an outlier. Phylotype IV appeared in demographic equilibrium, as indicated by the external to internal branch length ratio test (Inext) (Table 3 and Supplementary Figure S3). Within phylotype II, the structure was more complex with four clades identified. First, two Brazilian strains isolated from potato (43 and 81) appeared to be closely related to the common ancestor. Phylotype IIA formed (1) clade 2, gathering the Moko (equivalent to sequevars 6 and 24; Fegan and Prior, 2005) and BW-inducing strains, and (2) clade 3 (equivalent to sequevar 7) with strains originating from Southeastern United States (65, corresponding to the type strain K60; 1526 and 1527), Kenya (55=ICMP7963) and Caribbean (560, 544). Phylotype IIB was composed of (1) clade 4 (equivalent to sequevar 4) with Moko-inducing and ‘emerging’ strains isolated from Anthurium and Heliconia, and (2) clade 5, containing ‘potato brown rot’ strains (equivalent to sequevars 1 and 2) and Moko disease-causing strains (equivalent to sequevar 3). Importantly, phylotypes IIA and IIB differed in demographic parameters. The Inext of phylotype IIA (1.918, P=1.6 × 10−3) was indicative of a recent population expansion (Table 3 and Supplementary Figure S3), whereas phylotype IIB was inferred as being near population equilibrium (Inext=1.362, P=0.037).
Finally, in a third complex (Figure 4a) consisting of phylotype I (clade 1) and phylotype III (clade 6), the strain 76, isolated from tobacco in Zimbabwe, appeared as an outlier whereas it was assigned within phylotype III both using egl typing and in our MLSA scheme. The remaining strains were organized in two clonal complexes, both displaying short branches (Figure 3), and significant Inext values (Inext 2.528 and 2.647 for phylotypes I and III, respectively), indicating a recent and rapid expansion of population size, possibly following a bottleneck (indicated by significantly negative Tajima's D values, Supplementary Table S6) because of host adaptation or acquisition of a new life history trait. Phylotype I displayed an epidemic population structure, with a single ancestor from which derived mulberry-infecting strains 90, 91 (He et al., 1983) and strains from China (337, 339), and other clonal lines. Phylotype III contained strains all sampled from Africa. Apart from an outlier strain (39) sampled from eggplant in Burkina-Faso, this group also displayed an epidemic population structure.
Recombination and mutation among phylotypes
The nine loci displayed different per site recombination/mutation rate ratios (ρ/θ), falling in two broad groups: (1) more mutation than recombination with ρ/θ below 1, for gdhA, mutS, leuS, rplB (chromosome) and egl (megaplasmid) genes; and (2) more recombination than mutation with ρ/θ ranging from 2.64 to 3.99, for ppsA, adk, gyrB (chromosome) and fliC (megaplasmid). Interestingly, the flagellin-coding gene fliC, containing a well-known microbe-associated molecular pattern (Felix et al., 1999), displayed one of the highest ρ/θw ratios (2.80). Considering the entire R. solanacearum population, the standardized index of association, Ias (Maynard Smith et al., 1993; Haubold et al., 1998), was significantly different from 0, indicating a high level of linkage disequilibrium and a clonal genetic structure (Table 4). R. solanacearum as a whole had an intermediate relative effect of recombination and mutation (r/m=1.6), confirming previous results (Vos and Didelot, 2008). However, when calculated on the different phylotypes, Ias indicated that phylotype I population was in linkage equilibrium (Ias=0.024), suggesting a panmictic and freely recombining population structure. Phylotype III displayed a low but significant Ias (0.053), suggesting a nearly clonal structure, whereas phylotypes IV and II displayed high indices (0.247 and 0.277, respectively), evoking highly clonal structures. Within phylotype II, however, phylotype IIA Ias was lower than that of phylotype IIB; the clade 3 Ias (0.088) indicated a freely recombining structure. Considering either the relative rate of recombination and mutation (ρ/θw) or the relative effect of recombination and mutation (r/m) calculated using ClonalFrame, high recombination was inferred for phylotypes I, III and IV (Table 4). Conversely, phylotype II had moderate recombination, with a great contrast between the recombinogenic phylotype IIA and the clonal phylotype IIB.
Geographical origins of ancestral states
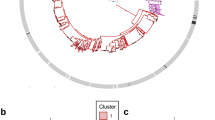
We used ancestral state reconstruction to infer the most probable patterns of migration that has shaped the current R. solanacearum geographical distribution. Interestingly, the different phylotypes and clades differed in geographical origin. Whereas within phylotypes III and IV, only one long-range migration each was inferred from the region they originated (Central/Western Africa and Indonesia/Australia for phylotypes III and IV, respectively), phylotype II, and phylotype I to a lesser extent, displayed complex patterns of migrations.
The common ancestor of all phylotype II strains probably emerged on the American continent (P=0.45), with a further split of the phylotype into subgroups IIA (Northern Latin America and Caribbean, P=0.40) and IIB (Austral Latin America, P=0.75). From those regions, intense migrations within America itself and throughout the world are inferred, explaining the worldwide distributions of this phylotype (particularly clade 5; Figure 5). Although some of the migrations may have resulted in spillovers (one strain only for some regions), subgroups of clades 2 and 3 have successfully established in Central America (see red branches within phylotype IIA) and Africa (green branches). Interestingly, the clade 3 most likely originated in the Caribbean, or the Caribbean border of Latin America, from which the Southeastern US populations were secondarily introduced.

Probability of origin of the different phylotypes and clades of Ralstonia solanacearum. (a) Map of the eight geographical location states, as defined by ‘hclust’ analysis (see Materials and methods). Black dots mark the countries sampled within each area. Cluster 1: Central and North America (Florida, North Carolina, Guatemala, Honduras); cluster 2: Austral Latin America (Peru, Brazil, Uruguay); cluster 3: North Latin America and Caribbean (Columbia, Venezuela, French Guyane, Trinidad, Grenada, Martinique, Guadeloupe); cluster 4: The Netherlands; cluster 5: Central and Western Africa (Cameroon, Nigeria, Burkina Faso, Guinea); cluster 6: Austral and Eastern Africa, Indian Ocean (Angola, Kenya, Zimbabwe, Madagascar, Reunion Island); cluster 7: Northeastern Asia (China, Taiwan, Japan, Philippines); cluster 8: Indonesia (Java, Borneo, Sumatra, Celebes) and Australia (Northern territory). (b) Maximum clade credibility tree constructed from the nine loci-concatenated sequence sets. Branches are coloured according to the most probable location state (1–8) of their descendant nodes, with the corresponding probability marked at the node. Strains are identified by their RUN ID or reference names (see Supplementary Table S1).
Phylotype I most probably originated either in the North-Asian or the Austral–African region, with probabilities of 0.58 and 0.37, respectively (Supplementary Figure S4). Within the phylotype I, several migrations of strains between Eastern Africa and the East Asian region are inferred, with secondary migrations to America, Europe, Indonesia and Western Africa (Figure 5).
Discussion
Recombination induces conflicting phylogenies across genes within the R. solanacearum species complex
Whereas R. solanacearum phylogeny was up to now mainly based on egl sequence analysis (Fegan and Prior, 2005, 2006; Ivey et al., 2007; Jeong et al., 2007; Ji et al., 2007; Wicker et al., 2007; Mahbou Somo Toukam et al., 2009), our study demonstrated the presence of bias in the phylogenetic replacement (in particular for the strains RUN1359, RUN0076 and RUN0064) because of widespread recombination within the R. solanacearum species complex.
The strain ACH732 (RUN0014) was the most striking case as it was not detected as a recombinant. Assigned to phylotype IV using maximum likelihood method (Figure 2), this strain was considered a single ancestral genotype using the coalescent approach (Figure 4 andSupplementary Figure S3). However, ClonalFrame genealogies reconstructed on different gene sets (total data set, housekeeping genes, genes exhibiting low or high ρ) placed ACH732 either basal to the genealogy or within phylotype IV (data not shown). Incongruence of the phylogenetic assignment of this strain was already reported: the first published egl-based classification placed it within phylotype I (Poussier et al., 2000a), whereas intergenic transcribed tracer-based classification (Fegan et al., 1998) and mutS-based classification considered it a single phylogenetic group (Prior and Fegan, 2005). Comparative genomic hybridization results (Remenant et al., 2010) established that ACH732 contained an original gene content, although placed within phylotype IV (Supplementary Figure S6). To date, exact position of ACH732 thus remains unsolved.
Recombination is ubiquitous within the R. solanacearum species complex
The observation of incongruent phylogenies inferred from partial sequences of individual genes strongly suggested that recombination is widespread within and across phylotypes, and occurs across seven of the nine genes studied here, both in housekeeping (gyrB, rplB, adk, ppsA, gdhA) and virulence-associated genes (egl and fliC). The number of recombination events (n=21) detected was a little higher than that observed (n=13) in the highly recombining soil-dwelling Streptomyces spp. (Doroghazi and Buckley, 2010); comparison with bacterial taxa of similar ecology and lifestyle remains difficult because of data scarcity in the literature. When it comes to comparing recombination contributions of the different phylotypes, it must be borne in mind that recombination between distant evolutionary lineages is easier to detect than between closely related ones. It is therefore plausible that recombination events between I and III, IIA and IIB, and within phylotypes, remain undetectable. Nevertheless, it was clearly apparent from our analysis that phylotype IV is an important gene source, with seven vents of gene exchange with other phylotypes.
Assessment of recombination contribution to R. solanacearum evolutionary dynamics highlighted other major differences between phylotypes. Three recombination estimates (linkage disequilibrium, ρ/θ and r/m) report that phylotype I is a recombinogenic lineage, but gave contradictory results for the three other phylotypes. Indeed, phylotypes II, III and IV appeared in linkage disequilibrium (thus rather clonal) whereas their r/m and ρ/θ values were high, indicating a predominant role of recombination; the only clonal group being phylotype IIB. Our study thus demonstrates that there is significant recombination going on (as evidenced by the ‘very high’ r/m values sensu; Vos and Didelot, 2008) within phylotypes I, IIA, III and IV, although its effect was not sufficient to erase all clonal structure (as evidenced by linkage disequilibrium). The r/m values inferred on the whole R. solanacearum species complex (1.6 with 95% confidence interval 1.04–2.32) was higher than that obtained by Castillo and Greenberg (2007) (1.1 with 95% confidence interval 0.7–1.6), but within the range reported so far on plant pathogenic bacteria (P. syringae, P. viridiflava, X. campestris pv. campestris) (Vos and Didelot, 2008) using MLSA data (Silva et al., 2005; Yan et al., 2008; Doroghazi and Buckley, 2010; Fargier et al., 2011).
Although our data and the data reported previously (Castillo and Greenberg, 2007) are broadly consistent on observed mutation rates θw as well as on low linkage disequilibrium observed on phylotype III, they differ widely on their conclusions regarding the extent of recombination within the R. solanacearum species complex. Whereas Castillo and Greenberg (2007) concluded that phylotypes I and IV are clonal complexes, we show here that contribution of recombination is predominant towards mutation in the evolution of phylotypes I, IIA, III and IV. Besides the differences in gene sets (we used two additional genes leuS and rplB and did not use the hrpB gene), these contrasting conclusions are most likely because of the difference in the data set composition: the collection of Castillo and Greenberg (2007) mainly consisted of race 3/biovar 2 strains (phylotype IIB/sequevar 1; clade 5 in this study), whereas phylotype I, III and IV sample sizes were much lower than in our study.
Evolutionary history of the R. solanacearum species complex
Coalescent genealogy and maximum likelihood recombination-free phylogeny led to consider R. solanacearum as a species complex constituted of five evolutionary lineages (I, IIA, IIB, III and IV) and eight clades, displaying different evolutionary and phylogeography dynamics. Gene flow orientation, topology and branch lengths and phylogeography predictions support previous work (Fegan, 2005) that proposed an Australian/Indonesian origin for R. solanacearum. Therefore, the highly diverse, universal gene donor but poorly expanding phylotype IV probably gathers remnants of the ancestral complex. From this region, and probably because of a change of ecological niche providing the ability to disseminate (through colonization of rhizomes, tubers, and perhaps seeds), a subgroup of the ancestral species probably spread throughout regions corresponding to present Austral–Eastern Africa and Madagascar, and differentiated later in phylotype III (predicted with a Western–Central African origin) and phylotype I (predicted with an East African/Asian origin), possibly at a time predating the Gondwanian fragmentation. Whereas phylotypes I and III share a high level of recombination and an expanding pattern, phylotype III was only sampled in Africa. Reasons for this difference remain to be determined, but may be related to differential ecological fitness for establishment between I and III, as situations of I–III sympatry in African lowlands are dominated by phylotype I (Elphinstone, 2005; Mahbou Somo Toukam et al., 2009).
Regarding phylotype II, lines of evidence indicate an origin in the region corresponding to the actual Brazil. Phylotype II afterwards differentiated into the sub-phylotypes IIA and IIB at a time similar to that of the phylotype I/III differentiation. Interestingly, the evolutionary patterns displayed by these two sub-phylotypes are highly contrasted, with phylotype IIB being nearly clonal and phylotype IIA being recombinogenic, highly diverse and expanding. Both sub-phylotypes, after emergence in South America, have recently migrated worldwide, probably through contaminated banana and ornamentals cuttings and potato tubers (Buddenhagen and Kelman, 1964; Milling et al., 2009).
Although each geographic region defined in our study appeared infected by a large diversity of clades (as shown by the multicolour phylogeography reconstruction on Figure 5b), multiple long-range migration and worldwide dissemination have been only observed for the Asian-African phylotype I and the South-American phylotype IIA. The geographical overlap of phylotype I with nondisseminating phylotypes tends to indicate that this lineage presents some specific genetic characteristics driving their worldwide emergence, related to not only dissemination capacity but also ecological fitness to establish in new environments. Adaptation to some particular hosts and capacity to induce latent infections in plant material may have been a key factor for dissemination (and a possible demographic bottleneck). Plant-mediated dissemination was mostly associated with banana, ginger and potato (Buddenhagen, 1986; Coutinho, 2005), and strains intercepted in global surveys on latently infected plant material were mostly of phylotype IIB, not of phylotype I (Norman et al., 2009). This contradiction can, however, be explained by the fact that this survey was largely performed on imports of South America, where IIB strains are prevalent.
This study highlighted that phylotype I possesses many features of a relatively recent (as evidenced by its low molecular diversity) and recombinogenic lineage that has spread over long distances very rapidly, most probably as latent infections in plant material. Its actual worldwide distribution in Asia, Africa, central and South America and Oceania, including relatively recent islands (Buddenhagen, 1986; Elphinstone, 2005), also suggests its better fitness to establish in tropical conditions. Now, phylotype I (historically known as biovars 3, 4, and 5) is known as the evolutionary lineage affecting the highest number of hosts (Hayward, 1994), including woody perennial hosts, rhizomes of Heliconia, Anthurium and ginger; it is capable of latently infecting potato tubers in the field (Ciampi et al., 1980), and can survive up to 6 months in 4 °C-stored tubers (Milling et al., 2009). Moreover, it contains three of the six pathoprofiles (virulence patterns on resistant tomato, eggplant and pepper accessions) that were defined for R. solanacearum (Lebeau et al., 2011). Its high ability to recombination (suggested by Coupat et al., 2008 and confirmed in this study), its pattern of dissemination, along with its large host range and virulence plasticity, place phylotype I as possessing the highest evolutionary potential (as defined by Mc Donald and Linde, 2002).
As the large dissemination of phylotype I is most likely explained with its large host range, it is important to assess what are the factors promoting host adaptation and the dynamics by which it evolves. Examples from the Moko-, brown rot-, BDB- and BW-causing strains can be quite informative. It is clearly apparent that within R. solanacearum clades coexist strains that are phenotypically very different: phylotype IIA (clade 2) and phylotype IV (clade 7) both gather banana-specialized (Moko or BDB) and BW-inducing strains; phylotype IIB (clade 4) contains banana-pathogenic (Moko) and ‘emerging’ banana-not pathogenic strains (Wicker et al., 2007). The polyphyletic nature of the Moko ecotype suggests that pathogenicity to banana lies on a very restricted number of genes (or even allelic forms of the same genes) that may be easily transferable through horizontal gene transfer. It also suggests that host preference is not a major driving force in structuring the ecotypes.
By addressing demographic and recombination aspects of evolutionary history of plant-associated R. solanacearum, our study revealed significant gaps to be investigated in the future, regarding (1) pathways and circuits of dissemination of the distinct phylotypes and (2) their ecological dynamics. In fact, as our collection is composed of plant-isolated strains, we cannot exclude that the overall genetic diversity has been underestimated. The sequencing of large amounts of R. solanacearum strains from environmental samples (soil, rhizosphere) using metagenomics sequencing approach will greatly improve our understanding of the whole R. solanacearum species complex. These researches will help delineate the genetic repertoire of adaptation factors shared in the Ralstonia complex along with its dynamics within and between populations. As suggested by Remenant et al. (2010, 2011), whether R. solanacearum lineages will eventually evolve in distinct species remains an open question. With the intensification of cropping and trade, increase of geographical dispersion may increase situations of phylotype sympatry and promote higher exchange of key factors for host adaptation available in their common genetic pool.
References
Achtman M . (2008). Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol 62: 53–70.
Ait Tayeb L, Lefevre M, Passet V, Diancourt L, Brisse S, Grimont PAD . (2008). Comparative phylogenies of Burkholderia, Ralstonia, Comamonas, Brevundimonas and related organisms derived from rpoB, gyrB and rrs gene sequences. Res Microbiol 159: 169–177.
Bertolla F, Frostegard A, Brito B, Nesme X, Simonet P . (1999). During infection of its host, the plant pathogen Ralstonia solanacearum naturally develops a state of competence and exchanges genetic material. Mol Plant Microbe Interact 12: 467–472.
Bruen T, Phillipe H, Bryant D . (2006). A quick and robust statistical test to detect the presence of recombination. Genetics 172: 2665–2681.
Buddenhagen I, Kelman A . (1964). Biological and physiological aspects of bacterial wilt caused by Pseudomonas solanacearum. Annu Rev Phytopathol 2: 203–230.
Buddenhagen IW . (1986). Bacterial wilt revisited. In: Persley GJ (ed.) Bacterial Wilt Disease in Asia and the Pacific. ACIAR: Canberra, Australia, pp 126–143.
Buddenhagen IW, Sequeira L, Kelman A . (1962). Designation of races in Pseudomonas solanacearum. Phytopathology 52: 726.
Castillo JA, Greenberg JT . (2007). Evolutionary dynamics of Ralstonia solanacearum. Appl Environ Microbiol 73: 1225–1238.
Ciampi L, Sequeira L, French ER . (1980). Latent infection of potato tubers by Pseudomonas solanacearum. Am J Potato Res 57: 377–386.
Cohan FM . (2002). What are bacterial species? Annu Rev Microbiol 56: 457–487.
Cohan FM . (2006). Towards a conceptual and operational union of bacterial systematics, ecology, and evolution. Philos Trans R Soc Biol 361: 1985–1996.
Coupat B, Chaumeille-Dole F, Fall S, Prior P, Simonet P, Nesme X et al. (2008). Natural transformation in the Ralstonia solanacearum species complex: number and size of DNA that can be transferred. FEMS Microbiol Ecol 66: 14–24.
Coupat-Goutaland B, Bernillon D, Guidot A, Prior P, Nesme X, Bertolla F . (2011). Ralstonia solanacearum virulence increased following large interstrain gene transfers by natural transformation. Mol Plant Microbe Interact 24: 497–505.
Coutinho TA . (2005). Introduction and prospects on the survival of R. solanacearum. In: Allen C, Prior P, Hayward AC (eds) Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex. APS Press: St Paul, MN, pp 29–38.
den Bakker HC, Didelot X, Fortes ED, Nightingale KK, Wiedmann M . (2008). Lineage specific recombination rates and microevolution in Listeria monocytogenes. BMC Evolut Biol 8: 277.
Didelot X, Falush D . (2007). Inference of bacterial microevolution using multilocus sequence data. Genetics 175: 1251–1266.
Didelot X, Maiden MCJ . (2010). Impact of recombination on bacterial evolution. Trends Microbiol 18: 315–322.
Doroghazi JR, Buckley DH . (2010). Widespread homologous recombination within and between Streptomyces species. ISME J 4: 1136–1143.
Elphinstone JG . (2005). The current bacterial wilt situation: a global overview. In: Allen C, Prior P, Hayward AC (eds) Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex. APS Press: Saint Paul, MN, pp 9–28.
Fall S, Mercier A, Bertolla F, Calteau A, Gueguen L, Perrière G et al. (2007). Horizontal gene transfer regulation in bacteria as a “spandrel” of DNA repair mechanisms. PLoS One 2: e1055.
Fargier E, Fischer-Le Saux M, Manceau C . (2011). A multilocus sequence analysis of Xanthomonas campestris reveals a complex structure within crucifer-attacking pathovars of this species. Syst Appl Microbiol 34: 156–165.
Fegan M . (2005). Bacterial wilt diseases of banana: evolution and ecology. In: Allen C, Prior P, Hayward AC (eds) Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex. APS Press: Saint Paul, MN, pp 379–386.
Fegan M, Prior P . (2005). How complex is the ‘Ralstonia solanacearum species complex’. In: Allen C, Prior P, Hayward AC (eds) Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex. APS Press: Madison, pp 449–462.
Fegan M, Prior P . (2006). Diverse members of the Ralstonia solanacearum species complex cause bacterial wilts of banana. Australas Plant Pathol 35: 93–101.
Fegan M, Taghavi M, Sly LI, Hayward AC . (1998). Phylogeny, diversity and molecular diagnostics of Ralstonia solanacearum. In: Prior P, Allen C, Elphinstone J (eds) Bacterial Wilt Disease - Molecular and Ecological Aspects. Springer, INRA Editions: Berlin, Paris, pp 19–33.
Feil EJ, Holmes EC, Bessen DE, Chan M-S, Day NPJ, Enright MC et al. (2001). Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc Natl Acad USA 98: 182–187.
Felix G, Duran JD, Volko S, Boller T . (1999). Plants have a sensitive perception system for the most conserved domain of bacterial flagellin. Plant J 18: 265–276.
Gillings MR, Fahy P . (1994). Genomic fingerprinting: towards a unified view of the Pseudomonas solanacearum species complex. In: Hayward AC, Hartman GL (eds) Bacterial Wilt: The Disease and Its Causative Agent, Pseudomonas solanacearum. CAB International: Wallingford, pp 95–112.
Guidot A, Coupat B, Fall S, Prior P, Bertolla F . (2009). Horizontal gene transfer between Ralstonia solanacearum strains detected by comparative genomic hybridization on microarrays. ISME J 3: 549–562.
Guidot A, Prior P, Schoenfeld J, Carrere S, Genin S, Boucher C . (2007). Genomic structure and phylogeny of the plant pathogen Ralstonia solanacearum inferred from gene distribution analysis. J Bacteriol 189: 377–387.
Guindon S, Gascuel O . (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704.
Haubold B, Hudson RR . (2000). LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16: 847–848.
Haubold B, Travisano M, Rainey PB, Hudson RR . (1998). Detecting linkage disequilibrium in bacterial populations. Genetics 150: 1341–1348.
Hayward AC . (1964). Characteristics of Pseudomonas solanacearum. J Appl Bacteriol 27: 265–277.
Hayward AC . (1991). Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Annu Rev Phytopathol 29: 65–87.
Hayward AC . (1994). The hosts of Pseudomonas solanacearum. In: Hayward AC, Hartman GL (eds) Bacterial Wilt -The Disease and Its Causative Agent, Pseudomonas solanacearum. CAB International: Wallingford, pp 9–24.
He LY, Sequeira L, Kelman A . (1983). Characteristics of strains of Pseudomonas solanacearum from China. Plant Dis 67: 1357–1361.
Huson DH, Bryant D . (2006). Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267.
Ivey MLL, Gardener BBM, Opina N, Miller SA . (2007). Diversity of Ralstonia solanacearum infecting eggplant in the Philippines. Phytopathology 97: 1467–1475.
Jeong Y, Kim J, Kang Y, Lee S, Hwang I . (2007). Genetic diversity and distribution of Korean isolates of Ralstonia solanacearum. Plant Dis 91: 1277–1287.
Ji PS, Allen C, Sanchez PA, Yao J, Elphinstone JG, Jones JB et al. (2007). New diversity of Ralstonia solanacearum strains associated with vegetable and ornamental crops in Florida. Plant Dis 91: 195–203.
Kunin V, Goldovsky L, Darzentas N, Ouzounis CA . (2011). The net of life: reconstructing the microbial phylogenetic network. Genome Res 15: 954–959.
Lebeau A, Daunay M-C, Frary A, Palloix P, Wang J-F, Dintinger J et al. (2011). Bacterial wilt resistance in tomato, pepper, and eggplant: genetic resources respond to diverse strains in the Ralstonia solanacearum species complex. Phytopathology 101: 154–165.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Mahbou Somo Toukam G, Cellier G, Wicker E, Guilbaud C, Kahane R, Allen C et al. (2009). Broad diversity of Ralstonia solanacearum strains in Cameroon. Plant Dis 93: 1123–1130.
Maiden MC . (2006). Multilocus sequence typing of bacteria. Annu Rev Microbiol 60: 561–588.
Martin DP, Beiko RG . (2010). Genetic recombination and bacterial population structure. In: Ashley Robinson D, Falush D, Feil EJ (eds) Bacterial Population Genetics in Infectious Disease. John Wiley & Sons: Hoboken, NJ, pp 61–86.
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P . (2010). RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26: 2462–2463.
Maynard Smith J, Smith NH, O′Rourke M, Spratt BG . (1993). How clonal are bacteria ? Proc Natl Acad Sci USA 90: 4384–4388.
McDonald BA, Linde C . (2002). Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol 40: 349–379.
McVean G, Awadalla P, Fearnhead P . (2002). A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241.
Milling A, Meng FH, Denny TP, Allen C . (2009). Interactions with hosts at cool temperatures, not cold tolerance, explain the unique epidemiology of Ralstonia solanacearum race 3 biovar 2. Phytopathology 99: 1127–1134.
Moodley M, Linz B, Yamaoka Y, Windsor HM, Breurec S, Wu J-Y et al. (2009). The peopling of the Pacific from a bacterial perspective. Science 323: 527–530.
Nei M, Gojobori T . (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426.
Norman DJ, Zapata M, Gabriel DW, Duan YP, Yuen JMF, Mangravita-Novo A et al. (2009). Genetic diversity and host range variation of Ralstonia solanacearum strains entering North America. Phytopathology 99: 1070–1077.
Ochman H, Lawrence JG, Groisman EA . (2000). Lateral gene transfer and the nature of bacterial evolution. Nature 405: 299–305.
Pagel M, Meade A, Barker D . (2004). Bayesian estimation of ancestral character states on phylogenies. Syst Biol 53: 673–684.
Pérez-Losada M, Browne EB, Madsen A, Wirth T, Viscidi RP, Crandall KA . (2006). Population genetics of microbial pathogens estimated from multilocus sequence typing (MLST) data. Infect Genet Evol 6: 97–112.
Poussier S, Prior P, Luisetti J, Hayward AC, Fegan M . (2000a). Partial sequencing of the hrpB and endoglucanase genes confirms and expands the known diversity within the Ralstonia solanacearum species complex. Syst Appl Microbiol 23: 479–486.
Poussier S, Trigalet Demery D, Vandewalle P, Goffinet B, Luisetti J, Trigalet A . (2000b). Genetic diversity of Ralstonia solanacearum as assessed by PCR-RFLP of the hrp gene region, AFLP and 16S rRNA sequence analysis, and identification of an African subdivision. Microbiology 146: 1679–1692.
Prior P, Fegan M . (2005). Recent development in the phylogeny and classification of Ralstonia solanacearum. In: Momol T, Jones JB (eds) Proceedings of the First International Symposium on tomato diseases. ISHS-Acta Horticulturae, pp 127–136.
R Development Core Team. (2010). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
Remenant B, Coupat-Goutaland B, Guidot A, Cellier G, Wicker E, Allen C et al. (2010). Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics 11: 1–16.
Remenant B, de Cambiaire JC, Cellier G, Jacobs JM, Mangenot S, Barbe V et al. (2011). The genomes of the insect-transmitted plant pathogens Ralstonia syzygii and Blood Disease Bacterium reveal a recent evolutionary origin within the R. solanacearum species. PLoS One 6: e24356.
Schierup MH, Wiuf C . (2010). The coalescent of bacterial populations. In: Robinson DA, Falush D, Feil EJ (eds) Bacterial Population Genetics in Infectious Disease. John Wiley & Sons: Hoboken, NJ, pp 3–18.
Silva C, Vinuesa P, Eguiarte LE, Souza V, Martinez-Romero E . (2005). Evolutionary genetics and biogeographic structure of Rhizobium gallicum sensu lato, a widely distributed bacterial symbiont of diverse legumes. Mol Ecol 14: 4033–4050.
Vos M, Didelot X . (2008). A comparison of homologous recombination rates in bacteria and archaea. ISME J 3: 199–208.
Wicker E, Grassart L, Coranson-Beaudu R, Mian D, Guilbaud C, Fegan M et al. (2007). Ralstonia solanacearum strains from Martinique (French west indies) exhibiting a new pathogenic potential. Appl Environ Microbiol 73: 6790–6801.
Wilson DJ, Gabriel E, Leatherbarrow AJH, Cheesbrough J, Gee S, Bolton E et al. (2009). Rapid evolution and the importance of recombination to the gastroenteric pathogen Campylobacter jejuni. Mol Biol Evol 26: 385–397.
Yan S, Liu H, Mohr TJ, Jenrette J, Chiodini R, Zaccardelli M et al. (2008). Role of recombination in the evolution of the model plant pathogen Pseudomonas syringae pv. tomato DC3000, a very atypical tomato strain. Appl Environ Microbiol 74: 3171–3181.
Acknowledgements
We are grateful to Xavier Didelot for helpful discussions on the recombination estimators and to Mark Fegan and Olivier Pruvost for their critical comments on a previous version of this manuscript. This work was funded by the European Regional Development Fund (FEDER) of the European Union and the Conseil Régional de la Réunion, as part of the BIORISK and GENETOM programs; financial support was also provided by the Federation Nationale des Producteurs de Plants de Pomme de terre, Mission-DAR, Grant-7124 of the French Ministry of Food, Agriculture and Fisheries.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Wicker, E., Lefeuvre, P., de Cambiaire, JC. et al. Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J 6, 961–974 (2012). https://doi.org/10.1038/ismej.2011.160
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.160
Keywords
This article is cited by
-
Analysis of the speciation process suggests a dual lifestyle in the plant pathogen Ralstonia solanacearum species complex
European Journal of Plant Pathology (2023)
-
Identification and characterization of resistance quantitative trait loci against bacterial wilt caused by the Ralstonia solanacearum species complex in potato
Molecular Breeding (2022)
-
Identification of candidate type 3 effectors that determine host specificity associated with emerging Ralstonia pseudosolanacearum strains
European Journal of Plant Pathology (2022)
-
On the assessment of the sources of inoculum of bacterial wilt in Brazil
Tropical Plant Pathology (2022)
-
Phylotype and sequevar analysis of a Ralstonia pseudosolanacearum causing wilt in marigold (Tagetes erecta)
Journal of Plant Pathology (2022)
