
Abstract
Dyslexia is one of the most common learning disorders affecting about 5% of all school-aged children. It has been shown that event-related potential measurements reveal differences between dyslexic children and age-matched controls. This holds particularly true for mismatch negativity (MMN), which reflects automatic speech deviance processing and is altered in dyslexic children. We performed a whole-genome association analysis in 200 dyslexic children, focusing on MMN measurements. We identified rs4234898, a marker located on chromosome 4q32.1, to be significantly associated with the late MMN component. This association could be replicated in an independent second sample of 186 dyslexic children, reaching genome-wide significance in the combined sample (P=5.14e−08). We also found an association between the late MMN component and a two-marker haplotype of rs4234898 and rs11100040, one of its neighboring single nucleotide polymorphisms (SNPs). In the combined sample, this marker combination withstands correction for multiple testing (P=6.71e−08). Both SNPs lie in a region devoid of any protein-coding genes; however, they both show significant association with mRNA-expression levels of SLC2A3 on chromosome 12, the predominant facilitative glucose transporter in neurons. Our results suggest a possible trans-regulation effect on SLC2A3, which might lead to glucose deficits in dyslexic children and could explain their attenuated MMN in passive listening tasks.
Similar content being viewed by others


Introduction
Dyslexia is one of the most prevalent learning disorders, affecting about 5–7% of school-aged children. It can have a large impact on the psychosocial development of the affected children, for example resulting in early school drop out or psychiatric disorders as a consequence of chronic experience of school failure.1 Understanding its etiology is a critical step to develop efficient therapeutic strategies and remedial help for dyslexic children, adolescents, and adults.
Molecular genetic work has had a strong impact on the understanding of dyslexia.2 So far, six candidate genes have been identified, four of which have a function in neuronal migration.3 These genes are expressed in brain regions that show functional relevance for cognitive processes related to reading and spelling development.
Investigating brain functions using event-related potentials (ERP) reveals convincing evidence for altered cortical networks and neuro-cognitive functions in dyslexic individuals.4, 5, 6, 7, 8, 9, 10 Recent findings report altered perception of speech sounds in bilateral superior temporal cortices,11 delayed, and reduced activation of brain areas that are activated by word recognition12 or delayed grapheme–phoneme association in the temporal–parietal cortex.5 An important process that has been repeatedly found to be impaired in dyslexic subjects is the perception of speech sounds, and one of the most commonly studied correlates of speech perception is the mismatch negativity (MMN).
The MMN is generated by the automatic response of the brain to any change in auditory stimulation. This process is pre-attentive; hence, no attention is needed when performing the task, and, therefore, a practical method when studying children. In the classical passive oddball paradigm, a standard stimulus is presented with a high probability (at 85%) interspersed by single deviant stimuli with a probability of 15%. The MMN is only generated when a train of standard stimuli (at least 2–3) is presented,13 and the interstimulus interval between standard and deviant is below 12 s. This means that a short-term memory trace is necessary for generating a MMN; however, a long-term memory process when discriminating between speech sounds was also found to be involved.14 A high retest reliability15 and stability16 have also been reported. The MMN is a negative curve, obtained by subtracting the ERP to the standard stimuli from the ERP to the deviant stimuli. This negativity is predominately seen in the fronto-central and central scalp electrodes, peaking at 150–250 ms from change onset.17, 18 This fronto-centrally predominant scalp distribution of the MMN is mainly explained by the sum of generator activity in supratemporal cortices of both hemispheres.19, 20 Experiments using complex stimuli such as speech sounds, as opposed to simpler sounds such as sinus tones, detect a later MMN component, with a latency from 300 to 600 ms8, 9, 10, 21, 22, 23, 24, 25 and a left lateralized topography of the speech MMN in young children, adolescents, and adults.4, 8, 11, 24 In children at risk for dyslexia, a bilateral topography of the late speech MMN was found suggesting lateralization differences of this component in dyslexic children24 and a predictor for word reading in fifth grade children.8 This late MMN component was suggested as a correlate for reduced auditory memory span25 and a more specific correlate for linguistic processing.21
Speech processing depends on the rapid integration of a complex set of acoustic features such as amplitude, frequency, duration, and rapid changes of speech signals. A speech-processing deficit has been found in a large subgroup of dyslexic individuals.4, 6, 9, 10, 26, 27, 28 Kraus et al.26 reported an attenuated MMN to consonant–vowel combinations (/da/-/ga/) in dyslexics. We9 have found an attenuated late MMN (300–600 ms; also referred to as the Late Discriminatory Negativitiy)7, 29 in dyslexic children, aged approximately 12.5 years, to /da/-/ba/ consonant–vowel stimuli. In a subsequent study, we also reported attenuated MMN component for /da/-/ga/ in dyslexic adults.10 Studies including babies at risk investigating speech perception repeatedly found evidence for an altered neurophysiological correlate for dyslexia.30, 31, 32 The fact that the speech MMN is also altered in adult dyslexic individuals10 strengthens the view that the speech MMN is an excellent candidate trait marker for an important perceptual process in dyslexia.
Evidence for investigating the MMN as a potential endophenotype in dyslexia comes from ERP studies in families of schizophrenic patients and from twin studies. Findings that the MMN amplitude is reduced in unaffected family members of schizophrenic patients,33 in children at risk for developing schizophrenia,34 and recent-onset patients35 strengthen the usage of this electrophysiological component as a potential endophenotype for dyslexia research. Furthermore, the heritability of the MMN in healthy populations was found to be high for the peak and mean amplitudes, 0.63 and 0.68, respectively.36 The authors also reported high retest reliabilities for both measures (peak amplitude: 0.67; mean amplitude: 0.66). In conclusion, the MMN can be a very useful endophenotype for research of susceptibility genes in dyslexia and for understanding their functions.
The aim of this study was to identify genetic correlates of the speech-processing deficit in dyslexia. We, therefore, investigated the auditory evoked potentials (speech MMN) in a large sample of 386 dyslexic children, using a genome-wide association-based approach. Our findings suggest a new candidate locus on chromosome 4q32.1 and allow us to formulate a patho-genetic model for neurophysiological endophenotypes of dyslexia.
Materials and methods
Subjects
Probands were recruited at the Departments of Child and Adolescent Psychiatry and Psychotherapy at the Universities in Marburg and Würzburg, Germany. Children with potential difficulties in reading or writing and children who had been diagnosed with dyslexia were referred to the investigators by parents, teachers, special educators, or practitioners. All parents and probands aged over 14 years gave informed written consent for participation in the study. All children participating in the study were of German descent. The ethics committees at each clinical site approved the study.
The proband's spelling ability was selected as inclusion criterion. This inclusion criterion was chosen because both clinical studies on dyslexia in Germany usually use spelling disorder as an inclusion criterion and our earlier findings are also based on this selection criterion (see Refs 37, 38, 39 and 40). In brief, spelling was measured using an age-appropriate spelling test (writing to dictation), and an observed spelling score was calculated on the basis of an assumed correlation between the proband's IQ and spelling of 0.4.38 Children were classified as ‘affected’ when this observed spelling score and the one expected according to IQ showed a discrepancy of ⩾2 s.d. in the initial sample (mean=2.65, s.d.=0.45, range=2.01–4.27) and a discrepancy of ⩾1 s.d. in the replication sample (mean=1.82, s.d.=0.29, range=1.10–2.87).
Families were excluded from the study when either the proband or a sibling presented symptoms of ADHD according to a standardized clinical interview with the mother.41 Furthermore, families were not included if the proband presented a bilingual education, an IQ <85, an uncorrected disorder of peripheral hearing or vision, a psychiatric or neurological disorder possibly having an impact on the development of reading and spelling ability, or an age >21 years.
Our initial study sample consisted of 200 dyslexic children, 150 male and 50 female, aged between 8 and 19 years (mean=12.53, s.d.=2.22). In addition, 186 dyslexic children aged between 8 and 18 years (mean=11.40, s.d.=2.19), 130 male and 56 female, were included in a replication sample. After the initial diagnosis of dyslexia, based on the spelling-discrepancy score, and inclusion in the study, probands were further assessed on their performance in different cognitive aspects of the dyslexia phenotype. A battery of psychometric tests targeted word reading fluency, phoneme awareness, phonological decoding, rapid naming, short-term verbal memory, and orthographic coding, as described earlier.40, 42 Both samples are also characterized by a low word reading (latency and accuracy) ability, by low phonological and orthographic processing, and by an impaired retrieval from the phonological lexicon as measured by a longer latency in rapid word naming. The replication sample, presumably based on the less strict inclusion criteria, is less impaired in comparison to the initial sample in these cognitive phenotypes. The distribution of these phenotypes in the two samples is shown in Supplementary Table 1.
ERP measures
EEG was recorded during a passive oddball paradigm with 1700 standard /da/ and 300 deviant /ba/ synthetic syllables.9, 22 The stimuli were produced using a Klatt synthesizer. Stimulus length was 240 ms and interstimulus interval was 740 ms. In contrast to our earlier MMN studies, we selected a longer interstimulus interval to reliably register the later MMN component in an expected latency window from 300 to 700 ms. Subjects were seated in a comfortable chair in a quiet room. They were specifically instructed not to attend to the presented stimuli, and to aid them in this, a self-selected silent film was shown on a TV monitor.
The EEG was recorded with a Neuroscan amplifier at 250 Hz sampling rate at 29 scalp sites Fp1, Fp2, F7, F3, Fz, F4, F8, FT7, FC3, FCz, FC4, FT8, T3, C3, Cz, C4, T4, TP7, CP3, CP4, TP8, T5, P3, Pz, P4, T6, O1, Oz, O2 plus two additional EOG electrodes below the subjects’ right and left eyes and an additional electrode at right mastoid. The reference electrode was placed at the left mastoid. The EEG was offline referenced to linked mastoids and filtered with a 0.53–40 Hz bandpass using the Brainvision Analyzer (www.brainproducts.com). Eye artifacts were corrected manually using independent component analysis, further artifacts were removed by excluding trials automatically with gradient and max–min criteria. Signals were averaged into epochs of 1100 ms, including a prestimulus baseline of 100 ms. The MMN was calculated as the difference between the averages of deviant and standard trials. Two components were identified through the grand average of the whole sample: MMNa (188–300 ms) and MMNb (300–710 ms) (Figure 1). Peak maximum amplitude in the above time window for MMNa and mean amplitude for MMNb were exported and analyzed with SAS software. For the two MMN components, the mean of nine electrodes over fronto-central sites (F3, Fz, F4, FC3, FCz, FC4, C3, Cz, C4) went into further analysis. The correlation between the two components, MMNa and MMNb, is low (r=0.32 initial sample, r=0.38 replication sample, Bravais–Pearson-correlation coefficient). We did not choose the first component of the difference curve because the first component, although overlapped by the MMN, is an exogenous N1 component.

MMN curves. Grand average of (a) the standard /da/ (dotted line) and deviant /ba/ (dashed line) curves and (b) the MMN demonstrating the MMNa (time window 188–300 ms) and the MMNb (time window 300–710 ms).
SNP selection and genotyping
We performed a whole-genome association analysis in the initial sample using Illumina HumanHap300k Bead Chips. Each chip contained 317.503 genomic markers (single nucleotide polymorphism, SNPs) with a mean spacing of 7.9 kb between neighboring SNPs. Data were analyzed using BeadStudio, a software package provided by the manufacturer. SNPs with either a minor allele frequency <5%, a call rate <98%, or a deviation from Hardy–Weinberg equilibrium (−log10 (P-value) higher than 5 in the exact test) were removed from further analysis. A total of 297.086 SNPs (93.6%) passed these quality criteria and were considered for statistical analysis. As all individuals in the initial sample presented with a genotype call rate of at least 98%, no samples had to be excluded.
In the replication study, candidate SNPs were genotyped using a MALDI-TOF (matrix-assisted-laser desorption/ionization time-of-flight, Sequenom) system. For monetary reasons, replication SNPs as well as SNPs for different projects were multiplexed into four iPlex assays and analyzed on chips in 384-sample format. Genotype data were obtained using SpectroTYPER software v3.1 (Sequenom). To ensure data consistency, the same quality parameters as in the initial sample were administered. Two individuals had a genotype call rate below 98% and were excluded from further analyses. Additional SNPs for haplotype and interaction analysis were also genotyped using Sequenom iPlex reaction. All sequences and protocols are available on request.
Statistical analysis
Statistical association analysis was performed using analysis of variance as implemented in the WG-Permer software, with at least five individuals per genotypic group (http://www.wg-permer.org/), based on the quality criteria described above. Interaction P-values and F-tests for comparison of variances were assessed in R (http://www.r-project.org/). Haplotype tests were performed with PLINK (http://pngu.mgh.harvard.edu/~purcell/plink/).
Expression analysis
Databases
Possible evidence for association of rs4234898 and rs1110040 with regulatory effects on gene expression was checked in three publically available databases: the mRNA-by-SNP-browser (http://www.sph.umich.edu/csg/liang/asthma/) from Dixon et al.43 and Moffat et al.44, the GENEVAR database (http://www.sanger.ac.uk/humgen/genevar/) from Stranger et al.,45 and the data from Myers et al.46 (http://labs.med.miami.edu/myers/LFuN/data.html).
Cell culture
EBV-transformed B-lymphoblastoid cell lines from 17 dyslectic children were grown on RPMI-1640 medium (Biochrom AG, Germany) in the presence of FCS (27%), L-glutamine (2 mM), sodium pyruvate (1 mM), penicillin/streptomycin and amphotericin B. DNA was extracted using Blood&Cell Culture DNA Mini Kit (Qiagen). RNA was isolated three times at three independent time points for each sample, using the RNeasy Micro Kit (Qiagen). In addition, we had access to DNA and RNA extracted from adult blood donor controls. For each sample, quantity and quality for both, RNA and DNA, were assessed on a NanoDrop ND-1000 spectrophotometer. We sequenced genomic DNA to ensure genotypes for rs4234898. PCR products (F: 5′-cgaagaagctggaattttgg-3′, R: 5′-gtgcagacagcaggaatgaa-3′) were purified by AMPure (Agencourt) and subsequently amplified with the BigDye Terminator v3.1 Cyle Sequncing Kit (ABI), using the same primers as for the PCR (3.2 pmol). CleanSEQ-purified products (Agencourt) were analyzed on a capillary sequencer (3130 Genetic Analyzer, Applied Biosystems), and data analysis was performed using ChromasLite 2.0.
For each sample, 500 ng of RNA were used to synthesize cDNA by SuperScript III First-Strand Synthesis for RT–PCR (Invitrogen), using Oligo(dT) primers. cDNA was analyzed on the ABI Prism 7900HT Fast Real-Time PCR System (Applied Biosystems) using a customary designed TaqMan Gene Expression Assay (Applied Biosystems) for SLC2A3 and TaqMan Endogenous Controls Human Cyc (Cyclophilin, 4326316E). We performed the gene-expression assay in a 20 μl reaction volume applying 1 μl cDNA as template. For each of the samples, expression analysis was performed in three independent experiments. Each sample was assayed in triplicates and expression was normalized using cyclophilin. Relative quantities were calculated by the Ct (Cycle threshold) method as described by the manufacturer.
Results
Single-marker analysis
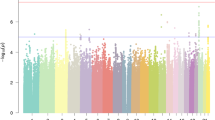
All SNPs that passed our quality control criteria were tested for association with the two MMN components, MMNa and MMNb, in the initial sample. Two association results with MMNa were genome-wide significant after correction for multiple testing (rs1365152: Pnominal=4.27e−08 allelic model, Pcorrected=0.013, rs2114167: Pnominal=1.17e−07 allelic model, Pcorrected=0.035). Both SNPs lie within the genomic locus of CLSTN2 on 3q23. LD between the SNPs is high (D′=1, r2=0.91) indicating that they depict the same effect. MMNb was not associated with any of these two SNPs. None of the other SNPs withstood correction for multiple testing. We identified 17 additional SNPs with nominal P-values <1e−05 with at least one of the two components of the MMN.
A total of 19 SNPs with nominal P-values <1e−05 was taken forward into a replication step. Information on the chosen SNPs is shown in Supplementary Table 2. In the replication sample, only rs4234898 was nominal significantly associated with MMN components, namely MMNb (P=1.46e−03 carrier T model) and withstood correction for testing of 19 SNPs (Pcorrected=0.028). None of the other SNPs were significantly associated in the replication sample (see Table 1).
To increase power, we combined initial and replication samples. For rs4234898 only, a lower P-value in the combined, compared with the initial sample, was obtained. This finding can be regarded as replication.47 In the combined sample, the association P-value for rs4234898 decreased to 1.44e−07 in the allelic and 5.14e−08 in the carrier T model. Both P-values were genome-wide significant (Pcorrected=0.043 allelic model, Pcorrected=0.015 carrier T model). The genotypic distribution for rs4234898 and MMNb is shown in Table 2. In both samples, individuals that are homozygous for the C-allele showed larger MMNb responses than individuals who carried at least one copy of the T allele (see Figure 2). Only 1% of the individuals were homozygous for the T allele. This is in concordance to the CEU individuals included in the HapMap database (http://www.hapmap.org/) in which this frequency is below 1%.

Boxplot for rs4234898 and MMNb in initial and replication sample. Boxplots for the performance in MMNb in relation to rs4234898 genotypes are given in initial (left) and replication (right) sample. For each group, the median (middle black line) and the quantiles (75%, top line; 25%, bottom line) are represented by the rectangle. The dashed vertical lines illustrate the distribution (minimum to maximum) of the performance scores. Outliers, showing >1.5-fold interquartile distance, are represented by a circle.
Multiple-marker analysis
To further explore the effect of rs4234898 on MMNb, we considered haplotypes and interactions with SNPs lying in its vicinity. For this, we chose 40 SNPs, which are located in direct distance to rs4234898 on the Illumina HumanHap300k, namely 20 SNPs in telomeric and 20 SNPs in centromeric direction of rs4234898.
We calculated interaction P-values for two-marker combinations between rs4234898 and each of those SNPs in the initial sample. SNPs with nominal significant interaction P-values were also genotyped and tested for interaction in the replication sample. No consistent, persuading results were found (data not shown).
Haplotypic effects of these SNPs were tested using a sliding window approach. We calculated P-values for all windows including rs4234898, up to a window size of five SNPs. The rs4234898 and rs11100040 formed the most significant haplotype in the initial sample (P=2.79e−06 for TT combination). In the replication sample, the P-value for the TT combination was also significant (P=5.77e−03). In the combined sample, the association P-value for the haplotype TT decreased to P=6.71e−08 (see Supplementary Table 3). Increasing the window size did not improve the significance.
Statistical dependence between rs4234898 and rs11100040 is present, but clearly not complete (r2=0.48 in both samples). In addition, in each of the samples, rs11100040 is significantly associated with MMNb (P=6.0e−04 in initial sample, P=0.03 in replication sample, P=5.31e−05 in the combined sample, allelic model). No significant interaction effect between both SNPs concerning MMNb is present (P=0.57 in initial sample, P=0.76 in replication sample).
Gene expression
Rs4234898 is located in a gene desert on chromosome 4q32.1. To assess possible regulatory effects of rs4234898, we used results from publically available data sets. Dixon et al.43 provide genome-wide Affymetrix-expression data from lymphoblastoid cell lines of 400 children, in correlation to their individual genotypes, assessed on Illumina 300K and Illumina Sentrix Human-1. These data are available online using the corresponding graphical interface ‘mRNA-by-SNP-browser’. In this dataset, probe 202497_x at showed the most significant association (P=1.1e−05) with rs4234898. It was shown that the C-allele of rs4234898 was associated with a higher abundance of the probe in the examined tissue. The 202497_x at detects a transcript of SLC2A3 on chromosome 12p13.3, a brain-expressed member of the facilitative glucose transporter family. Five other probes, also representing partial transcripts of SLC2A3, were also significantly associated with rs4234898, with P-values ranging from 4.7e−05 to 1.5e−04. Overall, seven individual probes for SLC2A3 were available. In addition to these, two combined probes, each averaged over three individual probes, were present and showed significant association with rs4234898 (8.5e−05 and 5.5e−06, respectively).
Those results pointed toward the presence of a trans-regulational effect of rs4234898. However, because of the large number of uncorrected genome-wide data, the findings needed to be replicated. We, therefore, aimed at clarifying the regulatory effects of rs4234898 on SLC2A3 expression in a cell culture experiment using EBV-transformed cell lines prepared from peripheral blood of our dyslexic children. In total, 17 cell lines were included in the functional assay (10 from individuals carrying the T allele CT/TT, 7 were homozygous CC for SNP rs4234898). We measured the abundance of SLC2A3 transcripts in these cell lines using quantitative Real-Time–PCR (TaqMan) and compared SLC2A3-expression levels between the two groups. For each individual, SLC2A3-expression levels measured in three different experiments were available. To be sure that the time point did not have any influence on the measurement, we performed an analysis of variance. We used a factor variable, coding the time point of the measurement, as the independent and measured expression levels as the dependent variable. This yielded a P-value of 0.13, indicating no association between time point and expression level. In the final analysis we, therefore, used the mean value of the three measured expression levels.
In our cell culture experiment, we detected a significant lower expression of SLC2A3 in individuals carrying at least one T allele, in comparison to non-T-carriers (P=0.028 Wilcoxon test). This is in concordance to Dixon et al.,43 in which individuals with at least one T allele showed a lower expression of SLC2A3 compared with individuals who were homozygous for the C-allele. To strengthen our result, we randomly permuted the carrier T-status in the 17 tested individuals and recalculated the P-value. In 226 out of 10 000 permutated data sets, the P-value was lower than in the real data set, resulting in an empirical P-value of 0.023 (95% CI=0.020–0.026). This result indicates that the significant difference in the expression level of SCL2A3 in both groups is a real signal and not a false-positive result.
We hypothesized that other SNPs might also influence expression of SCL2A3 and should also be associated with MMN in our sample. We, therefore, identified all SNPs that were significantly associated with expression of at least two probes for SLC2A3 in the mRNA-by-SNP-browser. These SNPs are listed in Supplementary Table 4. Besides rs4234898, only rs11100040 was significantly associated with different expression of six probes of SLC2A3, whereas all other SNPs showed association in less than six probes. This finding is in concordance with our earlier haplotype analysis, as both SNPs form a significant haplotype block in the association analysis of MMNb. We tested the same haplotype block for association with expression levels of SCL2A3 in our cell lines. Here, we compared individuals with an unambiguous TT haplotype for rs4234898–rs11100040 (n=4) to all other individuals (n=13). Individuals carrying a TT haplotype showed a significant lower expression (P=0.005, one-sided Wilcoxon test). However, as the group of individuals carrying the haplotype TT consisted of only four individuals, we permuted the haplotype TT status and again recalculated the P-values to exclude false-positive findings. A total of 55 of 10 000 permutations yielded better results than the original dataset. The empirical P-value of 0.005 (95% CI=0.0042–0.0073) indicates that SLC2A3 is expressed lower in carriers of the TT haplotype than in other individuals.
The present data suggest that a trans-regulational effect of rs4234898 and rs11100040 on SLC2A3 in dyslexic children might be present. To check whether this trans-regulation is still present in adults, we analyzed the expression levels of SCL2A3 in RNA from blood samples of 37 adult individuals. We could not detect any association of mRNA levels with rs4234898 in these data. A second adult data set45 yielded no positive results either, indicating that our findings might be restricted to children. In a third data set,46 neither rs4234898 nor rs11100040 was available. Other SNPs in the same genomic region show only low LD in the range of 0.01–0.5 to rs4234898 and rs11100040, making it difficult to use these data for confirmation of our results.
Discussion
In earlier studies, we and others found that the late component (300–600 ms) of the speech MMN, here called MMNb, is correlated with dyslexia4, 6, 9, 10, 26, 27 and with a risk for dyslexia.8, 24 MMNb was suggested to be a correlate of reduced auditory memory span, whereas the earlier component, here called MMNa, might index poor detection of differences in speech.25
We tested for association of central auditory speech processing (speech MMN) in a whole-genome data set of 200 dyslexic children. A total of 19 SNPs showing the most significant P-value were replicated in a second sample of 186 children with dyslexia.
The rs4234898 was significantly associated with MMNb in both samples (P=6.20e−06 initial sample, P=1.46e−03 replication sample). The association result in the combined sample withstood genome-wide correction for multiple testing (P=5.14e−08 carrier T model, Pcorrected=0.015). The rs4234898 lies within a genomic desert region, with the nearest genes (CTSO, TDO2, PDGFC) being >150 kb away. None of these genes show evidence for brain-specific expression or function, nor are any of these genes known to be related to neurophysiological phenotypes.
MMNb is a measurement, which is averaged over nine electrodes. To investigate whether the identified effect is only present in this region, we associated the measurement of each of the available 24 electrodes individually with the genotypic distribution of rs4234898. All nine electrodes of the original MMNb measurement were significantly associated with rs4234898, withstanding Bonferroni correction for 24 electrodes. All but one of the measurements for the other electrodes (F8, also a fronto-centrally located electrode) failed to meet this criterion. The identified effect, therefore, seems to be specific for the fronto-central electrodes chosen for MMN analysis. The corresponding p-map is shown in Supplementary Figure 1.
Two genome-wide significant SNPs, both associated with MMNa and in high LD to each other, were also identified. They are located in calsyntenin 2, CLSTN2, a post-synaptic modulator expressed in brain. However, this finding could not be replicated in our second, independent sample. On the basis of empirical evidence that severity of impairment has an impact on association results,40 we analyzed this potential influence on the components of the MMN and tested for association between severity of dyslexia, that is difference between expected and observed spelling score, and MMN components. Although no association between severity and MMNb is present (in initial sample P=0.80, in replication sample P=0.918), the degree of impairment has a significant influence on MMNa in the initial sample (P=0.034 in initial sample, P=0.965 in replication sample). We, therefore, conclude that the genome-wide significant effects on MMNa found in the initial sample might only be seen in children who are severely affected with dyslexia. As the children in the replication were less severely affected, the findings on MMNa could not be replicated. Nevertheless, CLSTN2 remains a promising candidate gene also for dyslexia because genetic variation within this gene has been found to be associated with verbal working memory,48 and this cognitive process is repeatedly found to be impaired in dyslexic subjects.49, 50, 51
We, therefore, concentrated on rs4234898 and to further elucidate its function, we checked whether alleles of rs4234898 were associated with significant expression level changes of some other genes, indicating a possible trans-regulatory impact. Expression analysis using public available data based on Dixon et al.43 yielded evidence for such an effect on SLC2A3, the predominant facilitative glucose transporter in neurons.52 We could confirm trans-regulation of SCL2A3 in EBV-transformed blood cell lines from dyslexic children, with individuals carrying at least one T allele showing lower expression of SLC2A3 (P=0.028 one-sided Wilcoxon test).
Apart from rs4234898, also rs11100040 was significantly associated with six probes of SLC2A3 in the data of Dixon et al.43 Interestingly, these two SNPs form a haplotype window, which is associated with MMNb (P=2.8e−06 for TT combination in initial sample, P=5.8e−03 in replication sample, P=6.7e−08 in combined sample). Statistical dependence between these SNPs is present but not complete. There is also no significant interaction effect between these SNPs.
In the EBV-transformed cell lines, individuals carrying the rs423489–rs11100040-TT haplotype also showed lower expression levels than individuals without the TT haplotype (P=0.005 one-sided Wilcoxon test). The two-marker haplotype is more significantly associated with the expression levels of SLC2A3 as compared with rs4234898 alone.
SLC2A3 (also called GLUT3) belongs to the family of facilitative glucose transporters strongly expressed in brain. Its gene product has been shown to be present in neurons and in region of neuropil in which it provides energy for synaptic transmission.53 In a mouse study, Mantych et al.54 presume that GLUT3 has a function in axonal and/or dendritic transport. Owing to the high sequence similarities within the SLC2A family and because of the sequence of the probes, not all of the probes used within Dixon et al.43 are exclusively binding at SLC2A3, instead they might also detect transcripts of other genes of the SCL2A group. Therefore, in our cell culture experiment, we designed the probe in a 5′ region that is specific for SLC2A3 and distinguishes this specific variant from other members of the family. Given the fact that we see similar effects with this specific probe, our hypothesis that real trans-regulation effects are present is further strengthened.
Although the findings from two studies, Dixon et al.'s43 and ours, provide convincing evidence for a regulatory impact of rs4234898 on SLC2A3 in children, we were not able to show this same effect in adults. Neither the data by Stranger et al.,45 nor the blood samples of 37 non-dyslexic adults yielded evidence for the presence of such an effect. This suggests that the trans-regulational effect of the rs4234898–rs11100040 haplotye is specifically restricted to children. This seems to be a striking finding, and there are indeed several arguments that strengthen this hypothesis of a children-specific effect.
First, it has already been shown in mice that the expression levels of SLC2A3 vary between developmental stages, with low amounts during prenatal phases and a maximum in the first 2 to 3 weeks after birth. Thereafter, mRNA levels of SLC2A3 in mice decline and then stay at a constant level during adulthood.55 Interestingly, the time point of maximal SLC2A3 expression is directly correlated to the stage in which synaptogenesis takes place in the murine brain (for example see Refs 56, 57). In human beings, synaptogenesis begins during embryonic development and continues at high rates until approximately 2 years of age. Thereafter, until adolescence, unused synapses are degraded and the general synaptic density steadily decreases.58, 59 However, the plasticity of synapses is reorganized during that time, with new branches that form and new synaptic connections are established (for example see Ref 60).
Second, studies using positron emission tomography in human beings have shown that the cerebral usage of glucose rises from birth to 4 years of age to a level, which is then maintained until approximately 10 years of age. Reaching age 10 and older, the metabolic rates of glucose usage gradually decline.61 SLC2A3 being the major glucose transporter in neurons, these findings give further support for a trans-regulational effect being limited to human childhood. Summarizing the present data, it can be suggested that cell migration processes such as axon elongation, branching of synapses, or formation of synaptic connections requires an increased demand of glucose in the human brain, and that these processes take place in 4–10-year old children and not in adults. The increased need of energy results in an elevated expression of SLC2A3, which is in concordance with earlier findings.62, 63 The restriction of the trans-regulation effect to the age of young children corresponds also to the neurophysiologic findings of speech perception in dyslexia. Even in 6-month old babies at risk for dyslexia, an altered ERP component elicited by consonant–vowel in a passive oddball paradigm enlarge the risk for later reading and spelling problems.31
The expression analyses conducted in this study or that are available in the databases refer to genetic expression in cells or cell lines that were extracted from blood. It is not clear whether the present direct effect of rs4234898 genotype on SLC2A3 expression levels can be directly applied to the human brain. Recently, the first study correlating genome-wide expression data with individual genome-wide genotypes in post-mortem human cortical brain samples was published.46 Although those data are based on adult samples and we, therefore, did not expect large trans-regulational effects, we checked whether SLC2A3 is differentially expressed in this database. Unfortunately, neither rs4234898 nor rs11100040 are available in this data set because of the different SNP contents of Illumina and Affymetrix chips. However, six other SNPs that are present in this database locate in the vicinity of our two associated SNPs, ranging from 500 bp to 13 kb in distance. The LD between either of these six SNPs and rs4234898 (respectively rs11100040) is very low, (0.001<r2<0.5), making it difficult to transfer findings from these data straightforward to our results.
An interesting aspect in our study is that genetic variation on chromosome 4 (rs4234898) regulates the expression of a gene (SLC2A3) located on chromosome 12. Recent genome-wide data revealed that trans-regulated genes can be found widespread in the human genome; however, their abundance compared with cis-effects is either low or can only be detected in larger samples.43, 46, 64 Several possible mechanisms for trans-effects have been suggested, for example interchromosomal interactions during gene expression. Here, loci or genes on different chromosomes can actually locate close to one another in their three-dimensional space of the nucleus. Therefore, loci and genes linked through trans-regulation might actually be not very far apart considering possible ‘chromosome kissing’.65 This being a direct effect of the two loci, it is also possible that our findings result from an indirect effect between genotype and gene. The rs4234898 could be responsible for a trans-regulational cis-effect by a so far unidentified mediator located on chromosome 4. This mediator might bind to promoter regions of SLC2A3, thus mediating the effect of rs4234898 indirectly. Such an unknown mediator or direct target of the rs4234898 genotype could be very restricted in spatial expression, for example limited to brain structures, which would explain why we did not detect another regulatory effect other than on SLC2A3 in the databases. The regulation of SLC2A3 expression by trans-activators has indeed already been described: Rajakumar et al.66 showed that the nuclear factors Sp1 and Sp3 regulate the transcription of GLUT3 in cultured murine neuroblast cells, and pCREB (phosphorylated cyclic AMP-regulatory element binding) and MSY-1 (mouse Y box-binding protein-1) were additionally identified as activators in vivo and in vitro of GLUT 3 during neurodevelopment.67 In addition, the brain-derived neurotrophic factor BDNF has also been found to increase GLUT3 expression in murine developing cortical neurons.68 BDNF, which is not expressed in blood and could, therefore, not be analyzed in our study, is thus one candidate gene for an indirect effect of rs4234898.
The significance of these findings for dyslexia research could be the identification of a memory-based perception deficit. The MMN depends on a short-term memory trace in the auditory cortex representing the repetitive aspects of the preceding auditory events.17 Thus, the MMN represents the correlate of a passive discrimination process in which the deviant event is found to be incongruent with the memory representation of the preceding stimuli. Interestingly, SLC2A3 is also expressed in the hippocampus52 in which neurons were postulated as functionally relevant for the MMN.69 The underlying neurobiological correlate of this memory-based speech perception deficit could be a reduced local cerebral glucose usage. This hypothesis is supported by animal studies and studies of patients suffering from Alzheimer disease. Liu et al.70 showed that a decreased level of GLUT3 might cause impairments in the glucose uptake and metabolism in the brain of patients suffering from Alzheimer disease.
Children who are homozygous for the C-allele at rs4234898 show a significantly higher variance in the expression of SLC2A3 than children who carry at least one T allele (P=0.0014, one-sided F-test for comparison of variances). The distributions of the expression of SLC2A3 are significantly different between the carrierT and non-carrierT-group (P<2.2e−16, two-sided Kolmogorov–Smirnov test). One might hypothesize that the T allele of rs4234898 keeps SLC2A3 on a uniform expression level, indicating that the gene cannot be regulated actively in these individuals. In adults, the differences in the variability in the expression levels of SLC2A3 is not that high anymore and depicts at most a trend (P=0.07 in the cell lines of healthy adults) or even goes in the different direction (P=0.20 in the Stranger dataset). These data lead to the hypothesis that in processes such as speech perception, which need a high level of glucose, SLC2A3 can possibly not be regulated upward and that this impairment is only present in children. Of course, further experiments will be needed to confirm this hypothesis, and would be ideally conducted in children of different ages.
The question is what consequences such a glucose deficit might have. Suh et al.71 conducted experiments with rats that had been exposed to hypoglycaemia. These animals showed slowing and increased amplitudes in the EEG and paroxysmal activity. Six weeks after hypoglycaemia, these rats had significant learning and memory deficits in a water maze experiment compared with rats that had not been exposed. Langan et al.72 associated hypoglycaemia with cognitive impairment in patients suffering from insulin-treated diabetes mellitus. Patients with five or more hypoglycaemic episodes were significantly slower in their decision and movement time.
These findings suggest that reduced glucose levels might influence the memory performances necessary for speech perception in dyslexic subjects. Children with reduced SLC2A3 levels possibly react slower to the odd stimuli and show a lower amplitude of the speech MMN because of glucose deficits. Larger samples will be needed to strengthen this hypothesis. Hypoglycaemia is a very general symptom, so it is probable that it affects not only MMN, but also other processes, which are based on memory performance and which require high levels of glucose, such as rapid naming. Another important point is that hypoglycaemia might of course not only be regulated by SLC2A3, but also other genes could be involved in this process. It would be interesting to study this in more detail in future experiments.
Taken all data together, our findings provide strong evidence for an association of rs4234898 and an electrophysiological endophenotype, namely MMNb, in dyslexic children. This study gives further evidence for a late MMN component to be relevant for the etiology of dyslexia and might explain whether speech perception in kindergarten children is predictive for later reading and spelling development.8 On a functional level, the locus is suggested to mediate its effect through regulation of the expression of SLC2A3, a member of the glucose transporters expressed in brain. It has been shown that the associated effect is relevant in human childhood, when an increased amount of glucose substrate is needed because of the formation of synaptic connections and branching. To our knowledge, this is the first genome-wide association study showing the interaction of gene expression and a neurophysiological endophenotype in dyslexia.
References
Shaywitz SE, Morris R, Shaywitz BA . The education of dyslexic children from childhood to young adulthood. Annu Rev Psychol 2008; 59: 451–475.
Paracchini S, Steer CD, Buckingham LL, Morris AP, Ring S, Scerri T et al. Association of the KIAA0319 dyslexia susceptibility gene with reading skills in the general population. Am J Psychiatry 2008; 165: 1505–1506.
Galaburda AM, LoTurco J, Ramus F, Fitch RH, Rosen GD . From genes to behaviour in developmental dyslexia. Nat Neurosci 2006; 9: 1213–1217.
Alonso-Bua B, Diaz F, Ferraces MJ . The contribution of AERPs (MMN and LDN) to studying temporal vs linguistic processing deficits in children with reading difficulties. Int J Psychophysiol 2006; 59: 59–167.
Bonte ML, Blomert L . Developmental dyslexia: ERP correlates of anomalous phonological processing during spoken word recognition. Brain Res Cogn Brain Res 2004; 21: 360–376.
Corbera S, Escera C, Artigas J . Impaired duration mismatch negativity in developmental dyslexia. Neuroreport 2006; 17: 1051–1055.
Korpilahti P . Electrophysiological correlates of auditory perception in normal and language impaired children, Thesis by Korpilahti P University of Turku: Turku, Finland, 1996.
Maurer U, Bucher K, Brem S, Benz R, Kranz F, Schulz E et al. Neurophysiology in preschool improves behavioral prediction of reading ability throughout primary school. Biol Psychiat 2009; 66: 341–348.
Schulte-Körne G, Deimel W, Bartling J, Remschmidt H . Auditory processing and dyslexia: evidence for a specific speech processing deficit. Neuroreport 1998; 9: 337–340.
Schulte-Körne G, Deimel W, Bartling J, Remschmidt H . Speech perception deficit in dyslexic adults as measured by mismatch negativity (MMN). Int J Psychophysiol 2001; 40: 77–87.
Hommet C, Vidal J, Roux S, Blanc R, Barthez MA, De Becque B et al. Topography of syllable change-detection electrophysiological indices in children and adults with reading disabilities. Neuropsychologia 2009; 47: 761–770.
Schulte-Körne G, Deimel W, Bartling J, Remschmidt H . Neurophysiological correlates of word recognition in dyslexia. J Neural Transm 2004; 111: 971–985.
Sams M, Paavilainen P, Alho K, Näätänen R . Auditory frequency discrimination and event-related potentials. Electroencephalogr Clin Neurophysiol 1985; 62: 437–448.
Näätänen R, Lehtokoski A, Lennes M, Cheour M, Huotilainen M, Iivonen A et al. Language-specific phoneme representations revealed by electric and magnetic brain responses. Nature 1997; 385: 432–434.
Light GA, Braff DL . Stability of mismatch negativity deficits and their relationship to functional impairments in chronic schizophrenia. Am J Psychiatry 2005; 162: 1741–1743.
Kathmann N, Frodl-Bauch T, Hegerl U . Stability of the mismatch negativity under different stimulus and attention conditions. Clin Neurophysiol 1999; 110: 317–323.
Näätänen R, Paavilainen P, Rinne T, Alho K . The mismatch negativity (MMN) in basic research of central auditory processing: a review. Clin Neurophysiol 2007; 118: 2544–2590.
Tiitinen H, May P, Reinikainen K, Näätänen R . Attentive novelty detection in humans is governed by pre-attentive sensory memory. Nature 1994; 372: 90–92.
Scherg M, Vajsar J, Picton TW . A source analysis of the late human auditory evoked potentials. J Cogn Neurosci 1989; 1: 336–355.
Jemel B, Achenbach C, Müller BW, Röpcke B, Oades RD . Mismatch negativity results from bilateral asymmetric dipole sources in the frontal and temporal lobes. Brain Topogr 2002; 15: 13–27.
Korpilahti P, Lang L, Aaltonen O . Is there a late-latency mismatch negativity (MMN) component? Electroencephalogr Clin Neurophysiol 1995; 95: 96.
Schulte-Körne G, Deimel W, Bartling J, Remschmidt H . The role of phonological awareness, speech perception, and auditory temporal processing for dyslexia. ECAP Suppl 1999; 8: 28–34.
Schulte-Körne G, Deimel W, Bartling J, Remschmidt H . Pre-attentive processing of auditory patterns in dyslexic human subjects. Neurosci Lett 1999; 276: 41–44.
Maurer U, Bucher K, Brem S, Brandeis D . Altered responses to tone and phoneme mismatch in kindergartners at familial dyslexia risk. Neuroreport 2003; 14: 2245–2250.
Stoodley CJ, Hill PR, Stein JF, Bishop DV . Auditory event-related potentials differ in dyslexics even when auditory psychophysical performance is normal. Brain Res 2006; 1121: 190–199.
Kraus N, McGee TJ, Carrell TD, Zecker SG, Nicol TG, Koch DB . Auditory neurophysiologic responses and discrimination deficits in children with learning problems. Science 1996; 273: 971–973.
Lachmann T, Berti S, Kujala T, Schröger E . Diagnostic subgroups of developmental dyslexia have different deficits in neural processing of tones and phonemes. Int J Psychophysiol 2005; 56: 105–120.
Sharma M, Purdy SC, Newall P, Wheldall K, Beaman R, Dillon H . Electrophysiological and behavioural evidence of auditory processing deficits in children with reading disorder. Clin Neurophysiol 2006; 117: 1130–1144.
Ceponiene R, Yaguchi K, Shestakova A, Alku P, Suominen K, Näätänen R . Sound complexity and ‘speechness’ effects on pre-attentive auditory discrimination in children. Int J Psychophysiol 2002; 43: 199–211.
Molfese VJ, Molfese DL, Modgline AA . Newborn and preschool predictors of second-grade reading scores: an evaluation of categorical and continuous scores. J Learn Disabil 2001; 34: 545–554.
Guttorm TK, Leppänen PH, Poikkeus AM, Eklund KM, Lyytinen P, Lyytinen H . Brain event-related potentials (ERPs) measured at birth predict later language development in children with and without familial risk for dyslexia. Cortex 2005; 41: 291–303.
van Leeuwen T, Been P, Kuijpers C, Zwarts F, Maassen B, van der Leij A . Mismatch response is absent in 2-month-old infants at risk for dyslexia. Neuroreport 2006; 17: 351–355. Erratum in: Neuroreport 2006 17: 1755.
Jessen F, Fries T, Kucharski C, Nishimura T, Hoenig K, Maier W et al. Amplitude reduction of the mismatch negativity in first-degree relatives of patients with schizophrenia. Neurosci Lett 2001; 309: 185–188.
Schreiber H, Stolz-Born G, Kornhuber HH, Born J . Event related potential correlates of impaired selective attention in children at high risk for schizophrenia. Biol Psychiat 1992; 32: 634–651.
Umbricht DS, Bates JA, Lieberman JA, Kane JM, Javitt DC . Electrophysiological indices of automatic and controlled auditory information processing in first-episode, recent-onset and chronic schizophrenia. Biol Psychiat 2006; 59: 762–772.
Hall MH, Schulze K, Rijsdijk F, Picchioni M, Ettinger U, Bramon E et al. Heritability and reliability of P300, P50 and duration mismatch negativity. Behav Genet 2006; 36: 845–857.
Schulte-Körne G, Grimm T, Nöthen MM, Müller-Myhsok B, Chichon S, Vogt IR et al. Evidence for linkage of spelling disability to chromosome 15. Am J Hum Genet 1998; 63: 269–282.
Schulte-Körne G, Deimel W, Remschmidt H . Diagnosis of reading and spelling disorder. Z Kinder Jugendpsychiatr Psychother 2001; 29: 113–116.
Ziegler A, König IR, Deimel W, Plume E, Nöthen MM, Propping P et al. Developmental dyslexia-recurrence risk estimates from a german bi-center study using the single proband sib pair design. Hum Hered 2005; 59: 136–143.
Schumacher J, Anthoni H, Dahdouh F, König IR, Hillmer AM, Kluck N et al. Strong evidence of DCDC2 as a susceptibility gene for dyslexia. Am J Hum Genet 2006; 78: 52–62.
Unnewehr S, Schneider S, Margraf J . Kinder-DIPS: Diagnostisches Interview bei Psychischen Störungen von Kindern und Jugendlichen. Springer: Berlin, Germany, 1998.
Schulte-Körne G, Ziegler A, Deimel W, Schumacher J, Plume E, Bachmann C et al. Interrelationship and familiality of dyslexia related quantitative measures. Ann Hum Genet 2007; 71: 160–175.
Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KCC et al. A whole-genome association study of global gene expression. Nat Genet 2007; 39: 1202–1207.
Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S et al. Genetic variants regulating ORMDL3 expression are determinants of susceptibility to childhood asthma. Nature 2007; 448: 470–473.
Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C et al. Population genomics of human gene expression. Nat Genet 2007; 39: 1217–1224.
Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L et al. A survey of genetic human cortical gene expression. Nat Genet 2007; 39: 1494–1499.
Skol AD, Scott LJ, Abecasis GR, Boehnke M . Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nature 2006; 38: 209–213.
Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV et al. Common Kibra alleles are associated with human memory performance. Science 2006; 314: 475–478.
Vellutino FR, Fletcher JM, Snowling MJ, Scanlon DM . Specific reading disability (dyslexia): what have we learned in the past four decades. J Child Psychol Psyc 2004; 45: 2–40.
Pickering SJ . Working memory in dyslexia. In: Alloway TP, Gathercole SE (eds). Working Memory and Neurodevelopmental Disorders. Psychology Press: Hove, UK, 2006, pp 7–40.
Swanson HL . Working memory and reading disabilities: both phonological and executive processing deficits are important. In: Alloway TP, Gathercole SE (eds). Working Memory and Neurodevelopmental Disorders. Psychological Press: Hove, UK, 2006, pp 59–88.
Maher F, Vannucci SJ, Simpson IA . Glucose transporter proteins in brain. FASEB J 1994; 8: 1003–1011.
McCall A, Van Bueren AM, Moholt-Siebert M, Cherry NJ, Woodward WR . Immunohistochemical localization of the neuron-specific glucose transporter (GLUT3) to neuropil in adult rat brain. Brain Res 1994; 659: 292–297.
Mantych GJ, James DE, Chung HD, Devaskar SU . Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology 1992; 131: 1270–1278.
Khan JY, Rajakumar RA, McKnight RA, Devaskar UP, Devaskar SU . Developmental regulation of genes mediating murine brain glucose uptake. Am J Physiol Regul Inter Comp Physiol 1999; 276: 892–900.
Aghajanian GK, Bloom FE . The formation of synaptic junctions in developing rat brain: a quantitative electron microscopic study. Brain Res 1967; 6: 716–727.
Sachs GM, Jacobson M, Caviness Jr VS . Postnatal changes in arborization patterns of murine retinocollicular axons. J Comp Neurol 1986; 246: 395–408.
Huttenlocher PR . Synaptic density in human frontal cortex—developmental changes and effects of aging. Brain Res 1979; 163: 195–205.
Huttenlocher PR, de Courten C . The development of synapses in striate cortex of man. Hum Neurobiol 1987; 6: 1–9.
Johnston MV . Brain plasticity in paediatric neurology. Eur J Paed Neur 2003; 7: 105–113.
Chugani HT . A critical period of brain development: studies of cerebral glucose utilization with PET. Prev Med 1998; 27: 184–188.
Deza L, Eidelberg E . Development of cortical electrical activity in the rat. Exp Neurol 1967; 17: 425–438.
Maher F, Simpson IA . Modulation of expression of glucose transporters, Glut 3 and Glut 1, by potassium and N-methyl-D-aspartate in cultured cerebellar granule neurons. Mol Cell Neurosci 1994; 5: 369–375.
Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT . Mapping determinants of human gene expression by regional and genome-wide association. Nature 2005; 43: 1365–1369.
Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA . Interchromosomal associations between alternatively expressed loci. Nature 2005; 435: 637–645.
Rajakumar RA, Thamotharan S, Menon RK, Devaskar SU . Sp1 and Sp3 regulate transcriptional activity of the facilitative glucose transporter isoform-3 gene in mammalian neuroblasts and trophoblasts. J Biol Chem 1998; 273: 27474–27483.
Rajakumar A, Thamotharan S, Raychaudhuri N, Menon RK, Devaskar SU . Trans-activators regulating neuronal glucose transporter isoform-3 gene expression in mammalian neurons. J Biol Chem 2004; 279: 26768–26779.
Burkhalter J, Fiumelli H, Allaman I, Chatton JY, Martin JL . Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J Neurosci 2003; 23: 8212–8220.
Rosburg T, Trautner P, Ludowig E, Schaller C, Kurthen M, Elger CE . Hippocampal event-related potentials to tone duration deviance in a passive oddball paradigm in humans. Neuroimage 2007; 37: 274–281.
Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX . Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 2008; 582: 359–364.
Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, Gum E et al. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci 2003; 23: 10681–10690.
Langan SJ, Deary IJ, Hepburn DA, Frier BM . Cumulative cognitive impairment following recurrent severe hypoglycemia in adult patient with insulin-treated diabetes mellitus. Diabetologia 1991; 34: 337–344.
Acknowledgements
We thank the children and their families for participating in the study. We also thank anonymous reviewers for their helpful comments. GSK, AW, HR, BMM, and MMN were supported by the Deutsche Forschungsgemeinschaft. GSK, BMM, MMN were funded by the EU in the Sixth Framework Program, LifeScienceHealth, project title Dyslexia genes and neurobiological pathways (Neurodys, 018696). MMN received support for this work from the Alfried Krupp von Bohlen und Halbach-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Rights and permissions
About this article
Cite this article
Roeske, D., Ludwig, K., Neuhoff, N. et al. First genome-wide association scan on neurophysiological endophenotypes points to trans-regulation effects on SLC2A3 in dyslexic children. Mol Psychiatry 16, 97–107 (2011). https://doi.org/10.1038/mp.2009.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2009.102
Keywords
This article is cited by
-
Brain structure, phenotypic and genetic correlates of reading performance
Nature Human Behaviour (2023)
-
Genome-wide association scan identifies new variants associated with a cognitive predictor of dyslexia
Translational Psychiatry (2019)
-
Effective instruction for persisting dyslexia in upper grades: Adding hope stories and computer coding to explicit literacy instruction
Education and Information Technologies (2018)
-
Neurogenetics of developmental dyslexia: from genes to behavior through brain neuroimaging and cognitive and sensorial mechanisms
Translational Psychiatry (2017)
-
Descriptive epidemiology of prenatal and perinatal risk factors in a Chinese population with reading disorder
Scientific Reports (2016)

{kind=link}