
Abstract
Diverse microbial consortia profoundly influence animal biology, necessitating an understanding of microbiome variation in studies of animal adaptation. Yet, little is known about such variability among fish, in spite of their importance in aquatic ecosystems. The Trinidadian guppy, Poecilia reticulata, is an intriguing candidate to test microbiome-related hypotheses on the drivers and consequences of animal adaptation, given the recent parallel origins of a similar ecotype across streams. To assess the relationships between the microbiome and host adaptation, we used 16S rRNA amplicon sequencing to characterize gut bacteria of two guppy ecotypes with known divergence in diet, life history, physiology and morphology collected from low-predation (LP) and high-predation (HP) habitats in four Trinidadian streams. Guts were populated by several recurring, core bacteria that are related to other fish associates and rarely detected in the environment. Although gut communities of lab-reared guppies differed from those in the wild, microbiome divergence between ecotypes from the same stream was evident under identical rearing conditions, suggesting host genetic divergence can affect associations with gut bacteria. In the field, gut communities varied over time, across streams and between ecotypes in a stream-specific manner. This latter finding, along with PICRUSt predictions of metagenome function, argues against strong parallelism of the gut microbiome in association with LP ecotype evolution. Thus, bacteria cannot be invoked in facilitating the heightened reliance of LP guppies on lower-quality diets. We argue that the macroevolutionary microbiome convergence seen across animals with similar diets may be a signature of secondary microbial shifts arising some time after host-driven adaptation.
Similar content being viewed by others
Introduction
Bacteria play a fundamental role in animal biology, and recent work has revolutionized our understanding of how these ubiquitous organisms affect their hosts’ development, physiology, ecology and evolution (Zilber-Rosenberg and Rosenberg, 2008; Lee and Mazmanian, 2010; Semova et al., 2012; Tremaroli and Backhed, 2012; McFall-Ngai et al., 2013). Across mammalian species, host phylogeny and diet correlate with the structure and diversity of gut bacterial communities (Ley et al., 2008). The concordance of gut microbiota and host phylogeny suggests host-derived factors can strongly shape the taxonomic composition of the gut microbiome (Rawls et al., 2006; Franzenburg et al., 2013), whereas dietary correlations reflect, partially, the importance of gut bacteria in the use of different foods. Yet, the types of consumed foods can mediate the influence of host-derived genetic factors on gut microbiota (Kashyap et al., 2013). Disentangling the influence of diet and host genetics in determining the composition of the gut microbiota remains an important challenge for understanding how hosts and their gut symbionts interact to shape the vast diversity in trophic strategies that so profoundly shape ecosystem structure and function.
Just as microevolutionary processes have been extrapolated to yield insight into macroevolutionary patterns (Kinnison and Hendry, 2001; Reznick and Ricklefs, 2009), studies of how gut communities vary among extant populations within a species can lend insights into the role of host genetic factors and trophic ecology in driving known interspecific patterns in gut microbiomes (Ley et al., 2008; Ochman et al., 2010). The positive relationship between host genetic and gut microbial divergence within mammalian species suggests an influence of host genotype on intraspecific gut microbiome variation (Zoetendal et al., 2001; Hildebrand et al., 2013; Linnenbrink et al., 2013). However, diet may also rapidly alter the gut microbiota (David et al., 2014) in ways that can be influenced by sex or host genetic background (Kashyap et al., 2013; Bolnick et al., 2014b). Through understanding how genes and the environment interact to structure gut communities, we stand to gain key knowledge on the potential for host–symbiont coevolution and the factors driving holobiont biodiversity. Furthermore, through studies of microbiome variation across intraspecific dietary gradients, we can ascertain whether macroevolutionary dietary correlations often originate because of symbiont-driven trophic diversification (see Hosokawa et al., 2007 and Chu et al., 2013 for examples in insects).
Although most research on vertebrate gut microbiota has focused on mammals, ray-finned fish are far more diverse, making up half of all living vertebrate species (Nelson, 2006). Fish were among the earliest jawed vertebrates to evolve many adaptive immune features that are hallmarks of mammalian systems (Flajnik and Du Pasquier, 2004) and, thus, the known interplay between immunity and mammalian gut bacteria (Hooper et al., 2012) suggests the potential for similar symbioses in ray-finned fish. Fish gut bacteria are generally related to animal-associated microbes, and marine herbivorous fish harbor communities that resemble those of other vertebrates, including gut-fermenting mammals (Mountfort et al., 2002; Sullam et al., 2012). Although community composition across fish species shows correlations with habitat salinity and feeding ecology (Ringø and Strøm, 1994; Ringø and Olsen, 1999; Sullam et al., 2012), a small number of recent surveys have provided glimpses into intraspecific variation in fish gut bacteria and have indicated that there are differences across more distantly related individuals and sampling locations and a sex-dependent effect of diet on gut microbiota (Wilson et al., 2008; Roeselers et al., 2011; Navarrete et al., 2012; Bolnick et al., 2014b). Overall, however, there are relatively few studies about fish gut microbiota, leaving open many questions regarding the symbiotic component of fish biodiversity.
Here, we explore the role of environmental, diet and host genetic factors in determining the gut microbiota of a vertebrate model system for rapid evolution in nature, the Trinidadian guppy (Poecilia reticulata). Guppies in Trinidadian streams are typically categorized into two different, predator regime-driven ecotypes (Magurran, 2005). In downstream, high-predation (HP) habitats, a diverse predator community preys upon guppies, whereas barrier waterfalls greatly reduce upstream predator dispersal and, thus, predation threats for guppies in the low-predation (LP), headwater stream habitats. Because various life history, sexually selected, morphological and physiological traits have evolved in parallel in different streams, P. reticulata serves as an important model system for studies of natural ecotype evolution in response to comparable ecological gradients (Reznick et al., 1996; Ghalambor et al., 2004; Torres Dowdall et al., 2012; Handelsman et al., 2013). In addition to these attributes, HP and LP ecotypes also diverge in trophic ecology, and HP guppies eat significantly more invertebrates than their derived LP counterparts that consume more detritus and diatoms (Zandonà et al., 2011). Dietary differences have also been observed in common-garden settings (Bassar et al., 2010) and appear related to differences in gut morphology and physiology (Sullam et al., 2014). Combined with known roles for microbial symbionts in dietary utilization and processing (Semova et al., 2012), this raises the expectation that HP and LP guppies may have undergone parallel changes in their gut microbiomes should bacterial shifts be integral to shifts in host diet. Such microbiome divergence could also occur if diet has a strong, proximate impact upon gut communities.
To better understand the variation in natural guppy gut communities and the potential for microbial roles in guppy divergence and adaptation, we utilized amplicon pyrosequencing of bacterial 16S rRNA genes from HP and LP guppies across populations from four streams. We sampled two of the streams across 2 years, enabling an assessment of the temporal stability of microbiome divergence within some populations. Comparisons of gut communities of field-caught fish to those reared in the lab on different diets and to communities from surrounding aquatic habitats were performed to yield further insight into the stability and origins of gut microbiota. Our results contribute the first glimpses into both the symbiotic component of this important evolutionary model and the nature of intraspecific microbiome variation in fish across multiple scales.
Materials and methods
Field collections
Our study consists of data from two field surveys and one dietary manipulation experiment. Both field surveys were conducted during the dry seasons in 2010 and 2011, respectively, and took place in mountain streams in the Northern Range of Trinidad, West Indies (Supplementary Table 1). The 2010 field survey was carried out to assess gut bacterial diversity across streams and guppy populations; from 20 April through 2 May 2010, 40 Trinidadian guppies were collected from previously categorized HP and LP habitats (Reznick et al., 1996; El-Sabaawi et al., 2012) that tend to differ in a number of ecosystem characteristics, including canopy coverage and primary productivity (Grether et al., 2001). Guppies from the two different habitat types can be divided into HP and LP ecotypes based on well-documented trait differences (Endler, 1995; Reznick and Bryga, 1996).
Protocols for collection and processing of wild guppies (2010 and 2011) were approved by Institutional Animal Care and Use Committee 18560 at Drexel University. In brief, we collected guppies with butterfly nets, starving them for 8–12 h in ambient stream water before killing with MS-222 (Sigma, St Louis, MO, USA) in order to decrease the probability of sampling transient bacteria introduced with dietary items (see Supplementary Text for details regarding sample collection and storage). In each of the four streams sampled in 2010, LP guppies were collected from one upstream (LP) site, whereas HP guppies were obtained from one downstream (HP) site, generating eight sites and populations, in total. Targeted streams, which were tributaries of the below-named rivers, included three drainages on the southern slope of the Northern Range: the Aripo and Guanapo Rivers, which are part of the Caroni drainage, and the Quare River, which is part of the Oropouche drainage. The fourth stream sampled was the Marianne, which is part of a separate drainage and found on the northern slope of the Northern Range, where guppy populations are estimated to have diverged from south slope populations several hundred thousand years ago (Willing et al., 2010). All guppies collected were adult females with a standard length >15 mm and did not differ significantly in standard length across sites. An additional 10–15 female guppies were collected from each site in 2010 for gut length analysis.
During the 2011 survey (27 March to 6 April 2011), an additional three guppies were collected from the LP and HP Aripo and Guanapo sites. These collections complemented those from 2010 and allowed for a temporal assessment of guppy gut microbiome stability over the span of 1 year. The 2011 Aripo HP site differed slightly from the 2010 Aripo HP site in that it was ∼750 m downstream from the 2010 locale, while the 2011 Guanapo HP and LP sites were the same as in 2010. An additional HP site (HP2) in the Guanapo was also added to the 2011 survey and was located ∼5 km downstream from the 2010 HP site. Three to four environmental samples (sediment and water) were collected from each of the Guanapo sites in 2011 to compare gut microbial communities with those of the ambient environment. For the environmental samples, Guanapo stream water was filtered through individual 25 mm diameter filters with 0.2 μm pore size Whatman Nuclepore Track-Etched Membranes (GE Healthcare, Piscataway, NJ, USA) using Sterile 60 cc Luer Lock Tip syringes (BD, Franklin Lakes, NJ, USA). Filters were subsequently cut into small pieces with sterile scissors before DNA extraction. Sediment samples were also taken from the three Guanapo sites by collecting ∼0.5 g of fine benthic organic matter/sediment from the streambed using a small hand scoop.
Dietary experiments
A subset of live guppies collected from Aripo HP and LP sites in 2011 were transported to Cornell University for a dietary manipulation study approved under Institutional Animal Care and Use Committee protocol 2008–0106 at Cornell University. In this experiment, 64 LP and HP guppies were randomly assigned to 16 tanks and fed a predominately spinach or brine shrimp diet. These diets roughly reflect the highly detrital and algal diet of LP guppies and invertebrate diet of HP guppies, previously found in the Aripo and Guanapo Rivers (Zandonà et al., 2011). A subset of reared females (n=14), each from a separate tank, was analyzed for gut microbes, whereas the remaining fish were analyzed for enzyme activities in another study (Sullam et al., 2014). Diet manipulations commenced on 7 June 2011 and proceeded for 10 weeks. Guppies were fed equal calories of the two diets (that is, 1.6 × more spinach diet by dry mass) at a level in excess of estimated daily caloric demands (Reznick, 1983), so that fish on the invertebrate diet received ∼6% more protein and 200% more lipids than fish on the spinach diet, whereas those on the spinach diet had 340% more carbohydrates than those on the invertebrate diet (Sullam et al., 2014).
DNA extraction and sequencing
Whole guts of individual guppies, which are each composed of a simple tubular digestive tract, were dissected with sterile instruments and then washed in 70% ethanol and sterile water before DNA extraction to help eliminate transient bacteria. DNA from these guts, plus that from sediment and water samples, was extracted separately with the MO BIO (Carlsbad, CA, USA) Power Soil Kit following the manufacturer’s protocol. An additional 10 min at 65 °C heating step was performed after the addition of lysis buffer for all but the dietary manipulation samples. Bacterial tag-encoded FLX-titanium amplicon pyrosequencing (bTEFAP) of 16S rRNA amplicons was then carried out on our DNA samples at Research and Testing Laboratories (Lubbock, TX, USA) using the following primers: 104F (5′-GGCGVACGGGTGAGTAA-3′) and 530R (5′-CCGCNGCNGCTGGCAC-3′). The resulting sequencing data have been submitted to NCBI SRA under Bioproject PRJNA259592.
Data analysis
Acacia (version 1.52; Bragg et al., 2012) was used to error-correct the amplicon 454 pyrosequencing reads with an average quality cutoff score of 30. The output from Acacia was then de-multiplexed using QIIME. The QIIME pipeline was followed with default parameters (Caporaso et al., 2010) using version 1.7 unless otherwise noted. Briefly, these steps included excluding sequences with any primer errors or more than one base pair error in the barcode, removing barcodes, truncating the sequences at the reverse primers, picking operational taxonomic units (OTUs) with UCLUST at 0.97 similarity, and aligning OTU reference sequences against the Greengenes reference alignment. Chimera checking was performed using ChimeraSlayer. Because sequences were aligned against a template file, regions of the 16S rRNA alignment were filtered according to the Greengenes Lane Mask (DeSantis et al., 2006). After filtering out low-quality sequences, chimeras, chloroplasts and sequences not classifying to bacteria, 234 725 of the original 462 193 amplicon reads remained. These sequences were classified using UCLUST with noted exceptions (see Supplementary Text for details). The phylogenetic tree, which was used for unweighted and weighted UniFrac analyses, was constructed in QIIME 1.8 using RAxML (Stamatakis et al., 2005). Because 97% OTU clusters can mask important within and between sample diversity (Eren et al., 2013; Sanders et al., 2014), we further examined genotype/strain variation within 97% OTUs among sampling sites and treatments (see Supplementary Text for analysis).
We used PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States, version 1.0.0), to predict metagenome function based on 16S rRNA sequences using a database of reference genomes (Langille et al., 2013). To prepare our data for PICRUSt analysis, we performed closed-reference 94% OTU picking against the Greengenes database released May 2013, after finding that many reads were not classified to existing 97% OTUs, thus marking large portions of our libraries for exclusion. After 94% OTU assignment, libraries were rarefied to 800 reads per sample. Subsequent analyses focused only on wild samples because 5 of the 14 lab fish libraries had <800 reads per library after closed-reference OTU picking. OTUs from wild guppies were normalized for predicted 16S rRNA copy number before predicting gene family abundance for each metagenome based on KEGG orthology groups (KOs) using the Kyoto Encyclopedia of Genes and Genomes (Kanehisa et al., 2012).
To assess the likely accuracy of the PICRUSt analysis, the Nearest Sequenced Taxon Index was calculated. This index indicates the average divergence between each 16S rRNA read within an OTU and the 16S rRNA gene of the closest reference organism with a sequenced genome. The average Nearest Sequenced Taxon Index values for our gut libraries (0.10±0.04) suggests comparable predictive accuracy for our communities as those achieved for mammalian guts and salamander skin (Langille et al., 2013; Loudon et al., 2014). In a second quality check, we found that after closed-reference 94% OTU assignment, our communities still showed strong taxonomic resemblance to those of our complete data set as seen in Figure 1 (data not shown). Combined, these analyses suggested fairly high accuracy of subsequent functional predictions.

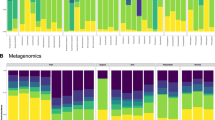
Taxonomic composition of gut bacterial communities. Results are based on 454 pyrosequencing across 55 Trinidadian guppies from the wild and 14 included in a dietary manipulation experiment. Stream, ecotype and year of collection are listed on the x-axis for wild-caught fish. Communities in the central box represent gut bacteria of guppies from three streams on the southern slope of the Northern range, and those in the leftmost box show gut bacteria of guppies from a stream on the northern slope. Columns in the rightmost box represent samples taken after the dietary manipulation experiment in the lab, for which fish collected in 2011 from the Aripo were used, and HP fish and LP fish were fed either an invertebrate (I) or spinach (S)-based diet. Bacteria are classified to order by color, with phyla indicated to the right of ordinal labels.
Statistical analyses
The 97% OTU table derived through the QIIME pipeline was rarefied to 800 reads per sample before the calculation of four different distance matrices used to analyze community similarity. Unweighted and weighted UniFrac distance matrices (Lozupone and Knight, 2005), which use phylogenetic information to calculate community similarity, were produced through the QIIME pipeline. To compare community similarity based on presence/absence of OTUs, the Jaccard index was used, and to compare community similarity based on OTU abundance, the Bray–Curtis dissimilarity matrix was made with Hellinger transformed data (Oksanen et al., 2013). Different principal coordinates were derived from the four different distance measurements. Analysis of variance was run on distance-based redundancy analyses using the four different principal coordinate values (Legendre and Anderson, 1999), and P-values were calculated for model attributes based on 9999 random permutations of all principal coordinates. All distance-based analyses and statistical tests on PICRUSt output (discussed below) were performed in R (v3.0.1; R Development Core Team, 2013). We also classified gut communities into partitions or ‘enterotypes’ using the function cascadeKM in vegan (Oksanen et al., 2013) on the rarified abundance matrix, and the Calinski–Harabasz criterion was used to evaluate the optimal number of partitions.
Network analyses were performed in QIIME and visualized using Cytoscape version 3.0.2 (Shannon et al., 2003) using the edge-weighted spring embedded algorithm to display the OTUs and sample nodes (Ley et al., 2008). A heatmap was constructed with all OTUs that had ≥500 reads represented in the entire dataset using heatmap.2 in R. The dendrogram of the samples shown in the heatmap was created with Ward’s hierarchical clustering of bacterial communities in hclust with sequence libraries rarefied to 800 reads per sample. OTUs were deemed to belong to the ‘core’ set if they were found within ≥50% of samples. Core sets were identified separately for the wild HP and LP fish as well as environmental samples. To better understand the lifestyles and origins of core gut bacteria, we retained the habitats of the three closest BLASTn hits (analyses performed on 24 September 2013) of each core OTU from gut and environmental samples. BLASTn hits were counted just once for each category (LP, HP and environment) in cases where multiple OTUs from a single category gave the same hit. Using this approach, we retained and characterized 81 unique hits for environmental core OTUs, 22 unique hits for HP guppy core OTUs and 33 unique hits for LP guppy core OTUs, with 11 hits shared between ecotypes.
We investigated whether the most prevalent OTUs (that is, those present in more than 12/80 samples and represented by ≥300 reads in the entire data set) differed in abundance across streams, ecotypes and years in the wild and across treatments in the dietary manipulation. To achieve this, either Kruskal–Wallis tests (for comparisons between >2 groups) or nonparametric t-tests (for comparisons between two groups) were used with QIIME’s group significance script (version 1.8) on OTU tables rarefied to 800 reads per sample. We also used Kruskal–Wallis tests on gene function from PICRUSt outputs (that is, % of the predicted metagenome made up by a given KEGG functional module) to compare differences between ecotypes, streams and enterotypes, while accounting for year. All multiple comparisons were corrected with the Benjamini–Hochberg false discovery rate method.
Results
Wild guppy gut bacterial communities
Gut bacterial surveys from across the four targeted streams revealed modestly diverse gut communities among the sampled wild fish compared with communities from water and sediment (Supplementary Figure 1). Gut communities showed a fairly high degree of interindividual variability, even within populations, yet a number of trends were evident at broader scales (Figure 1). For instance, guppies from the Marianne were mostly unique in their enrichment for Fusobacteriales, accounting for over 50% of the reads in 8/10 samples. In addition, guppies from the Guanapo harbored large proportions of Mycoplasmatales across two years (2010 and 2011), whereas Aripo fish showed enrichment for these bacteria mostly in 2011. Many other fish had a high prevalence of Proteobacteria in their gut bacterial communities, including Quare guppies and Aripo LP guppies from 2010 (5/10 and 5/5 with ≥50% Proteobacterial prevalence, respectively). Several different Proteobacterial orders showed high prevalence, with those from the Vibrionales and Aeromonadales being the most predominant.
Principal coordinates plots and statistics based on four different β-diversity metrics all showed differentiation among bacterial communities across streams, while further suggesting important stream × ecotype interactions. Effects of ecotype by itself were contrastingly subtle, although significant, varying mostly in a stream-dependent manner (see Table 1a for Bray–Curtis and unweighted UniFrac statistics; see Supplementary Table 2a for the Jaccard index and weighted UniFrac statistics; see Supplementary Figure 2 for principal coordinates analysis plots). Accordingly, several OTUs from the Firmicutes, Fusobacteria, Proteobacteria, Spirochaetes or Tenericutes varied in relative abundance across streams and years, yet we could not identify 97% OTUs driving differences between HP and LP ecotypes from across the four streams (Supplementary Table 3). We did observe genotypic, or strain, variation of two dominant OTUs across sites/populations, suggesting cryptic diversity invisible to analyses on more traditional 97% OTU delineations (Supplementary Figure 3, Supplementary Table 4). However, these patterns did not differentiate LP vs HP guppies in a consistent fashion.
Guppy gut bacterial community ‘enterotype’ groupings defined at the 97% OTU level (Figure 2 and Supplementary Table 5; discussed further below) also did not appear to be driven by ecotype, except for fish within the dietary manipulation that separated exclusively into LP and HP enterotypes in accordance with patterns seen at higher taxonomic levels (Figure 1). Groupings of wild fish showed mostly stream-specific patterns, due largely to frequent classification of Guanapo samples into enterotype 4 and of Marianne samples into enterotype 1. Temporal differences were also evident for guppy gut communities (Figures 1 and 3a), including their enterotype assignments, with an increased proportion of Aripo fish bearing enterotype 4 communities in 2011 (Supplementary Table 5).

Bacterial community composition and enterotype groupings in relation to 97% OTU distributions. A heatmap is shown for all OTUs with over 500 reads in the whole data set. The phylum associated with each OTU is listed above, and the asterisks indicate dominant OTUs that appear to be driving enterotype groupings (Supplementary Table 5). The shade of blue in the heatmap indicates the proportion of reads that come from the given OTU out of all reads for that sample. The color bar on the right of the heatmap indicates experiment/sample type—light gray: samples from the dietary manipulation experiment; dark gray: non-fish environmental samples; remaining colors: 2010 and 2011 field collections (yellow: Guanapo; red: Quare; green: Marianne; and blue: Aripo). Names on the left aid in further classifying samples, with ‘11’ denoting field samples obtained in 2011 (2010 samples have no year abbreviation), and LP/HP/HP2 abbreviations are used as described for Figure 1. The sample names on the left are also shaded in green according to their enterotype grouping (displayed to the left of the sample label). The dendrogram on the right shows community similarity based on hierarchical clustering using Ward’s method.

Effects of time, diet and population background on guppy gut microbiomes. (a) Similarity of gut communities among wild guppies from the Aripo and Guanapo Rivers during the 2011 and 2010 surveys. Communities from the two years and streams were significantly different from each other (Supplementary Table 7), although the stream effect was weak. Principal coordinates analyses (PCoAs) shown were generated from unweighted UniFrac distances, and PCoA 3 shows the strongest correlation with yearly variation. (b) Principal coordinates of guppy gut bacterial community similarity from experimental dietary treatments based on Bray–Curtis distances. Shape indicates treatment diet (invertebrate- or spinach-based diet) and color indicates guppy ecotype (HP or LP). Note that there are two overlapping LP spinach data points at (0.36, −0.17). Analyses of Bray–Curtis distances indicate a clear effect of ecotype on gut community composition. Subtle, yet significant, effects included both experimental diet and an ecotype by diet interaction (Table 1) because of a dietary difference in bacterial communities within HP but not LP guppies.
To investigate the relationship between diet and gut bacteria, we measured gut length of HP and LP ecotypes from the four sampled 2010 streams as a proxy for diet (Zandonà, 2010). HP populations in all of the streams except the Quare had significantly shorter guts than LP populations, as predicted based on prior findings of increased detritus and algae consumption by LP guppies (Supplementary Figure 4) (Zandonà, 2010). Yet, there was no correlation between gut microbial community composition and size-standardized gut length, consistent with the absence of LP versus HP community types when viewed across streams.
Guppy gut microbial communities from the dietary manipulation
After 10 weeks on controlled diets, HP and LP fish exhibited distinct gut bacterial communities regardless of their experimental diet (Figures 1 and 3b). All four community composition metrics revealed a significant impact of ecotype on gut bacterial communities, whereas three of four metrics suggested a significant effect of diet and a diet by ecotype interaction effect on gut community composition (see Table 1b for Bray–Curtis and unweighted UniFrac analyses and Supplementary Table 2b for the Jaccard index and weighted UniFrac results). Discrepancies among β-diversity metrics were supported by visual trends (Figure 1), suggesting that dietary plasticity in HP guppies was largely a function of shifting bacterial relative abundance between the two dietary treatments, and ecotype differences were more clearly the result of presence/absence effects driven by Entomoplasmatales (HP) versus Spirochaetales (LP) bacteria.
Analyses of individual OTUs (t-tests; Supplementary Table 3) accordingly showed that ecotype differences in lab experiments were driven by OTUs classified to the Entomoplasmatales and the genus Spirochaeta, although no OTUs varied significantly among dietary treatments for HP or LP guppies. Hierarchical clustering revealed a clear distinction among lab HP and LP communities (Figure 2), which were assigned to two distinct and exclusive lab enterotypes each comprising communities from both dietary treatments. Although gut communities of lab-reared fish differed greatly from those in the wild (Figure 1), the two most dominant OTUs in lab populations were found in some wild fish and showed weak tendencies to naturally associate with the same ecotypes they dominated in the laboratory.
The core gut microbiome
HP fish had 15 core OTUs present in over 50% of wild samples, whereas LP guppies had a core set of 14 OTUs. Six of these overlapped between HP and LP guppies. Environmental samples had a core set of 28 OTUs, none of which overlapped with guppy core microbes. By analyzing the top three BLASTn hits of the core guppy OTUs, we found that ∼40% of all unique top hits for LP and HP guppies matched with bacteria derived from other, non-guppy fish guts, in contrast to BLAST results for environmental samples (Figure 4a and Supplementary Table 6).

Relationships between guppy gut microbes and those from other habitats. (a) Closest relatives of core bacterial OTUs. Core OTUs were separately identified for LP fish (14 OTUs), HP fish (15 OTUs) and environmental (28 OTUs) samples when they occurred in >50% of the samples from one of these categories. Each OTU was then searched using BLASTn against the NCBI database and its three closest hits were categorized based on their environment of isolation, and proportions of these categories are shown. (b) Network analysis of all gut microbes derived from guppies and environmental samples. Square nodes correspond to 97% OTUs, with colors indicating whether these were found in one or more samples. Purple reveals the OTUs found in both gut and environmental samples. Black symbols correspond to guppy gut (lab vs field) vs environmental (water and sediment) samples.
Certain core gut microbiota were enriched in the guts of wild guppies, depending on stream, including (1) a Tenericutes bacterium from the Mycoplasmataceae family (OTU 2023) in the Guanapo, 2011 Aripo and a few Quare LP fish, (2) Cetobacterium somerae from the Fusobacteriaceae (OTU 4447), in Marianne fish; and (3) a Proteobacterium in the Aeromonadaceae family (OTU 5998) that dominated 2–3 fish from each the Quare, Marianne and Aripo streams. Each of these dominant OTUs had a close BLASTn hit in the GenBank database to bacteria from the guts of other fish species.
Comparison of environmental versus guppy gut bacteria
Gut bacterial communities were distinct from bacterial assemblages in both water and stream sediments, and only 5% of all OTUs were found in both guppy guts and the environment (Figure 4b). As mentioned above, core microbes from guppy guts were distinct from core microbes of environmental samples; however, two of the six dominant guppy OTUs were found in the environment at relatively low abundances (making up 0.14–1.49% of reads in 5 of 14 environmental samples). To examine whether guts from sites within the Guanapo were enriched with environmental bacteria more abundant in that site, we ran Kruskal–Wallis tests for each OTU found in both environmental and gut samples from our 2011 Guanapo survey, finding no significant differences in sampling sites when stratified according to sample type (environment or guppy gut). Therefore, the abundance of certain OTUs in guppy guts did not correlate with their abundance in the environment. Furthermore, the significant interaction between sampling type and site location indicates that within each sampling site, gut and environmental samples differ (Supplementary Table 7). Finally, network diagrams showed that guppy gut communities were no more similar to environmental bacteria from the same versus different Guanapo sites (Supplementary Figure 5). Combined, these findings reveal little evidence for a strong interplay between microbes in guppy guts and the environment.
Functional analysis
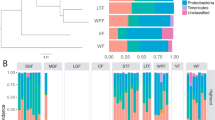
PICRUSt assignment of predicted metagenome content to Level 2 KOs suggested no significant functional differences between HP and LP ecotypes after correction for 39 multiple comparisons. However, almost all functional categories differed significantly among enterotypes (Figure 5), including KOs of likely symbiotic importance such as carbohydrate and amino-acid metabolism, vitamin biosynthesis and the degradation of xenobiotics.

Results of PICRUSt analysis showing predicted relative abundance of KEGG ortholog groups for guppy (a) ecotypes and (b) enterotypes. Asterisks on the right of each panel indicate significant differences among groups. No significant differences were found between ecotypes, whereas the majority of KEGG orthogroups differed among Enterotypes. The samples from the dietary manipulation (and their two associated enterotypes: 3 and 5) are not shown because an insufficient number of reads per sample (<800) clustered with the closed reference OTUs in approximately one third of the dietary manipulation samples.
Discussion
Drivers of natural microbiome composition
To assess the diversity of the gut microbiome in Trinidadian guppies, we sequenced bacterial 16S rRNA genes from whole guts of LP and HP ecotypes from each of four streams, finding interindividual variation at the phylum, 97% OTU and strain levels. Although we found notable intrasite variation, we also detected community differences between streams and habitats at each of these taxonomic scales, implicating local environmental or host genetic attributes as drivers of microbiome differentiation.
Previous research has identified significant genetic divergence based on single-nucleotide polymorphisms between guppies from different streams and the HP and LP sites contained within (Willing et al., 2010), with Marianne populations being the most divergent among those sampled here. Therefore, the pronounced divergence of gut microbial communities from Marianne guppies suggests some impact of host relatedness on microbiome similarity. Although selection could play a role in these patterns, they are also consistent with neutral evolutionary processes affecting host genes that shape gut microbial composition. Microbiome differences between lab-reared Aripo HP and LP guppies consuming the same diets in identical laboratory conditions further suggest that genetic divergence affects microbiome differences. However, because the fish used in our experiments were born in the wild, we cannot rule out the effect of plasticity stemming from early-life experience in nature.
In other systems ranging from hydra to fish and mice, closely related hosts harbor more similar microbial communities than do more distantly related individuals (Zoetendal et al., 2001; Friswell et al., 2010; Navarrete et al., 2012; Faith et al., 2013; Franzenburg et al., 2013). The ongoing discovery of particular loci that shape the gut microbiome in model organisms (Benson et al., 2010) presents an intriguing target for future studies across animal systems in both the lab and field. Such host encoded genes may affect aspects of host physiology or immunity, as illustrated by the recent finding that the types and diversity of major histocompatibility complex alleles are related to the composition and diversity of gut bacteria in freshwater stickleback fish (Bolnick et al., 2014a). Differences in these or other genetic attributes among guppy individuals and populations could be key to understanding the causes of their microbiome divergence.
Although host genetics and population divergence appear to be of some importance in the guppy system, populations showed some overlap in their gut communities, possibly suggesting incomplete population divergence in relevant genetic factors that control gut microbiota. Furthermore, population-level microbiome signatures did not appear fixed, as the microbiomes of Aripo guppies shifted in composition across just a single year. Guppies have been shown to exhibit rapid, local adaptation in response to transplantation in Trinidadian streams, including shifts in some traits such as age at maturity within only 4 years (Reznick et al., 1997). Although this timescale is longer than that observed here, a role for shifting host genotype in microbiome alterations could still possibly extend from impacts of genes involved in immunity, as such genes are often under strong positive selection (Yang and Bielawski, 2000).
Another conceivable source of microbiome differentiation is the availability of specialized gut bacteria in the surrounding aquatic habitat. Although there was minimal overlap between environmental and gut microbes, a few dominant OTUs from guppies were found at very low abundance in the environment, suggesting that environmental pools of bacteria are involved in seeding guppy guts. Colonization from the surrounding environment is thought to be one of the primary mechanisms of microbiota acquisition for fish (Nayak, 2010), and is found in many other animal-microbe symbioses, including the Hawaiian bobtail squid and their luminescent Vibrio fischeri symbionts. In that system, V. fischeri are found at very low abundances in the surrounding aquatic habitat (<0.1% of bacterioplankton), yet exclusive colonization of squid light organs is achieved through specific mechanisms employed by both symbionts and hosts (Nyholm and McFall-Ngai, 2004). Although Trinidadian guppies likely acquire their symbionts from their environment, we found no correlations between gut and environmental bacterial OTU abundance from the same location when examined across Guanapo stream sites. However, the few reads of guppy gut microbes found among environmental sequence libraries and our limited sampling design hinder our abilities to completely rule out effects of environmental abundance and priority effects in governing the variation in gut communities over space and time.
Among the remaining candidate drivers of microbiome divergence are a myriad of other temporally and spatially varying factors, including various environmental conditions, host immunological history and overall host physiological condition (Clemente et al., 2012). Research that takes these and the aforementioned factors into account will be important in pinpointing the sources of natural variation, enabling a better understanding of how the complex trait of the gut microbiome is mediated by host genetics, the environment and their intersection (Benson et al., 2010).
The functional implications of variable gut communities
The distinct community enterotypes we found in guppies were defined largely by the dominance of a single, distinguishing 97% OTU. The recurrence of such enterotypes, combined with our findings of additional core bacteria in a majority of HP or LP guppies, suggest the guppy gut is strongly selective, resembling selectivity demonstrated in zebrafish guts (Rawls et al., 2006). In addition, we found guppy core gut bacteria to be closely related to microbes from other fish, suggesting that such selectivity often favors microbes that have specialized, for some time, on a symbiotic lifestyle.
While there is some debate on the discrete and permanent nature of enterotypes within the human literature (Arumugam et al., 2011; Wu et al., 2011; Huse et al., 2012; Rajilić-Stojanović et al., 2013), our functional predictions of metagenome content suggest that differing community types in the guppy system have inherent functional differences. For instance, guppies categorized to enterotype 1 were estimated to have the greatest relative proportion of their metagenomes devoted to vitamin biosynthesis. Interestingly, the bacterium characterizing enterotype 1 was classified as Cetobacterium somerae, a B-12 vitamin-producing, obligate anaerobe found in several freshwater fish including zebrafish, goldfish, carp, tilapia and ayu (Sakata et al., 1981; Sugita et al., 1991; Tsuchiya et al., 2008; Roeselers et al., 2011). Other functional differences between enterotypes found in our study, including carbohydrate and amino-acid metabolism, are of likely nutritional or digestive relevance to guppy hosts, and more direct studies on functional implications of taxonomic differences in fish gut microbiomes may help elucidate their roles. Studies on the stability of different community types, through nondestructive sampling of individuals, would also help to determine whether any such functional differences are a hallmark of the individual, and perhaps of its genotype, or part of a plastic range of functional signatures that unfold throughout its lifetime. Certainly, human gut microbiomes show a capacity to change over time, with altered abundance of bacterial phyla having important impacts on energy extraction and weight gain (Ley et al., 2006; Ridaura et al., 2013).
Non-parallelism of gut microbiota and implications for trophic diversity across animals
Variation in gut bacteria has been found within and among animal populations (Cahill, 1990; Benson et al., 2010; Friswell et al., 2010; Linnenbrink et al., 2013), yet some of the best-cited work in the field of microbiome research has focused on macroevolutionary patterns. Across such a phylogenetic scale, related hosts indeed often harbor similar microbes (see, for example, Jones et al., 2013), as do those with similar diets (Ley et al., 2008). Well documented in the mammals and in several insects, specific symbioses related to particular trophic habits are widely argued to reflect the importance of bacteria in host dietary utilization (see, for example, Ley et al., 2008; Russell et al., 2009). Empirical and metagenomic evidence clearly indicate such an importance (Feldhaar et al., 2007; Douglas, 2009; Muegge et al., 2011). Hence, are shifts in bacteria essential for shifts in diet or trophic level?
Our findings suggest that the answer is ‘no’ within the guppy system, as dietary shifts between HP and LP ecotypes have not been preceded or accompanied by consistent changes in microbiome structure or predicted function. Despite dietary differences repeated across streams and years (Bassar et al., 2010; Zandonà et al., 2011), and differences in gut size and enzymatic activity (Sullam et al., 2014), gut bacterial communities did not show strong or clear convergence across independently derived LP ecotypes, nor did they show high similarity among the ancestral HP ecotypes from across the targeted streams.
The change in LP guppy gut length, however, is not a drastic structural shift in comparison, for example, to an introduced lizard population’s rapid cecal valve evolution with increased plant consumption (Herrel et al., 2008). Therefore, despite dietary differences in HP and LP guppies, both ecotypes still rely on high intake and fast dietary passage that may hinder the pressure for whole-sale changes in gut communities. A more ancient example of gut microbiota ‘resistance’ to dietary change includes pandas that show similar gut microbiota compared with their carnivorous relatives in spite of their extensive consumption of foliage (Ley et al., 2008). Their rapid gut processing of large volumes of plant material (Van Soest, 1994), rather than slow digestion and extensive fermentation seen for many mammalian herbivores, may have promoted the retention of their ‘ancestral’ microbiome instead of shifts to canonical fermenting gut communities (see Clements et al., 2014 for a similar discussion of pertinence to herbivorous fish).
Our work on a model system for microevolution in nature suggests that macroevolution of microbiome divergence across trophic levels may occur as part of a gradual process, whereby diet changes first, followed by major changes in gut morphology, physiology and symbiotic communities within only select lineages. The lack of microbiome parallelism in our system may, thus, partially stem from the absence of sufficient concurrent changes in guppy digestion. Of course, our results may also indicate the importance of elements beyond diet in shaping gut microbes in the guppy system. Indeed, not all environmental variables, such as agricultural inputs and flooding conditions, show parallel similarities among habitat types (Fitzpatrick et al., 2014), raising interesting considerations for future work on guppy gut microbes.
Conclusions
In delineating priority areas for future microbiome research across guppies and beyond, there is a clear need for studies on the genetic and environmental sources of gut community variation to elucidate just when and how we should expect microbiomes to diverge. These efforts should adopt a systems-level approach, interpreting the timing and mode of microbiome shifts in light of the ecological, morphological, physiological and phylogenetic divergence among the hosts. In addition, emphasis on the integration of microbes into host development, immunity and overall defense will be key to understanding both the causes and consequences of gut community variation beyond the realms of diet and life history. Through a better comprehension of the genetic components shaping variable gut communities and their major costs and benefits in an ecological context, we can gain powerful insights into the potential for microbially mediated animal adaptation and its broader impacts on biodiversity.
References
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR et al. (2011). Enterotypes of the human gut microbiome. Nature 473: 174–180.
Bassar RD, Marshall MC, López-Sepulcre A, Zandonà E, Auer SK, Travis J et al. (2010). Local adaptation in Trinidadian guppies alters ecosystem processes. Proc Natl Acad Sci USA 107: 3616–3621.
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 107: 18933–18938.
Bolnick DI, Snowberg LK, Gregory Caporaso J, Lauber C, Knight R, Stutz WE . (2014a). Major histocompatibility complex class IIb polymorphism influences gut microbiota composition and diversity. Mol Ecol 23: 4831–4845.
Bolnick DI, Snowberg LK, Hirsch PE, Lauber CL, Org E, Parks B et al. (2014b). Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat Commun 5: 4500.
Bragg L, Stone G, Imelfort M, Hugenholtz P, Tyson GW . (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat Methods 9: 425–426.
Cahill MM . (1990). Bacterial flora of fishes: a review. Microb Ecol 19: 21–41.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Chu C-C, Spencer JL, Curzi MJ, Zavala JA, Seufferheld MJ . (2013). Gut bacteria facilitate adaptation to crop rotation in the western corn rootworm. Proc Natl Acad Sci USA 110: 11917–11922.
Clemente Jose C, Ursell Luke K, Parfrey Laura W, Knight R . (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270.
Clements KD, Angert ER, Montgomery WL, Choat JH . (2014). Intestinal microbiota in fishes: what’s known and what’s not. Mol Ecol 23: 1891–1898.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505: 559–563.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microb 72: 5069–5072.
Douglas AE . (2009). The microbial dimension in insect nutritional ecology. Funct Ecol 23: 38–47.
El-Sabaawi RW, Zandonà E, Kohler TJ, Marshall MC, Moslemi JM, Travis J et al. (2012). Widespread intraspecific organismal stoichiometry among populations of the Trinidadian guppy. Funct Ecol 26: 666–676.
Endler JA . (1995). Multiple-trait coevolution and environmental gradients in guppies. Trends Ecol Evol 10: 22–29.
Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG et al. (2013). Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol Evol 4: 1111–1119.
Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al. (2013). The long-term stability of the human gut microbiota. Science 341: 1237439.
Feldhaar H, Straka J, Krischke M, Berthold K, Stoll S, Mueller M et al. (2007). Nutritional upgrading for omnivorous carpenter ants by the endosymbiont Blochmannia. BMC Biol 5: 48.
Fitzpatrick SW, Torres-Dowdall J, Reznick DN, Cameron KG, Funk WC . (2014). Parallelism isn’t perfect: could disease and flooding drive a life-history anomaly in Trinidadian guppies? AmNat 183: 290–300.
Flajnik MF, Du Pasquier L . (2004). Evolution of innate and adaptive immunity: can we draw a line? Trends Immunol 25: 640–644.
Franzenburg S, Walter J, Künzel S, Wang J, Baines JF, Bosch TCG et al. (2013). Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc Natl Acad Sci 110: E3730–E3738.
Friswell MK, Gika H, Stratford IJ, Theodoridis G, Telfer B, Wilson ID et al. (2010). Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice. PLoS One 5: e8584.
Ghalambor CK, Reznick DN, Walker JA . (2004). Constraints on adaptive evolution: the functional trade-off between reproduction and fast-start swimming performance in the Trinidadian guppy (Poecilia reticulata). Am Nat 164: 38–50.
Grether GF, Millie DF, Bryant MJ, Reznick DN, Mayea W . (2001). Rain forest canopy cover, resource availability, and life history evolution in guppies. Ecology 82: 1546–1559.
Handelsman CA, Broder ED, Dalton CM, Ruell EW, Myrick CA, Reznick DN et al. (2013). Predator-induced phenotypic plasticity in metabolism and rate of growth: rapid adaptation to a novel environment. Integr Comp Biol 53: 975–988.
Herrel A, Huyghe K, Vanhooydonck B, Backeljau T, Breugelmans K, Grbac I et al. (2008). Rapid large-scale evolutionary divergence in morphology and performance associated with exploitation of a different dietary resource. Proc Natl Acad Sci USA 105: 4792–4795.
Hildebrand F, Nguyen TLA, Brinkman B, Yunta R, Cauwe B, Vandenabeele P et al. (2013). Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol 14: R4.
Hooper LV, Littman DR, Macpherson AJ . (2012). Interactions between the microbiota and the immune system. Science 336: 1268–1273.
Hosokawa T, Kikuchi Y, Shimada M, Fukatsu T . (2007). Obligate symbiont involved in pest status of host insect. Proc R Soc B Biol Sci 274: 1979–1984.
Huse SM, Ye Y, Zhou Y, Fodor AA . (2012). A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7: e34242.
Jones RT, Sanchez LG, Fierer N . (2013). A cross-taxon analysis of insect-associated bacterial diversity. PLoS One 8: e61218.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M . (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109–D114.
Kashyap PC, Marcobal A, Ursell LK, Smits SA, Sonnenburg ED, Costello EK et al. (2013). Genetically dictated change in host mucus carbohydrate landscape exerts a diet-dependent effect on the gut microbiota. Proc Natl Acad Sci USA 110: 17059–17064.
Kinnison M, Hendry A . (2001). The pace of modern life II: from rates of contemporary microevolution to pattern and process. In: Hendry AP, Kinnison MT, (eds) Microevolution Rate, Pattern, Process. Springer: The Netherlands, pp 145–164.
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotech 31: 814–821.
Lee YK, Mazmanian SK . (2010). Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330: 1768–1773.
Legendre P, Anderson MJ . (1999). Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monographs 69: 1–24.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI . (2006). Human gut microbes associated with obesity. Nature 444: 1022–1023.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS et al. (2008). Evolution of mammals and their gut microbes. Science 320: 1647–1651.
Linnenbrink M, Wang J, Hardouin EA, Künzel S, Metzler D, Baines JF . (2013). The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Mol Ecol 22: 1904–1916.
Loudon AH, Woodhams DC, Parfrey LW, Archer H, Knight R, McKenzie V et al. (2014). Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). ISME J 8: 830–840.
Lozupone C, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Magurran AE . (2005) Evolutionary Ecology: The Trinidadian Guppy. Oxford University Press: New York.
McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci USA 110: 3229–3236.
Mountfort DO, Campbell J, Clements KD . (2002). Hindgut fermentation in three species of marine herbivorous fish. Appl Environ Microbiol 68: 1374–1380.
Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332: 970–974.
Navarrete P, Magne F, Araneda C, Fuentes P, Barros L, Opazo R et al. (2012). PCR-TTGE analysis of 16S rRNA from Rainbow Trout (Oncorhynchus mykiss) gut microbiota reveals host-specific communities of active bacteria. PLoS One 7: e31335.
Nayak SK . (2010). Role of gastrointestinal microbiota in fish. Aquac Res 41: 1553–1573.
Nelson JS . (2006) Fishes of the World 4th edn. John Wiley & Sons, Inc.: Hoboken, New Jersey.
Nyholm SV, McFall-Ngai M . (2004). The winnowing: establishing the squid-vibrio symbiosis. Nat Rev Micro 2: 632–642.
Ochman H, Worobey M, Kuo C-H, Ndjango J-BN, Peeters M, Hahn BH et al. (2010). Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol 8: e1000546.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB et al. (2013). vegan: Community Ecology Package. R package version 2.0-8. Available at: http://CRAN.R-project.org/package=vegan.
R Development Core Team . (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing V: Austria, Available at: http://www.R-project.org/.
Rajilić-Stojanović M, Heilig HGHJ, Tims S, Zoetendal EG, de Vos WM . (2013). Long-term monitoring of the human intestinal microbiota composition. Environ Microbiol 15: 1146–1159.
Rawls JF, Mahowald MA, Ley RE, Gordon JI . (2006). Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127: 423–433.
Reznick DN . (1983). The structure of guppy life histories: the tradeoff between growth and reproduction. Ecology 64: 862–873.
Reznick DN, Bryga HA . (1996). Life-history evolution in guppies (Poecilia reticulata: Poeciliidae). 5. Genetic basis of parallelism in life histories. Am Nat 147: 339–359.
Reznick DN, Rodd FH, Cardenas M . (1996). Life-history evolution in guppies (Poecilia reticulata: Poeciliidae). IV. Parallelism in life-history phenotypes. Am Nat 147: 319–338.
Reznick DN, Shaw FH, Rodd FH, Shaw RG . (1997). Evaluation of the rate of evolution in natural populations of guppies (Poecilia reticulata). Science 275: 1934–1937.
Reznick DN, Ricklefs RE . (2009). Darwin’s bridge between microevolution and macroevolution. Nature 457: 837–842.
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214.
Ringø E, Strøm E . (1994). Microflora of Arctic charr, Salvelinus alpinus (L.): gastrointestinal microflora of free-living fish and effect of diet and salinity on intestinal microflora. Aquac Res 25: 623–629.
Ringø E, Olsen RE . (1999). The effect of diet on aerobic bacterial flora associated with intestine of Arctic charr (Salvelinus alpinus L.). J Appl Microbiol 86: 22–28.
Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K et al. (2011). Evidence for a core gut microbiota in the zebrafish. ISME J 5: 1595–1608.
Russell JA, Moreau CS, Goldman-Huertas B, Fujiwara M, Lohman DJ, Pierce NE . (2009). Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc Natl Acad Sci USA 106: 21236–21241.
Sakata T, Sugita H, Mitsuoka T, Kakimoto D, Kadota H . (1981). Characteristics of obligate anaerobic bacteria in the intestines of freshwater fish. Bull Jap Soc Sci Fish 47: 421–427.
Sanders JG, Powell S, Kronauer DJC, Vasconcelos HL, Frederickson ME, Pierce NE . (2014). Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol Ecol 23: 1268–1283.
Semova I, Carten JD, Stombaugh J, Mackey LC, Knight R, Farber SA et al. (2012). Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12: 277–288.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504.
Stamatakis A, Ludwig T, Meier H . (2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21: 456–463.
Sugita H, Miyajima C, Deguchi Y . (1991). The vitamin B12-producing ability of the intestinal microflora of freshwater fish. Aquaculture 92: 267–276.
Sullam KE, Essinger SD, Lozupone CA, O’Connor MP, Rosen GL, Knight R et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol 21: 3363–3378.
Sullam KE, Dalton CM, Russell JA, Kilham SS, El-Sabaawi R, German DP et al. (2014). Changes in digestive traits and body nutritional composition accommodate a trophic niche shift in Trinidadian guppies. Oecologia e-pub ahead of print 28 November 2014; doi:10.1007/s00442-014-3158-5.
Torres Dowdall J, Handelsman CA, Ruell EW, Auer SK, Reznick DN, Ghalambor CK . (2012). Fine-scale local adaptation in life histories along a continuous environmental gradient in Trinidadian guppies. Funct Ecol 26: 616–627.
Tremaroli V, Backhed F . (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489: 242–249.
Tsuchiya C, Sakata T, Sugita H . (2008). Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Lett Appl Microbiol 46: 43–48.
Van Soest PJ . (1994) Nutritional Ecology of the Ruminant 2nd edn. Cornell University Press: Ithaca, USA.
Willing E-M, Bentzen P, Van Oosterhout C, Hoffmann M, Cable J, Breden F et al. (2010). Genome-wide single nucleotide polymorphisms reveal population history and adaptive divergence in wild guppies. Mol Ecol 19: 968–984.
Wilson B, Danilowicz B, Meijer W . (2008). The diversity of bacterial communities associated with Atlantic Cod Gadus morhua. Microb Ecol 55: 425–434.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334: 105–108.
Yang Z, Bielawski JP . (2000). Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15: 496–503.
Zandonà E . (2010) The trophic ecology of guppies (Poecilia reticulata) from the streams of Trinidad. Ph.D thesis, Drexel University: Philadelphia.
Zandonà E, Auer SK, Kilham SS, Howard JL, López-Sepulcre A, O’Connor MP et al. (2011). Diet quality and prey selectivity correlate with life histories and predation regime in Trinidadian guppies. Funct Ecol 25: 964–973.
Zilber-Rosenberg I, Rosenberg E . (2008). Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol Rev 32: 723–735.
Zoetendal EG, Akkermans ADL, Akkermans-van Vliet WM, de Visser JAGM, de Vos WM . (2001). The host genotype affects the bacterial community in the human gastronintestinal tract. Microbial Ecol Health Dis 13: 129–134.
Acknowledgements
We acknowledge funding from NSF-Frontiers in Integrative Biological Research grant (DEB-0623632EF) to ASF and an NSF-Doctoral Dissertation Improvement Grant (DEB- 1210695) to JAR, SSK and KES, a Sigma Xi Grant-in-aid to KES and the Betz Chair Endowment for funding this research. We thank Eugenia Zandonà for her invaluable assistance in the field, contribution of knowledge regarding field sites and helpful discussion. We also thank David Reznick and FIBR team for support, Keeley MacNeill for fieldwork assistance, Drew McQuade and Mauri Ren for laboratory assistance and Amy McCune for laboratory space at Cornell University. We thank two anonymous reviewers for their constructive comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Sullam, K., Rubin, B., Dalton, C. et al. Divergence across diet, time and populations rules out parallel evolution in the gut microbiomes of Trinidadian guppies. ISME J 9, 1508–1522 (2015). https://doi.org/10.1038/ismej.2014.231
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.231
This article is cited by
-
Geography and elevation as drivers of cloacal microbiome assemblages of a passerine bird distributed across Sulawesi, Indonesia
Animal Microbiome (2023)
-
The effects of host quantitative genetic architecture on the gut microbiota composition of Chinook salmon (Oncorhynchus tshawytscha)
Heredity (2023)
-
Contrasting patterns of bacterial communities in the rearing water and gut of Penaeus vannamei in response to exogenous glucose addition
Marine Life Science & Technology (2022)
-
Gut Microbiome of Wild Baltic Salmon (Salmo salar L.) Parr
Microbial Ecology (2022)
-
Flexibility and resilience of great tit (Parus major) gut microbiomes to changing diets
Animal Microbiome (2021)
