
Abstract
Tibet is one of the most threatened regions by climate warming, thus understanding how its microbial communities function may be of high importance for predicting microbial responses to climate changes. Here, we report a study to profile soil microbial structural genes, which infers functional roles of microbial communities, along four sites/elevations of a Tibetan mountainous grassland, aiming to explore the potential microbial responses to climate changes via a strategy of space-for-time substitution. Using a microarray-based metagenomics tool named GeoChip 4.0, we showed that microbial communities were distinct for most but not all of the sites. Substantial variations were apparent in stress, N and C-cycling genes, but they were in line with the functional roles of these genes. Cold shock genes were more abundant at higher elevations. Also, gdh converting ammonium into urea was more abundant at higher elevations, whereas ureC converting urea into ammonium was less abundant, which was consistent with soil ammonium contents. Significant correlations were observed between N-cycling genes (ureC, gdh and amoA) and nitrous oxide flux, suggesting that they contributed to community metabolism. Lastly, we found by Canonical correspondence analysis, Mantel tests and the similarity tests that soil pH, temperature, NH4+–N and vegetation diversity accounted for the majority (81.4%) of microbial community variations, suggesting that these four attributes were major factors affecting soil microbial communities. On the basis of these observations, we predict that climate changes in the Tibetan grasslands are very likely to change soil microbial community functional structure, with particular impacts on microbial N-cycling genes and consequently microbe-mediated soil N dynamics.
Similar content being viewed by others


Introduction
As the roof of the world, the Tibet plateau is considered the third ‘pole’ (Qiu, 2008). Rich in glaciers and permafrost, the region has Earth’s third largest ice store after Antarctic and Arctic (Qiu, 2010). Recent decades have witnessed strong effects of climatic warming as this region has been experiencing three times the average global warming rate since 1960 (Li and Tang, 1988). It is predicted that the Tibetan alpine tundra could decrease to 20% of its current level with further climate warming (Walker et al., 2001). These observations and in silico modeling render Tibet as one of the most threatened regions by climate warming.
Tibetan ecosystems are particularly vulnerable to climate warming, which can partially be attributed to the pronounced role of climate in structuring alpine system (for example short growth season of plants) (Klein et al., 2004) and the snow-albedo or ice-albedo feedback that amplifies climate warming (Déry and Brown, 2007). The alpine grassland, accounting for roughly 35% of Tibet plateau, is a representative landscape of the Tibetan region (Cao et al., 2004). Studies have been carried out to analyze vegetation (Klein et al., 2004, 2007) and soil biogeochemical processes (Cao et al., 2001; Hastings et al., 2005; Wang et al., 2009b). However, soil microbial communities remain little understood. This knowledge gap calls for attention as it has been well documented that soil microbes drive a variety of biogeochemical processes such as carbon (C), nitrogen (N), phosphorus (P) and sulfur (S) cycling, and consequently provide critical ecosystem services such as soil fertility and greenhouse gas mitigation (Whitman et al., 1998; Green et al., 2008).
The recent, rapid development of high-throughput metagenomics technologies has enabled scientists to conduct detailed investigation of microbial communities. Although advancements have been achieved (Huber et al., 2007; Pointing et al., 2009; Dick and Tebo, 2010), most studies of this kind focus on microbial taxonomy. However, it is crucial to examine microbial functional signatures such as structural genes relevant to metabolic pathways, energetics and regulatory circuits, which directly address microbial functional potentials related to ecosystem processes. To fulfill this need, a microarray-based metagenomics tool named GeoChip is an excellent choice and has been widely adopted to examine microbial gene diversity in various environments (Van Nostrand et al., 2009; Waldron et al., 2009; Wang et al., 2009a; He et al., 2010; Lu et al., 2012). The most advanced version of GeoChip, GeoChip 4.0 can target a wide array of 152 414 microbial genes belonging to 410 gene families.
In this study, we used GeoChip 4.0 to survey microbial gene diversity along the elevation/temperature gradient of a mountainous grassland of the Tibet plateau. The observational investigations along an elevation/temperature gradient reveal potential microbial responses to climate changes, using a strategy of space-for-time substitution (Dunne et al., 2004). Although there have been numerous studies on profiling microbial distribution at the phylogenetic level (Bryant et al., 2008; Fierer et al., 2011; Wang et al., 2012), there has been no such study at the functional gene level. Here we define microbial genes present in all of the elevations as ubiquitous genes and those present at only one elevation as endemic genes. We are interested in testing the following hypotheses: (i) the functional structure of microbial community differs among elevations, given high soil heterogeneity and variations of vegetation and climatic conditions, and soil and vegetation attributes contribute significantly in shaping the functional structure; (ii) variations of microbial stress genes are consistent with the increasing alpine stress at higher elevations; (iii) variations of C- or N-cycling genes are significantly linked to soil C and N dynamics in the alpine grassland; and (iv) the pattern of endemic gene abundances not only differs from that of ubiquitous gene abundances but also differs among elevations. The study is based on field investigations of four fenced sites/elevations in a mountainous grassland located at the Haibei Alpine Meadow Ecosystem Research Station of Qinghai province, which has typical climate and vegetation of Tibetan alpine grasslands (Wang et al., 2009b, 2010a, 2010b).
Materials and methods
Site and sampling
Sampling was carried out at the Haibei Alpine Meadow Ecosystem Research Station in Qinghai province, located in the northeast of Qinghai-Tibet Plateau (37°37′N, 101°12E’) in a valley surrounded by the Qilian Mountains. The local climate is highland continental, characterized to be cold and long in winter but warm and short in summer. The annual mean air temperature and precipitation are −1.7 °C and 560 mm, respectively (Zhao et al., 2006). Soil type at the research sites is dominated by Mat Cryic Cambisols typical of alpine grassland soil, with pH value of 7.3 and 7.4 at depths of 10 cm and 20 cm, respectively. The growing season of vegetation is from May to September, during which more than 80% of the precipitation occurs. Vegetation reaches its maximal aboveground biomass level in late July and early August and starts withering in early October. Approximately 87% of vegetation species at the sites use the C3 photosynthetic pathway (Zhao et al., 2006).
The study sites, as part of a comprehensive project to examine the effects of artificial warming and/or livestock grazing on Tibetan grasslands, were initiated at four sites/elevations of 3200, 3400, 3600 and 3800 m in May 2006. The spatial distances between adjacent elevations are 6.2 km (3200–3400 m), 4.2 km (3400–3600 m) and 1.3 km (3600–3800 m), respectively. These sites were selected for their representative vegetation and soil attributes for the elevations. The plant community at 3200 m is dominated by Kobresia humilis, Elymus nutans, Stipa aliena, Potentilla anserine and Thalictrum alpinum. The plant community at 3400 m consists primarily of a species of hardy deciduous flowering shrub (Potentilla fruticosa) with 50–60% total coverage and beneath grass species of Kobresia humilis, Elymus nulan and Festuca ovina with 80% total coverage. The plant community at 3600 m site is dominated by Kobresia humilis, Potentilla nivea, Thalictrum alpinum, Carex atrofusca, Poa crymophila and Potentilla fruticosa. The plant community at 3800 site is dominated by Kobresia humilis, Poa crymophila, Androsace mariae, Polygonum macrophyllum and Kobresia pygmaea. Owing to the alpine environment, the plant communities are characterized by short growth period, low primary production and diversity.
At each elevation, three plots comprising typical vegetation and soil attributes were fenced to minimize disturbance. The dimensions of the plots were 1.0 m, 1.0 m and 0.3 m in length, width and height, respectively. The distance between adjacent plots was roughly 0.6 meter. In August 2009, soil samples were collected from all of the plots. Five soil cores at a depth of 0–20 cm and diameter of 1.5 cm were randomly taken from each plot and mixed thoroughly to generate a soil sample representing the plot. Soil samples were kept on ice when transporting to laboratory and sieved with 2 mm mesh to remove roots and stones. Soil samples were preserved at −80 °C before DNA extraction.
DNA extraction, purification and quantitation
Soil genomic DNA was extracted using a FastDNA spin kit for soil (MP Biomedical, Carlsbad, CA, USA) following the manufacturer’s instructions. To purify it, the DNA extract was mixed with 2.5 volume of 100% ice-cold ethanol and 0.1 volume of 3 M NaOAc (pH 5.2) before overnight incubation at −20 °C. DNA was precipitated by centrifugation for 30 min at 13 000 g. Then supernatant was decanted and washed with 1 ml of 70% ethanol. DNA was air-dried and dissolved in 50 μl of nuclease-free water. DNA quality and quantity were measured using NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA) and with PicoGreen (Ahn et al., 1996) using a FLUOstar Optima (BMG Labtech, Jena, Germany), respectively.
GeoChip 4.0 experiments
As previously described (Yang et al., 2013), DNA samples were labeled with the fluorescent dye Cy-5 using a random priming method and purified using the QIA quick purification kit (Qiagen, Valencia, CA, USA). Then DNA was dried in a SpeedVac (ThermoSavant, Milford, MA, USA) at 45 °C for 45 min. The hybridization was carried out at 42 °C for 16 h on a MAUI hybridization station (BioMicro, Salt Lake City, UT, USA). After purification, GeoChip microarrays were scanned by a NimbleGen MS200 scanner (Roche, Madison, WI, USA) at 633 nm using a laser power and photomultiplier tube gain of 100% and 75%, respectively.
Data analyses
Signal intensities were quantified and processed using the data analysis pipeline as previously described (Yang et al., 2009; He et al., 2010; Yang et al., 2013). Then processed GeoChip data were analyzed using the following steps: (i) remove the poor quality spots, which were flagged as 1 or 3 by ImaGene (Arrayit, Sunnyvale, CA, USA) or with a signal to noise ratio of less than 2.0; (ii) normalize the signal intensity of each spot by dividing the signal intensity by the total intensity of the microarray followed by multiplying by a constant; (iii) transform the data to the natural logarithmic form; and (iv) remove genes detected in only one out of three samples from the same elevation.
Principal component analysis was used to determine overall functional changes in the microbial communities. Bray-Curtis distance was used to obtain dissimilarity matrices in the adonis algorithm of the dissimilarity test for comparing GeoChip data of four elevations. All the analyses were performed using functions in the Vegan package (v.1.15-1) in R v. 2.8.1 (RDC Team, 2006).
The similarity test, Mantel test, Canonical correspondence analysis (CCA) and Variation partitioning analysis were used to evaluate the linkages between microbial gene compositions and environmental attributes. In the similarity test, Euclidean distance was used to calculate the distance between samples, followed by calculation of Pearson correlation coefficient. To select attributes in CCA modeling, we used variation inflation factors to examine whether the variance of canonical coefficients was inflated by the presence of correlations with other attributes. If an attribute had a variation inflation factor value larger than 20, we deemed it to be affected by other attributes and consequently removed it from the CCA modeling. The Variation partitioning analysis analysis resulted in five soil attributes (soil pH, moisture, NH4+–N, NO3−–N and soil temperature at the depth of 5 cm) and two vegetation attributes (Shannon diversity and total coverage of vegetation). As temperature was measured per site, one way analysis of variance was not conducted. All the analyses were performed using functions in the Vegan package (v.1.15-1) in R v. 2.8.1 (RDC Team, 2006).
Soil and vegetation attribute measurements
At the time of soil sampling, soil temperature was measured by type-K thermocouples (Campbell Scientific, Logan, UT, USA) coupled to a CR1000 datalogger at depths of 5, 10 and 20 cm. Soil moisture at depths of 5 and 10 cm were recorded by time domain reflectometry (Model Diviner-2000, Sentek Pty Ltd., Stepney, SA, Australia).
In the lab, total soil organic C and N were quantified by a TOC-5000 A analyzer (Shimadzu Corp., Kyoto, Japan) and a Vario EL III Elemental Analyzer (Elementar, Hanau, Germany), respectively. To measure NH4+–N and NO3−–N, 10 g of fresh soil was suspended in 50 ml of 2 mol l−1 KCl solution, then shaken at room temperature for 1 hour and placed for 30 min under unstirred conditions. Subsequently, clear supernatant was passed through a filter paper of 30–50 μm pore size and analyzed using continuous flowing SAN++ Analyzer (SKALAR, Breda, the Netherlands).
The greenhouse gases (CO2, N2O and CH4) were measured by chamber per plot setting at the 5 cm depth of soil as previously described (Wang et al., 2009b). During sampling, chambers were closed for half an hour. Hundred milliliters of air samples were taken by plastic syringes every 10 min and transported to the lab. Greenhouse gas concentrations were measured by gas chromatograph (Agilent 4890D, Agilent Co., Santa Clara, CA, USA) equipped with a flame ionization detector and an electron capture detector as previously described (Yuesi and Yinghong, 2003). As there were many rat holes in the soil at the elevation of 3600 m, the measurement of the greenhouse gas fluxes data (CO2, CH4 and N2O) was unreliable. Therefore, we excluded those points in the correlation analysis with the greenhouse gas fluxes.
One quadrant of 1 × 1 m was selected at each plot to measure vegetation attributes. The vegetation attribute indices included vegetation species number, diversity, total coverage and aboveground biomass, which were all recorded according to established protocols (Klein et al., 2007). Vegetation was weighted instantly for aboveground biomass data after mowing. Vegetation diversity was calculated by Shannon index.
Results
Distribution patterns of soil microbial communities along the elevation gradient
A total of 49 520 genes were detected by GeoChip 4.0. Detected genes were the fewest at the 3200 m site (23 369 genes) and roughly doubled at the 3400 m site (45 685 genes). Slightly fewer genes were detected at higher elevations than those at the 3400 m site (Supplementary Table S1). Alpha-diversities, as shown by Shannon and Simpson indices, varied concurrently with gene numbers. Alpha-diversities differed significantly (P<0.05) among 3200, 3400 and 3600 m sites, which was supported by the dissimilarity test using adonis algorithm (Supplementary Table S2). However, microbial communities at 3600 and 3800 m sites appeared to be similar by the statistical test (P>0.05). Consistently, principal component analysis with the whole GeoChip data set showed that samples of 3200 and 3400 m sites were well separated from the others, but samples of 3600 and 3800 m sites were similar (Figure 1).

Principle component analysis (PCA) of GeoChip data. The values of Axis 1 and 2 are percentages of total variations that can be attributed to the corresponding axis.
Gene overlap between elevations was also calculated. Higher percentages of shared genes were observed among 3400, 3600 and 3800 m sites (81.1–83.8%) (Supplementary Table S3), whereas only 50.3–53.4% of the genes were shared between 3200 m and other sites. The highest percentages of shared genes (82.7–92.3%) were observed for the plots within the same elevation (data not shown), suggesting that the within-elevation variation was small.
Selected gene categories
Genes involved in stress response, N and C cycling were examined below. Only selected genes were presented or described.
Stress response
Considerable variability of stress genes was observed (Figure 2). Cold shock genes, CspA, cspB, desK and desR genes, which are cold shock genes required for adaption to cold environment (Ermolenko and Makhatadze, 2002), were more abundant at higher elevations. In contrast, heat shock genes (dnaK, groEL, groES, grpE and hrcA) remained unchanged.

Total gene abundances, normalized to the abundance at 3200 m, of stress gene categories at each elevation. All data are presented as mean±s.e. calculated from biological triplicates. Significant (P<0.05) differences among elevations are indicated by alphabetic letters above the bars.
N cycling
N-cycling genes differed substantially among the four elevations (Figure 3). For example, total abundances of ureC and gdh genes involved in ammonification, napA genes involved in dissimilatory N reduction and nosZ genes involved in denitrification exhibited distinct differences for each site, whereas no significant difference was observed for the abundances of nifH and nrfA genes. Notably, changes of ureC and gdh abundances were largely reverse, which was consistent with their opposite functional role in N mineralization. nirS and nosZ genes involved in denitrification were more abundant at higher elevations but amoA gene involved in nitrification was less abundant. Consistently, soil NO3−–N and NH4+–N contents were increased and decreased at higher elevations, respectively (Supplementary Table S4).

Total gene abundances, normalized to the abundance at 3200 m, of N genes at each elevation. All data are presented as mean±s.e. calculated from biological triplicates. Significant (P<0.05) differences among elevations are indicated by alphabetic letters above the bars.
Grassland nitrous oxide (N2O) emission is a microbe-mediated process. Thus, we explored whether N-cycling genes were correlated with N2O flux. The results showed that total abundances of ureC and amoA were positively correlated (P<0.01) with N2O flux, whereas those of gdh (P<0.01), nosZ (P<0.01) and nirS (P<0.1) were negatively correlated (Figure 4).

Correlations between N2O flux and the abundance of N-cycling genes.
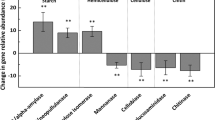
C cycling
Variability was apparent for C cycling genes (Supplementary Figure S1). For C-fixation genes, the abundance of rubisco (ribulose-1, 5-bisphosphate carboxylase/oxygenase) was the lowest at 3200 m, but no other C-fixation genes exhibited similar patterns. For C degradation (starch, hemicellulose, cellulose, aromatics, chitin and lignin degradation), isopullulanase and exoglucanase were more abundant at higher elevations, whereas exochitinase and glx were less abundant. For methane (CH4) cycling, our results indicated that methane-producing gene mcrA and methane-oxidizing gene mmoX were more abundant at higher elevations, whereas methane-oxidizing gene pmoA remained unchanged. Although these results might suggest that functional potentials of CH4 cycling were enhanced, no correlation was observed between CH4 flux and abundances of mmoX (r=−0.05, P>0.11), mcrA (r=−0.10, P>0.25) and pmoA (r=−0.43, P>0.84) genes. Similarly, there was no correlation between CO2 flux and abundances of C-degradation genes (r=0.29, P>0.36).
Ubiquitous and endemic genes
A total of 21842 microbial genes were present in all of the four sites, whereas 5962 genes were present at only one site. That is, ubiquitous and endemic genes accounted for 44.1% and 12.0% of total detected genes, respectively (Supplementary Figure S2). Ubiquitous genes were predominantly derived from the phyla of Proteobacteria, Actinobacteria and Firmicute, which was consistent with observations in a wide range of soil types that these bacteria were prevalent in soil (Roesch et al., 2007; Monroy et al., 2012; Yergeau et al., 2012).
The average abundance of endemic genes was roughly 33% of that of ubiquitous genes, suggesting that endemic genes tended to be rare genes that were typically low in abundance. Several gene categories enriched in the endemic genes (Figure 5a). Among them, there were a total number of 1718 endemic stress genes, including 151 σ24 genes responsive to extreme temperature stress, 122 glnA genes encoding a glutamine synthetase required for N-limiting conditions (Merrick and Edwards, 1995) and 162 fnr genes encoding a transcriptional activator required for oxygen-limiting conditions (Spiro and Guest, 1987).

Comparison of percentage changes of (a) each gene category of endemic genes to that of ubiquitous genes and (b) microbial domains or phyla of endemic genes to those of ubiquitous genes.
It was also noted that endemic genes enriched genes derived from viruses, archaea and fungi (Figure 5b). Major endemic viruses included Enterobacteria phages and Bacillus phages. Major endemic archaea included Halobacteriaceae, Methanosarcinaceae and Sulfolobaceae, and major endemic fungi included Aspergillus niger and Gibberella zeae.
The numbers of endemic genes differed substantially among the four elevations, with the highest at the 3400 m site and the lowest at the 3200 m site (Supplementary Table S5). At higher elevations, stress, N cycling and S cycling genes occupied larger portions of endemic genes as compared with the 3200 m site, suggesting that they were needed to cope with the environments at higher elevations (Supplementary Figure S3). In contrast, C cycling, metal resistance and bacterial phage occupied smaller portions. Endemic stress genes included chaperone dnaK, groEL, hsp10/groES and grpE, response regulators desR and hrcA of temperature stress, heme catalase katE, response regulator oxyR of oxygen stress, proline transporter proU of osmotic stress and GTPase obgE of radiation stress involved in final steps of the 50S ribosomal subunit assembly and glnA and glnR genes responsive to N stress.
The tight linkage between the microbial gene composition and environmental attributes
All the environmental attributes were generated by soil geochemical measurements, and four vegetation attributes including total coverage of vegetation, vegetation species number, vegetation diversity were calculated with the Shannon index and vegetation biomass (Supplementary Table S4). To explore the linkages between the microbial gene composition and environmental attributes, the similarities between microbial gene composition and aboveground vegetation and soil attributes were examined, which in turn showed very strong and significant correlations (P<0.01) (Figure 6). To verify it, Mantel tests (Table 1) were conducted with the whole data set of vegetation and soil attributes, showing that the microbial community was significantly (P<0.01) correlated with vegetation and soil attributes with r values of 0.75 and 0.67, respectively.

The correlations between (a) microbial community and vegetation and (b) microbial community and soil attributes. Euclid distances among samples were calculated and plotted to generate Pearson correlation values.
CCA was used to identify the major environmental attributes controlling soil microbial gene composition. The results of CCA, a significant model at the confidence level of P<0.01, indicated that soil pH, temperature, NH4+–N and vegetation diversity were important environmental attributes controlling the microbial community structure as they were significantly correlated with Axis 1 (P<0.01), which represented the major variations among microbial communities (Figure 7a). The importance of these environmental attributes was verified by Mantel tests on the basis of their significant correlation (P<0.01) with GeoChip data (Table 1).

(a) CCA of GeoChip data and geochemistry attributes: soil pH, NO3−–N, NH4+–N, the temperature of 5 cm depth in soil (T5), the total coverage of vegetation (TCOV), the alpha diversity of vegetation (diversity), soil moisture (moisture). The percentage of variation explained by each axis is shown and the relationship is significant (P<0.05). (b) Partitioning of microbial diversity variance among important geochemical attributes, soil chemistry including soil pH, moisture, ammonia and nitrate (S), soil climate attributes including soil temperature and soil moisture of 5 cm depth (C), vegetation including total coverage and diversity (V), and their interactions (S × V, S × C and V × C). The values in parentheses are P-values.
To partition the contributions of environmental attributes, Variation partitioning analysis was carried out. The results showed that a small set of seven environmental attributes accounted for 81.4% of microbial community variations of GeoChip data (Figure 7b), indicating that they were major factors shaping microbial gene composition. Soil climatic, geochemical and aboveground vegetation attributes appeared to impose strong effects on microbial community. In addition, interaction between vegetation and soil geochemical attributes was substantial (13.2%).
Discussion
Our first hypothesis, namely that the functional structure of a microbial community differs across elevations and that soil and vegetation attributes contribute significantly to shaping the functional structure, was verified in part, as significant differences were detected except for 3600 and 3800 m samples. The number of detected microbial genes at the 3200 m site was roughly half of those at the other sites, suggesting that its microbial gene diversity was substantially different. This observation can be explained by soil heterogeneity linking to geographic distance, as the distance between 3600 and 3800 m sites is the shortest (1.3 km), and the 3200 m site is distant from the other sites (6.2–11.5 km). Notably, soil pH might have an adverse effect on microbial gene diversity of the 3200 m site, as it was the highest (pH of 7.9). Alternatively, it might be caused by aboveground vegetation as the vegetation diversity was the lowest at this site. Vegetation affects soil microbial community through a number of mechanisms (Wardle et al., 2004). As producer, it supplies organic C and other resources to soil microbial community, which comprises most of the decomposer system. The quantity and quality of resources depend on vegetation species, thus individual vegetation species can influence the composition and function of soil microbial communities. However, effects of the vegetation diversity and productivity on soil biodiversity depend on context and vary substantially between locations (Wardle, 2002). In our case, productivity appeared not to be the determinant of microbial gene diversity. Rather, the results of CCA (Figure 7a) and Mantel tests (Table 1) demonstrated that the vegetation diversity had an important role in controlling microbial community structure of the Tibetan grassland. Also, we found that the vegetation diversity appeared to be strongly correlated (r=0.58, P<0.04) with the microbial gene diversity (Supplementary Figure S4), which provided further support.
Soil attributes of pH, temperature and NH4+–N were crucial for shaping microbial community structure, as shown by CCA (Figure 7a) and Mantel tests (Table 1). Consistently, soil pH and temperature were key drivers in shaping bacterial communities along elevation gradients (Wang et al., 2012; Shen et al., 2013). It has also been observed that the productivity of alpine grassland was mainly controlled by the available N pool (Baumann et al., 2009). Generally, microbial abundance and composition depended on environmental attributes of soil nutrient availability (Lozupone and Knight, 2007; Jesus et al., 2009). These previous studies demonstrated that the soil pH, temperature and N were important for shaping phylogenetic compositions of microbial communities, whereas our results supported it at the level of microbial functional genes.
Notably, the two axes of CCA together (Figure 7a) explained 64.6% of community variance, which was more substantial than most other GeoChip studies (Liang et al., 2011; Lu et al., 2012). It has recently been documented that harsh environments could reduce microbial community variation and consequently correlate microbial community with environmental attributes well (Pointing et al., 2009; Yergeau et al., 2012). Consistently, our recent experiments to examine effects of anthropogenic perturbation on this site indicated that microbial community structure was fragile (Yang et al., 2013). However, it should be noted that a limitation in our study exists in the use of a small area as a proxy to assess the Tibetan alpine grassland. Although, we take comfort in the fact that the study site, located at Haibei Alpine Meadow Ecosystem Research Station, has a representative landscape of a large portion of Tibetan grasslands (Wang et al., 2009b, 2010b), there will be exceptions in other alpine grasslands, given the typically high soil heterogeneity.
Our second hypothesis, namely that variations of microbial stress genes are consistent with the increasing alpine stress at higher elevations, was verified by our observation that microbial stress genes responsible for coping with a cold environment were more abundant at higher elevations (Figure 2), which was apparently a response to low temperature. However, our third hypothesis, namely that variations of C- or N-cycling genes are significantly linked to soil C and N dynamics in the alpine grassland, was only partially supported by the correlation between several N-cycling genes and N2O flux (Figure 4), but no correlation was detected for CO2 and CH4 fluxes. An explanation was that the C/N ratio of plants was higher than that of microbes, rendering microbial communities sensitive to N as a limiting factor. For N-cycling genes, ureC converted urea into the substrate of N2O flux (NH4+–N), whereas gdh reversed the reaction. Accordingly, ureC and gdh were positively and negatively correlated to N2O flux, respectively. Interestingly, a positive correlation was observed for amoA gene involved in nitrification, whereas negative correlations were observed for nirS and nosZ involved in denitrification. This finding might implicate that nitrification, but not denitrification, was the major process of N2O emission of the alpine grassland. As a consequence, the change of N2O emission negatively affected soil NO3−–N content, which in turn affected the abundance of denitrification genes nirS and nosZ. In support of this view, previous studies indicated that nitrification was the dominant mechanism influencing grassland N2O emission (Ryden, 1981; Klemedtsson et al., 1988). Meanwhile, it is not surprising to detect no correlation between C-cycling genes and CO2 flux because the latter includes soil autotrophic respiration, making it unlikely to correlate CO2 flux with soil microbial genes. However, it remains unclear why no correlation was detected for CH4 flux.
Our findings supported our fourth hypothesis that the pattern of endemic gene abundances not only differs from that of ubiquitous gene abundances but also differs among elevations. Distribution patterns vary by microbial functional or taxonomic groups (Horner-Devine et al., 2004; Zhou et al., 2008). Thus, we examined endemic genes to identify those with relatively high habitat specificity, which reflected sensitivity to environmental variations. Interestingly, endemic genes were usually low in abundance, raising the possibility that they are part of a ‘rare biosphere’. Meanwhile, bearing in mind a caveat that the identification of endemic genes is subjected to the detection level of GeoChip, we noted the enrichment of several microbial groups. Enrichment of stress genes (Figure 5a) in the endemic group implicates their important roles in coping with different alpine environments characterized by low temperature, oxygen and nutrient levels and high UV irradiation. For phylogenetic groups, the enrichment of bacteriophage was not surprising, as the appearance of new bacteria induced new bacteriophage species. For fungi, the narrow dispersal has been reported (Herlemann et al., 2011; Yergeau et al., 2012), which is attributed to its relatively large size compared with bacteria. However, size could not explain the enrichment of archaea, which are of comparable size to bacteria. Notably, there were reports that archaea were sensitive to certain environments such as N-limited or dry environment (Rothrock and Garcia-Pichel, 2005; Pointing et al., 2009).Our results showed that many endemic genes differed among elevations, which reflected habitat specificity. It was noted that vegetation biomass was the highest for 3200 m (Supplementary Table S4), which might require additional C-cycling genes to utilize C deposited to soil. Meanwhile, stress of temperature, oxygen and nutrient differs by elevations, which induced different stress genes.
Global climate warming effects are most pronounced in the polar and alpine regions, where ecosystems are fragile (Li and Tang, 1988; Hansen, 2000). It is thus necessary to document the ecosystem, particularly the poorly understood microbial communities, because they may encounter catastrophic shifts, should warming continue to occur. Here we report the profiling of microbial distribution at the gene level, which provides valuable insights for potential effects of climate changes on this alpine ecosystem. On the basis of the strategy of space-for-time substitution, we predict that the warming will upshift vegetation and microbial communities along the elevation, thus affecting microbial community structures as well as the particularly sensitive N cycling in the Tibetan grasslands.
References
Ahn SJ, Costa J, Emanuel JR . (1996). PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res 24: 2623–2625.
Baumann F, JINS HE, Schmidt K, KÜHN P, Scholten T . (2009). Pedogenesis, permafrost, and soil moisture as controlling factors for soil nitrogen and carbon contents across the Tibetan Plateau. Glob Change Biol 15: 3001–3017.
Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL . (2008). Colloquium paper: microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc Natl Acad Sci USA 105 (Suppl 1): 11505–11511.
Cao G, Li Y, Zhang J . (2001). Effect of soil circumstances biogeochemical factors on carbon dioxide emission from mollic-cryic cambisols. Acta Agrestia Sinica 4: 307–312.
Cao G, Tang Y, Mo W, Wang Y, Li Y, Zhao X . (2004). Grazing intensity alters soil respiration in an alpine meadow on the Tibetan plateau. Soil Biol Biochem 36: 237–243.
Déry SJ, Brown RD . (2007). Recent Northern Hemisphere snow cover extent trends and implications for the snow-albedo feedback. Geophys Res Lett 34: L22504.
Dick GJ, Tebo BM . (2010). Microbial diversity and biogeochemistry of the Guaymas Basin deep-sea hydrothermal plume. Environ Microbiol 12: 1334–1347.
Dunne JA, Saleska SR, Fischer ML, Harte J . (2004). Integrating experimental and gradient methods in ecological climate change research. Ecology 85: 904–916.
Ermolenko D, Makhatadze G . (2002). Bacterial cold-shock proteins. Cell Mol Life Sci CMLS 59: 1902–1913.
Fierer N, McCain CM, Meir P, Zimmermann M, Rapp JM, Silman MR et al (2011). Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92: 797–804.
Green JL, Bohannan BJ, Whitaker RJ . (2008). Microbial biogeography: from taxonomy to traits. Science 320: 1039–1043.
Hansen J . (2000). GISS analysis of surface temperature change (vol 104, pg 30,997, 1999). J Geophys Res-Atmos 105: 12517–12517.
Hastings SJ, Oechel WC, Muhlia-Melo A . (2005). Diurnal, seasonal and annual variation in the net ecosystem CO2 exchange of a desert shrub community (Sarcocaulescent) in Baja California, Mexico. Glob Change Bio 11: 927–939.
He ZL, Deng Y, Van Nostrand JD, Tu QC, Xu MY, Hemme CL et al (2010). GeoChip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME J 4: 1167–1179.
Herlemann DP, Labrenz M, Jurgens K, Bertilsson S, Waniek JJ, Andersson AF . (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5: 1571–1579.
Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM . (2004). A taxa–area relationship for bacteria. Nature 432: 750–753.
Huber JA, Welch DBM, Morrison HG, Huse SM, Neal PR, Butterfield DA et al (2007). Microbial population structures in the deep marine biosphere. Science 318: 97–100.
Jesus ED, Marsh TL, Tiedje JM, Moreira FMD . (2009). Changes in land use alter the structure of bacterial communities in Western Amazon soils. ISME J 3: 1222.
Klein JA, Harte J, Zhao XQ . (2004). Experimental warming causes large and rapid species loss, dampened by simulated grazing, on the Tibetan Plateau. Ecol Lett 7: 1170–1179.
Klein JA, Harte J, Zhao XQ . (2007). Experimental warming, not grazing, decreases rangeland quality on the Tibetan Plateau. Ecol Appl 17: 541–557.
Klemedtsson L, Svensson B, Rosswall T . (1988). Relationships between soil moisture content and nitrous oxide production during nitrification and denitrification. Biol Fertility Soils 6: 106–111.
Li CQ, Tang MC . (1988). The climate change of Qinghai-xizang plateau and its neighborhood in the recent 30 years. Plateau Meteorol 1: 332–341.
Liang Y, Van Nostrand JD, Deng Y, He Z, Wu L, Zhang X et al (2011). Functional gene diversity of soil microbial communities from five oil-contaminated fields in China. The ISME J 5: 403–413.
Lozupone CA, Knight R . (2007). Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104: 11436–11440.
Lu Z, Deng Y, Van Nostrand JD, He Z, Voordeckers J, Zhou A et al (2012). Microbial gene functions enriched in the Deepwater Horizon deep-sea oil plume. ISME J 6: 451–460.
Merrick M, Edwards R . (1995). Nitrogen control in bacteria. Microbiol Rev 59: 604–622.
Monroy F, van der Putten WH, Yergeau E, Mortimer SR, Duyts H, Bezemer TM . (2012). Community patterns of soil bacteria and nematodes in relation to geographic distance. Soil Biol Biochem 45: 1–7.
Pointing SB, Chan Y, Lacap DC, Lau MC, Jurgens JA, Farrell RL . (2009). Highly specialized microbial diversity in hyper-arid polar desert. Proc Natl Acad Sci USA 106: 19964–19969.
Qiu J . (2008). China: The third pole. Nature 454: 393–396.
Qiu J . (2010). Measuring the meltdown. Nature 468: 141–142.
RDC Team . (2006) R: A language and environment for statistical computing In: Computing RFfS (ed) Vienna, Austria.
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD et al (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290.
Rothrock Jr MJ, Garcia-Pichel F . (2005). Microbial diversity of benthic mats along a tidal desiccation gradient. Environ Microbiol 7: 593–601.
Ryden J . (1981). N2O exchange between a grassland soil and the atmosphere.
Shen C, Xiong J, Zhang H, Feng Y, Lin X, Li X et al (2013). Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol Biochem 57: 204–211.
Spiro S, Guest JR . (1987). Regulation and over-expression of the fnr gene of Escherichia coli. J Gen Microbiol 133: 3279–3288.
Van Nostrand JD, Wu WM, Wu LY, Deng Y, Carley J, Carroll S et al (2009). GeoChip-based analysis of functional microbial communities during the reoxidation of a bioreduced uranium-contaminated aquifer. Environ Microbiol 11: 2611–2626.
Waldron PJ, Wu LY, Van Nostrand JD, Schadt CW, He ZL, Watson DB et al (2009). Functional gene array-based analysis of microbial community structure in groundwaters with a gradient of contaminant levels. Environ Sci Technol 43: 3529–3534.
Walker MD, Gould WA, Chapin FS . (2001). Scenarios of biodiversity changes in arctic and alpine tundra. Global Biodiversity in a Changing Environment 152: 83–100.
Wang FP, Zhou HY, Meng J, Peng XT, Jiang LJ, Sun P et al (2009a). GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent. Proc Natl Acad Sci USA 106: 4840–4845.
Wang J, Soininen J, He J, Shen J . (2012). Phylogenetic clustering increases with elevation for microbes. Environ Microbiol Rep 4: 217–226.
Wang SP, Lin XW, Ma XZ, Xu GP, Luo CY, Li YN et al (2009b). Fluxes of CO(2), CH(4), and N(2)O in an alpine meadow affected by yak excreta on the Qinghai-Tibetan plateau during summer grazing periods. Soil Biol Biochem 41: 718–725.
Wang SP, Hu YG, Chang XF, Lin XW, Wang YF, Duan JC et al (2010a). Effects of warming and grazing on N(2)O fluxes in an alpine meadow ecosystem on the Tibetan plateau. Soil Biol Biochem 42: 944–952.
Wang SP, Luo CY, Xu GP, Chao ZG, Lin XW, Hu YG et al (2010b). Effect of warming and grazing on litter mass loss and temperature sensitivity of litter and dung mass loss on the Tibetan plateau. Glob Change Biol 16: 1606–1617.
Wardle DA . (2002) Communities and Ecosystems: Linking the Aboveground and Belowground Components. Princeton University. Press: Princeton, NJ, USA.
Wardle DA, Bardgett RD, Klironomos JN, Setala H, van der Putten WH, Wall DH . (2004). Ecological linkages between aboveground and belowground biota. Science 304: 1629–1633.
Whitman WB, Coleman DC, Wiebe WJ . (1998). Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95: 6578–6583.
Yang Y, Harris DP, Luo F, Xiong W, Joachimiak M, Wu L et al (2009). Snapshot of iron response in Shewanella oneidensis by gene network reconstruction. BMC Genomics 10: 131.
Yang Y, Wu L, Lin Q, Yuan M, Xu D, Yu H et al (2013). Responses of the functional structure of soil microbial community to livestock grazing in the Tibetan alpine grassland. Glob Change Biology 19: 637–648.
Yergeau E, Bokhorst S, Kang S, Zhou J, Greer CW, Aerts R et al (2012). Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J 6: 692–702.
Yuesi W, Yinghong W . (2003). Quick measurement of CH 4, CO 2 and N 2 O emissions from a short-plant ecosystem. Adv Atmos Sci 20: 842–844.
Zhao L, Li Y, Xu S, Zhou H, Gu S, Yu G et al (2006). Diurnal, seasonal and annual variation in net ecosystem CO2 exchange of an alpine shrubland on Qinghai-Tibetan plateau. Glob Change Biol 12: 1940–1953.
Zhou J, Kang S, Schadt CW, Garten CT . (2008). Spatial scaling of functional gene diversity across various microbial taxa. Proc Natl Acad Sci 105: 7768.
Acknowledgements
We thank Haibei Research Station staff for sampling assistance, Madelyn Ball for manuscript editing and the anonymous reviewers and the editor for constructive comments and suggestion to make this manuscript greatly improved. This research was supported by grants to Yunfeng Yang from National Science Foundation of China (41171201) and National High Technology Research and Development Program of China (2012AA061401). To Shiping Wang from the National Basic Research Program (2010CB833502), to Jizhong Zhou from the United States Department of Energy, Biological Systems Research on the Role of Microbial Communities in C Cycling Program (DE-SC0004601) and Oklahoma Bioenergy Center (OBC). The GeoChips and associated computational pipelines used in this study were supported by ENIGMA-Ecosystems and Networks Integrated with Genes and Molecular Assemblies through the Office of Science, Office of Biological and Environmental Research, of the US Department of Energy under Contract No. DE-AC02–05CH11231 and by the United States Department of Agriculture (Project 2007-35319-18305) through NSF-USDA Microbial Observatories Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Yang, Y., Gao, Y., Wang, S. et al. The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J 8, 430–440 (2014). https://doi.org/10.1038/ismej.2013.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.146
Keywords
This article is cited by
-
Soil bacterial community composition and function play roles in soil carbon balance in alpine timberline ecosystems
Journal of Soils and Sediments (2024)
-
Microbial Catabolic Activity: Methods, Pertinence, and Potential Interest for Improving Microbial Inoculant Efficiency
Microbial Ecology (2023)
-
Effect of tillage practices on soil CO2 emissions, microbial C-fixation, and C-degradation functional gene abundance in Northeast China
Journal of Soils and Sediments (2023)
-
Soil pH and dissolved organic carbon shape microbial communities in wetlands with two different vegetation types in Changdu area, Tibet
Journal of Mountain Science (2023)
-
Soil microbial community diversity and distribution characteristics under three vegetation types in the Qilian Mountains, China
Journal of Arid Land (2023)
