
Abstract
Cutaneous malignant melanoma is a highly aggressive and frequently chemoresistant cancer, the incidence of which continues to rise. Epidemiological studies show that the major aetiological melanoma risk factor is ultraviolet (UV) solar radiation, with the highest risk associated with intermittent burning doses, especially during childhood1,2. We have experimentally validated these epidemiological findings using the hepatocyte growth factor/scatter factor transgenic mouse model, which develops lesions in stages highly reminiscent of human melanoma with respect to biological, genetic and aetiological criteria, but only when irradiated as neonatal pups with UVB, not UVA3,4. However, the mechanisms underlying UVB-initiated, neonatal-specific melanomagenesis remain largely unknown. Here we introduce a mouse model permitting fluorescence-aided melanocyte imaging and isolation following in vivo UV irradiation. We use expression profiling to show that activated neonatal skin melanocytes isolated following a melanomagenic UVB dose bear a distinct, persistent interferon response signature, including genes associated with immunoevasion. UVB-induced melanocyte activation, characterized by aberrant growth and migration, was abolished by antibody-mediated systemic blockade of interferon-γ (IFN-γ), but not type-I interferons. IFN-γ was produced by macrophages recruited to neonatal skin by UVB-induced ligands to the chemokine receptor Ccr2. Admixed recruited skin macrophages enhanced transplanted melanoma growth by inhibiting apoptosis; notably, IFN-γ blockade abolished macrophage-enhanced melanoma growth and survival. IFN-γ-producing macrophages were also identified in 70% of human melanomas examined. Our data reveal an unanticipated role for IFN-γ in promoting melanocytic cell survival/immunoevasion, identifying a novel candidate therapeutic target for a subset of melanoma patients.
Similar content being viewed by others


Main
Mechanisms associated with UV-mediated alterations to melanocytes and their microenvironment have been inscrutable because they cannot be adequately studied in cultured cells. Moreover, melanocytes represent only ∼1% of skin cells, and bear few specific cell-surface markers permitting efficient isolation. To enable detailed study of melanocyte biology in vivo, we generated a mouse model in which expression of the reverse tetracycline-activated transactivator rtTA2s-M2, characterized by minimal leakiness and background, was regulated by the melanocyte-specific dopachrome tautomerase (Dct) gene promoter (Supplementary Fig. 1a). Dct-rtTA mice bred with transgenic mice bearing a histone H2B–GFP fusion construct controlled by the tetracycline response element (TRE) created Dct-rtTA/TRE-H2B–GFP bi-transgenic mice (hereafter iDct–GFP) (Supplementary Fig. 1b). iDct–GFP mice showed an inducible GFP profile from embryonic to adult stages consistent with known Dct expression patterns. GFP expression was observed in embryonic neural crest, retinal pigment epithelium and telencephalon, as expected (Fig. 1a and Supplementary Fig. 2). Neonatal and adult skin GFP+ cells were strictly localized to hair follicles, where most GFP+ cells were in bulb regions, with smaller numbers in the outer root sheath and bulge regions, harbouring melanocyte precursors5 (Fig. 1b). Co-localization of GFP and anti-Dct antibody by immunohistochemistry unequivocally identified GFP+ cells as melanocytes (Fig. 1c). No background GFP expression was detectable without doxycycline. Full GFP induction was achieved within 12–18 h of a single intraperitoneal injection of a non-toxic doxycycline dose in neonatal or adult mice (Supplementary Fig. 1c).

a, Embryonic day 11.5 iDct–GFP embryo showing GFP+ cells in neural crest (red arrow) and telencephalon (white arrow); magnification, ×10. b, In 7-day old pup skin GFP+ cells are located in the bulb (lower arrow) and bulge (upper arrow) regions of hair follicles. Blue, 4′,6-diamidino-2-phenylindole (DAPI); magnification, ×40. D, dermis; E, epidermis. c, Immunohistochemistry with anti-Dct antibody shows co-localization with GFP in iDct–GFP skin; magnification, ×100. d, UVB-induced activation of melanocytes, characterized by proliferation and migration towards epidermis. Dorsal skins were examined at 1 day (at age PD2) and 6 days (PD7) after irradiation. Scale bars, 40 µm. e, Schematic of the regime for isolating GFP+ melanocytes by FACS. Pups are irradiated at PD1, and dorsal skins harvested at either PD2 (24 h post-UV) or PD7 (6 days post-UV). Doxycycline (Dox) injections are always given after irradiation, 24 h before skin harvest.
Reasoning that new clues to the molecular mechanism(s) underlying UV-induced melanomagenesis would be found within the genomic response of melanocytes to UV radiation, we used the iDct–GFP mouse to examine the responses to UVB versus UVA of melanocytes residing in situ, within their natural morphological and physiological microenvironment. Precisely defined wideband wavelengths and physiologically relevant doses4 (see Methods) of UV (Supplementary Fig. 3) were used to irradiate postnatal day 1 (PD1) iDct–GFP mice, and skins were harvested at certain time points after irradiation. To avoid potential toxicity from chronic expression and interference with UV absorption, GFP was doxycycline-induced after UV irradiation, 24 h before skin harvest. GFP+ melanocytes were phenotypically activated, characterized by raised melanocyte numbers and migration towards the epidermis as previously reported6, 24–48 h after exposure to UVB, but not UVA, peaking at 3 days and lasting at least 10 days after irradiation (Fig. 1d and Supplementary Fig. 4a). UVB-induced melanocyte activation was specific to neonatal irradiation; adult mice irradiated at PD29 did not show this response (Supplementary Fig. 4b).
We performed an expression microarray study on melanocytes isolated from dorsal skin of iDct–GFP pups irradiated at PD1 with UVB or UVA (Fig. 1e). Doxycycline-induced GFP-labelled melanocytes were isolated via fluorescence-activated cell sorting (FACS) at 1 day and 6 days after irradiation (ages PD2 and PD7, respectively); arrays from 1 day post-UV would reflect the acute UV stress response of in vivo melanocytes, whereas the 6 days post-UV time point should uncover responses persisting after the acute stress response subsides. FACS isolation consistently yielded >95% melanocyte enrichment (Supplementary Fig. 5). Gene expression profiling produced robust data with good reproducibility among biological triplicates (Fig. 2a), and confirmed the absence of detectable levels of contaminating skin cell types, including keratinocytes, fibroblasts and adipocytes (Supplementary Fig. 6).

a, Unsupervised clustering of complementary DNA (cDNA) microarray analysis of gene expression in FACS-sorted melanocytes from 1 day (PD2) or 6 days (PD7) after UVB or UVA irradiation, and respective unirradiated controls. The expanded heatmap (right) shows the delayed induced gene subset, which includes multiple genes known to be induced by IFN-γ. Primary mouse keratinocytes (PrKC) were included as controls. All groups included biological triplicates. b, qRT–PCR validation of expression of 4 genes (n = 3 samples each) from IFN signature (error bars = s.e.m.). c, Antibody-mediated blockade of interferons by treating pups with intraperitoneal injections of anti-IFN-αR1, anti-IFN-γ, or both in combination, 1 h before and 3 days after UVB irradiation at PD1. The dorsal skins were harvested (n = 3 each group) and analysed for melanocyte activation. Representative images are shown. D, dermis; E, epidermis. Scale bars, 18 µm.
UVB elicited a potent, transient, acute stress response in melanocytes, including increased expression of p53 target genes (for example, p21Waf1/Cip1 , cyclin G1 and reprimo), whereas UVA-associated changes were subtle (Fig. 2a and Supplementary Fig. 7). Intriguingly, a small subset of genes showed a delayed response evident at 6 days after UVB exposure (Fig. 2a), including a putative IFN-responsive gene signature7 (Fig. 2a and Supplementary Table 1). Four upregulated genes (Ccl8, Ctla4, H2-K1 and H2-T23) from this group were validated by quantitative reverse transcription–polymerase chain reaction (qRT–PCR) (Fig. 2b). The response was neonate specific (Supplementary Fig. 8), and included genes implicated in conferring immunoevasiveness (that is, Ctla4, H2-T23, H2-M3, Cfb (also known as H2-Bf) and C4a (also known as Slp)).
To determine whether IFN signalling has a biologically significant role in UVB-induced melanocyte activation, we blocked both type-I (IFN-α and IFN-β) and type-II (IFN-γ) interferons by neonatal administration of anti-IFN-α receptor 1 (IFN-αR1) and anti-IFN-γ antibodies, respectively. Melanocytes from 6-day post-UVB skin were activated in the presence of isotype control antibody, whereas the anti-IFN-αR1+anti-IFN-γ antibody combination completely abolished this response (Fig. 2c). Moreover, although antibody-mediated blockade of type-I IFN-α/β signalling alone failed to overtly affect UVB-mediated activation, blockade of IFN-γ alone markedly inhibited this response (Fig. 2c). These results were corroborated by flow cytometric quantification of GFP+ skin cells from each antibody-treated group (Supplementary Fig. 9). We next isolated GFP+ melanocytes from UV-irradiated neonates blocked with either anti-IFN-γ or anti-IFN-αR1, and compared their expression patterns to control. Anti-IFN-γ and anti-IFN-αR1 antibodies repressed expression of a common gene set associated with the IFN response (Supplementary Fig. 10). However, IFN-γ inhibition also more potently repressed expression of several non-classical major histocompatibility complex (MHC) class Ib antigens (for example, H2-T23 and H2-Q2), as well as Psmb9, Gbp2, Icam, Irf1 and Fosb; members of this gene subset should be responsible for the observed melanocytic phenotypes. Notably, IFN-γ blockade exclusively and potently inhibited expression of chemokine Ccl8 (also known as MCP-2).
We determined the IFN-γ source by interrogating 6-day post-UVB-irradiated skin for immune-cell infiltration. Immunohistochemistry failed to detect T cells, B cells, dendritic cells, natural killer (NK), or NK-T cells; however, CD11b+ cells of myeloid origin were evident (Supplementary Fig. 11). Anti-F4/80 and anti-Gr-1 antibodies identified these as macrophages (F4/80+Gr-1−), not neutrophils (Fig. 3a, upper panel). Nearly 90% of CD11b+ cells were also F4/80+ (Supplementary Fig. 12). Notably, the adult response was distinct; dorsal skins from PD35 mice 2 days after UVB showed predominant Gr-1+ cell infiltration, and minimal F4/80+ cells (Fig. 3a, lower panel), as reported8.

a, Immunohistochemistry with anti-F4/80 and anti-Gr-1 antibodies in dorsal skins of UVB-irradiated and unirradiated neonatal (upper panel) and adult (lower panel) mice. Scale bars, 40 µm. b, qRT–PCR for IFN-α, IFN-β and IFN-γ expression in F4/80+ and CD11b+ cells FACS-isolated from neonatal dorsal skins UVB-irradiated at PD1 and examined at PD7, compared with non-activated RAW264.7 macrophages. c, Flow cytometric analysis of PD1 UVB-irradiated (PD7 examined) skin-cell-suspension-identified IFN-γ+ macrophages. PE, phycoerythrin. d, Flow cytometric analysis of macrophage (F4/80+) infiltration into skin, 2 days after UVB irradiation in Ccr2-deficient pups (irradiated at PD1), as compared to wild-type pups. **P < 0.01; one-way ANOVA test with post-hoc Tukey analysis. NS, not significant. e, F5061 melanoma cells ectopically expressing Ccl8 chemoattract F4/80+ macrophages (red), but not Gr-1+ cells, to sites of subcutaneous inoculation in syngeneic FVB/N mice. One vector-transfected and two Ccl8-transfected clone cells were used. Blue, DAPI.
To determine if infiltrating macrophages expressed IFN-γ, as has been suggested9, type-I and type-II interferon mRNAs were quantified from FACS-purified CD11b+ and F4/80+ cells. Both had upregulated IFN-α and IFN-γ expression, and to a lesser extent IFN-β (Fig. 3b). Flow cytometry demonstrated that 28% of both CD11b+ and F4/80+ cells expressed IFN-γ (Fig. 3c). Our data support the notion that UVB-recruited, infiltrating macrophages secrete IFN-γ, inducing the IFN signature detected in activated melanocytes. Although we were unable to detect NK cell markers in FACS-isolated CD11b+ cells by qRT–PCR (Supplementary Fig. 13), we cannot rule out a possible contribution by undetectably low numbers of NK or other inflammatory cells.
That UVB-induced chemokines were responsible for neonatal macrophage recruitment, particularly Ccr2/Ccr5 ligands, was indicated by our melanocyte microarray data and confirmed by qRT–PCR (Supplementary Fig. 14a, b). These results were further corroborated through treatment of cultured melan-c normal immortalized melanocyte cell line with non-cytocidal UV (Supplementary Fig. 14c). In contrast, upregulation of these chemokines was not detected in UV-irradiated whole skin (Supplementary Fig. 15) or isolated keratinocytes10. Ccr2 and Ccr5 were highly expressed in skin-infiltrating macrophages, but not in non-activated RAW264.7 macrophages (Supplementary Fig. 16). Finally, mice deficient in Ccr2 were significantly inhibited in their ability to recruit F4/80+ macrophages into neonatal skin (Fig. 3d); in contrast, Ccr5-deficient mice showed no significant difference (data not shown).
The arrival of IFN-γ-expressing macrophages coincided with a >100-fold upregulation in melanocyte expression of the Ccr2 ligand Ccl8, a known IFN-γ-response gene11, whereas expression of other Ccr2 ligands had returned to baseline. We propose that recruited IFN-γ+ macrophages enhance melanocyte Ccl8 expression, reinforcing macrophage–melanocyte interactions and fueling an inflammatory positive feedback loop. To confirm its ability to potently chemoattract macrophages, Ccl8 was ectopically expressed in F5061 cells—established from a UV-induced melanoma from an immunocompetent hepatocyte growth factor/scatter factor (HGF/SF)-transgenic mouse. F5061-Ccl8 cells were subcutaneously inoculated into syngeneic FVB/N mice, markedly enhancing macrophage infiltration into the transplantation site (Fig. 3e). Conditioned media from F5061-Ccl8 cells also significantly elevated transmembrane migration of RAW264.7 macrophages (Supplementary Fig. 17).
Macrophages show either anti- or pro-tumorigenic properties12. F4/80+ macrophages were isolated from 6-day post-UVB neonatal skin, admixed at a ratio of 1:5 with F5061 melanoma cells and transplanted subcutaneously into FVB/N mice. Admixed transplants showed significantly increased growth relative to controls (Fig. 4a and Supplementary Fig. 18), indicating that these activated macrophages were pro-tumorigenic. In contrast, macrophages isolated from spleens of unirradiated control pups did not affect tumour growth (Supplementary Fig. 19). Ki-67 immunohistochemistry showed no difference in proliferation between admixed tumours versus controls (Supplementary Fig. 20); however, TdT-mediated dUTP nick end labelling (TUNEL) assays revealed significantly less apoptosis in admixed tumours (Fig. 4b and Supplementary Fig. 21), demonstrating that UVB-recruited macrophages promote melanoma cell survival. Immunohistochemistry confirmed that a subset of tumour-associated macrophages maintained IFN-γ expression (Supplementary Fig. 22). Moreover, macrophage presence strongly correlated with enhanced Ctla4 expression in F5061 melanoma cells (Supplementary Fig. 23), recapitulating the functional consequence of macrophage infiltration observed in neonatal skin melanocytes (Fig. 2a, b).

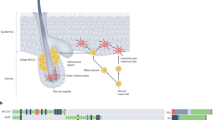
a, Mean volumes (± s.e.m.) of F5061 melanomas admixed with skin macrophages isolated from PD1 UVB-irradiated (PD7-examined) pups (n = 10), versus F5061-only controls (n = 9), following subcutaneous transplantation in syngeneic FVB/N mice. **P < 0.01. b, Percentages of TUNEL plus cells (± s.e.m.) in admixed and control tumours (n = 6 each). ***P < 0.001. c, Antibody-mediated blockade of IFN-γ significantly inhibits pro-tumorigenic effects of macrophages. Cntrl Ab, control antibody. *P < 0.05; **P < 0.01; NS, not significant. One-Way ANOVA test with post-hoc Tukey analysis. d, Dual immunohistochemistry with anti-IFN-γ (red) and anti-CD68 (green) antibodies on a human melanoma tissue microarray shows IFN-γ-expressing macrophages (yellow). Representative tumour is shown. e, Schematic representation of the UVB-induced inflammatory cascade leading to IFN-γ-mediated immunoevasion and survival of melanocytes during sunburn-associated remodelling. UV induces release of CCR2 ligands (1); that activate CCR2+ macrophages (2); which are recruited to neonatal skin (3); macrophages secrete IFN-γ (4); which activates melanocytes, inducing expression of genes that include CCL8, fueling inflammation (5); and immunoevasion (6).
Despite its well-documented anti-tumorigenic activity13, IFN-γ has also been implicated as a pro-tumorigenic factor14,15. To determine if macrophage-secreted IFN-γ was responsible for the enhanced melanoma growth observed in admixed tumours, we included intraperitoneal administration of either anti-IFN-γ or control antibodies. Whereas admixed melanomas in the control group showed the expected enhanced growth, those in mice given anti-IFN-γ antibody showed significantly reduced growth (Fig. 4c). Immunophenotyping of tissue microarrays containing UVB-induced mouse melanomas showed that most (66%) were macrophage-rich, with fewer having T cells (59%) and B cells (32%) (Supplementary Fig. 24). To determine if human melanoma-associated macrophages produce IFN-γ, we performed dual immunohistochemistry for CD68 and IFN-γ using human melanoma tissue microarrays. We discovered that 19 of 27 (70%) melanomas examined contained abundant macrophages (CD68+), of which all 19 demonstrated CD68+IFN-γ+ dual positivity (Fig. 4d and Supplementary Fig. 25).
In this study we show that UV incites melanomagenesis not only through DNA mutagenesis, but also by altering interactions between melanocytes and their microenvironment to regulate remodelling of UV-damaged skin. On the basis of our results, a model implicating a neonatal-specific UV-induced pro-melanomagenic inflammatory cascade emerges (Fig. 4e). In accordance with their relative tumorigenicity in albino HGF/SF-transgenic mice4, UVB not UVA induces melanocytic expression of multiple chemoattracting Ccr2 ligands (Ccl2, Ccl7, Ccl8, Ccl12), recruiting Ccr2+ macrophages into neonatal skin. IFN-γ from recruited macrophages stimulates melanocyte proliferation and migration, and the expression of genes implicated in immunoevasion/survival.
Erythemal neonatal UVB causes robust macrophage infiltration and is melanomagenic; adult skin responds with a rapid, short-lived neutrophil influx, but no melanoma. We propose that mechanisms underlying neonatal UVB-induced melanomagenesis operate within the immunoediting paradigm13: UVB-activated mutant neonatal melanocytes—particularly progenitors16—exposed to inflammation evade immune-mediated elimination, persisting through an extended equilibrium phase before evolving into clinically significant melanoma. Macrophage-induced melanocyte proliferation would more efficiently fix UV-induced mutations in prospective melanoma cells, whereas enhanced melanocyte migration could facilitate UVB-associated long-term tolerance to melanocytic antigens17 by promoting aberrant melanocyte–immune-cell interactions. Moreover, enduring inflammation-associated epigenetic alterations occur in transformed cells18, and perhaps in long-lived macrophage subpopulations19, indefinitely extending their biological effects.
Notably, our systemic antibody blockade experiments demonstrating the importance of physiologically relevant IFN-γ in UVB-induced melanocyte activation and melanoma cell survival strongly support the notion that IFN-γ can be pro-tumorigenic as well as anti-tumorigenic, depending on the context, intensity and durability of the IFN-γ signal14. In fact, an association between IFN-γ-induced genes and early mouse melanocytic lesions has been reported15, and serum IFN-γ has been implicated as an independent prognostic indicator for melanoma recurrence20. We propose that IFN-γ-associated survival mechanisms operational in neonatal melanocytes are recapitulated in melanoma, contributing to the selection of more aggressive, therapeutically resistant phenotypes. Relevance to human melanoma is supported by the detection of macrophage-associated IFN-γ expression in most patient samples examined, and a clinical trial showing that IFN-γ may have adverse effects regarding melanoma patient relapse and mortality21. We provide the first evidence, to our knowledge, that IFN-γ-R signalling can facilitate melanoma progression, a remarkable discovery considering that high-dose IFN-α is used to treat melanoma, albeit with limited success22. Non-overlapping functions of type-I and type-II interferons are well described13, and strongly supported by our data.
The IFN ‘survival signature’ associated with UVB-activated mouse melanocytes contains genes involved in human melanoma immunoevasion, including non-classical MHC class Ib antigens (mouse H2-M3/human HLA-G; mouse H2-T23 (also known as Qa-1)/human HLA-E)23,24. HLA-E suppresses NK and cytotoxic T lymphocytes25,26. This IFN signature also features complement isoforms C4a and C4b, implicated in systemic autoimmunity suppression27, and CTLA4, a potent immune evasion facilitator. CTLA4 is also highly upregulated in mouse melanoma cells admixed with neonatal macrophages, and expressed on human melanoma cells, where it may be involved in immune escape28,29.
Here we identify novel cellular/molecular inflammatory mechanisms centred on IFN-γ signalling that may underlie the initiation, survival and/or outgrowth of UVB-induced melanoma cells. We propose that such mechanisms are highly relevant to strategies used by melanoma cells to evade immunosurveillance in patients. In what could prove to be a paradigm shift, our data strongly suggest that IFN-γ/IFN-γ-R or its downstream pathway members represent promising prognostic markers and/or efficacious therapeutic targets in an appropriate subset of melanoma patients.
Methods Summary
All mouse studies were performed under the strict guidelines of Animal Study Protocols approved by the Animal Care and Use Committees at the National Cancer Institute/National Institutes of Health and the George Washington University Medical Center. The Dct-rtTA transgenic mice express rtTA2s-M2 under the control of the Dct (Trp2) gene promoter. Thirty-one founder transgenic mice were generated by standard microinjection techniques, individually crossed with the TRE-H2B–GFP transgenic mice, and screened for a line that produced no leaky background expression of GFP without doxycycline and quickly responded to a single intraperitoneal doxycycline injection. One such line was selected to make a double homozygous Dct-rtTA/TRE-H2B–GFP transgenic mouse line in albino FVB/N background, and males were crossed with wild-type FVB/N females to produce litters that were 100% double heterozygous (iDct–GFP). One day after birth, the pups were irradiated with UV radiation (UVB, UVA, or sham)4 (Supplementary Fig. 3). Dorsal skins were harvested at 24 h and 6 days after irradiation (or sham irradiation). Twenty-four hours before mice were killed and dorsal skins harvested, neonates were injected intraperitoneally with doxycycline at 80 µg g−1 body weight to activate GFP expression. Skins from 6–8 pups of a litter were pooled to prepare single-cell suspensions using a published protocol30, followed by isolation of GFP+ melanocytes via FACS. RNA samples isolated from these melanocytes were subjected to cDNA microarray on Illumina Murine Beadchips v. 2.0 (Illumina). qRT–PCR validation was performed using primers listed in Supplementary Table 2. Immunohistochemical and flow cytometric analyses were performed using antibodies listed in Supplementary Table 3.
Online Methods
Generation of Dct-rtTA and iDct–GFP mice
DNA fragments from the following plasmids were used to construct the plasmid pDct-rtTA-βGlo, which harbours the transgene fragment used to make the Dct-rtTA transgenic mice. Firstly, phβGlobin: the vector backbone is pBluescript II KS+ and the insert is a BamHI/PstI genomic fragment from the human β-globin gene, which is comprised of partial exon 2, full intron 2, full exon 3 and poly A signal. Secondly, pPDct: this plasmid contains the 3.4-kb BamHI/Eco47III fragment from the Dct gene promoter31. Thirdly, pBS/IRES-M2: this plasmid contains the rtTA2s-M2 fragment. It is a variant of pUHrT62-132, and was obtained from C. Toniatti and H. Bujard. The Dct-rtTA transgenic mice were generated using standard microinjection techniques in a FVB/N background strain. Dct-rtTA mice were bred with the TRE-H2B–GFP transgenic mice33 to generate the bi-transgenic iDct–GFP mice.
Mice
Wild-type FVB/N female breeders, Ccr2−/− mice (strain B6.129S4-Ccr2tm1Ifc /J), and Ccr5−/− mice (strain B6.129P2-Ccr5tm1Kuz /J) were obtained from Jackson Laboratories and housed under the strict guidelines of the Institutional Animal Care and Use Committee (IACUC)-approved protocols. Transportation of mice between National Cancer Institute–Frederick and George Washington University Medical Center (GWUMC) animal facilities was performed by special animal courier service approved by IACUC.
In vivo UV irradiation
iDct–GFP pups were irradiated with UV radiation (UVB, UVA, or sham) as previously described4 (Supplementary Fig. 3). The standard erythemal dose (SED) is used to compare the sunburning effectiveness of different UV-emitting sources. These sources include UV-emitting lamps in suntanning beds, welder’s arcs and sunlight among others. By determining the SED one is able to compare how efficient the UV-emitting sources are at inducing sunburn or reddening in skin relative to each other. In experimental UV studies, many laboratories use different types of UV sources for a variety of experiments. Thus, determining the SED allows one to produce equivalent amounts of ‘sunburning’ radiation regardless of the UV spectral output of the different sources used. The SED is produced by multiplying the spectral output or irradiance (W m−2 of the UV-emitting source) with the Commission Internationale de l'Eclairage (CIE) standard erythemal action spectrum (note that 1 W = 1 J s−1). The CIE standard erythemal action spectrum is thus used to ‘weight’ the incoming UV radiation on an erythemal wavelength basis. The product curve produced from this convolution will give the ‘erythemal effective’ irradiance (Werythemal effective m−2) upon integration of the area under this curve. The erythemal effective irradiance is used to calculate the erythemal or sunburning dose by the equation: doseerythemal effective = irradianceerythemal effective × time (s). By definition, 1 SED = 100 J m−2.
Thus, our UVB source, which consists of a UVB bandpass interference filter with a half-band width of ±5 nm (dimensions: 5.1 cm × 5.1 cm) blocked to 10−3 to 10−4 outside the main bandpass (280–320 nm). This custom-made filter when coupled to our 2.5 kW Xenon arc allows for very clean wavelength resolution. The 100 cm2 exposure area is large enough to accommodate up to 12 neonates (1–3 days old), which are situated in a three-chambered animal holder covered with a quartz lid that allows for UV transmission. Holes are drilled into the sides to allow for air exchange. This unit sits on top of a turntable rotating at approximately 3 r.p.m. to average out beam uniformity. The irradiance is measured with a spectroradiometer (StellarNet) and under standard conditions regularly produces an irradiance of approximately 0.30 CIE-effective W m−2. The exposure time is constant at 90 min. These conditions regularly produce a CIE-effective dose of approximately 14–16 SED. This level of CIE-effective irradiance agrees well with our previous calculations using a radiation transfer algorithm over a global latitude/longitude grid at Northern or Southern summer mid-latitudes34. Direct terrestrial measurements at these latitudes indicate that under summer sunlight conditions on clear days in the Northern or Southern Hemisphere between 11:00 and 15:00 local time one can measure SEDs in the range of 10 to 20 depending on exposure conditions. As an example and as part of a project to estimate the number of SEDs one might receive during the day using, in this case, cyclists training in Spain during summer and winter, a recent paper shows just such levels in good agreement with our measurements35.
FACS
Single-cell suspensions from pooled skin samples were prepared using a published protocol30. FACS was performed on a FACSAria flow cytometer (BD Biosciences) equipped with a fixed-alignment cuvette flow cell and a Coherent Sapphire solid state laser providing 13 mW of 488-nm excitation wavelength. Processing rates averaged about 5,000 events per second at 20 psi. Cells of interest were gated by a combination of forward and orthogonal light scatter, and GFP fluorescence was captured in the detector with a 530/30 bandpass filter. GFP-positive cells suspended in PBS plus 1% bovine serum albumin (BSA) were sorted as bulk populations into 12 × 75 mm polypropylene tubes. Dorsal skins from a combined litter of 6–8 pups gave a disaggregated cell suspension containing about 30–50 × 106 cells. FACS resulted in collection of 1–3 × 105 GFP-positive melanocytes per litter. Following FACS collection of cells into PBS plus 1% BSA solution, the cells were centrifuged at 4 °C, and the pelleted cells were lysed in Trizol Reagent (Invitrogen) for isolation of RNA. Total RNA was isolated from cells by Trizol organic extraction followed by RNeasy Micro Kit (Qiagen Sciences) procedure. Skin macrophages were isolated by immunostaining with anti-F4/80-Alexa Fluor 647 antibody (BioLegend)
Microarray analysis of gene expression
Three biological replicates for each group were used for cDNA microarray analyses. RNA was quantified fluorimetrically and assayed for integrity using the Agilent Bioanalyser (Agilent Technologies). One-hundred nanograms of RNA was converted to biotinylated cRNA using one round of amplification with the Illumina Labelling Kit (Illumina) and one round of T7 polymerase amplification and hybridized to Illumina Murine Beadchips v. 2.0. After hybridization and staining, the arrays were scanned in an Illumina Bead Station, and the images processed using Illumina Bead Studio software.
Statistical analysis of microarray data
The raw microarray data were extracted from the BeadStudio software and imported into the R statistical programming environment (R Development Core Team)36 and Bioconductor37. A variance stabilization transformation38 followed by quantile normalization and quality assessment were performed using the lumi package38. Differential expression between groups was calculated using a linear model and empirical Bayes-moderated false discovery rates (FDR) for each treated versus untreated line were calculated using the limma package39. A heatmap including probes showing differential expression between UVB-treated and untreated with a FDR of 0.05 was generated (Fig. 2a and Supplementary Fig. 7). Hierarchical clustering of genes was performed using single linkage and Pearson correlation distance metric. Samples were simply ordered naturally.
qRT–PCR
The most highly significant genes identified through the statistical analysis of microarray data were validated by qRT–PCR. Reverse transcription was performed on 0.1–1.0 μg RNA with Superscript III RT system (Invitrogen) following the manufacturer’s protocol. Real-time PCR was performed with Quantitect SYBR Green PCR system (Qiagen Sciences) on a 7900HT Real-Time PCR machine (Applied Biosystems). 18S ribosomal RNA was used as the normalizer. The primers used in qRT–PCR are listed in Supplementary Table 2.
Histology and immunohistochemistry
Skin and tumour samples were preserved in optimal cutting temperature (OCT) compound and stored frozen at −80 °C. Five-micrometre sections were cut using a cryostat (Leica Microsystems) and were observed under a fluorescence microscope (Nikon Instruments) with DAPI (Vectashield, Vector Laboratories) counterstaining. The antibodies for fluorescence immunohistochemistry were obtained as listed in Supplementary Table 3. Haematoxylin and eosin stainings were performed using standard protocols. Select haematoxylin and eosin stained sections are shown in Supplementary Figure 26.
In vivo antibody-mediated blockade
For the IFN-blockade experiment in pups, the anti-murine IFN-γ neutralizing antibody was the rat IgG clone XMG-6 (3.19 mg ml−1)40. The anti-murine IFN-αR1 antibody was the mouse IgG1 clone MARI-5A3 (purified monoclonal antibodies, 4 mg ml−1)41. The control antibodies were rat IgG1 clone GL113 (1.52 mg ml−1) ascites. The in vivo applications of these antibodies have been described42,43. 0.1–0.2 mg antibody was injected per pup intraperitoneally, 1–2 h before and 3 days after UVB irradiation. For IFN-γ blockade in the tumorigenesis experiment, the anti-IFN-γ antibody was XMG-6. The control antibody was rat IgG1 against horseradish peroxidase (HRPN; BioXcell). Antibodies were administered intraperitoneally as follows: 1 mg per mouse on days −1, 0, 1, 3, 6; and 0.5 mg per mouse on days 9, 12, and 15 after inoculation. Tumours were harvested at day 18.
Immunofluorescence flow cytometry and FACS
The immunofluorescence-based flow cytometry and FACS sorting were done using antibodies listed in Supplementary Table 3, using standard procedures.
In vitro UV irradiation
The melan-c melanocyte cell line (gift from V. Hearing) was irradiated with the Junggust Box, a hand-made enclosed module that houses two F20 sunlamps outputting 60% UVB and 38% UVA wavebands. A total UV dose of 175 J m−2 was given at a dose rate of 2.0 W m−2. The cells were irradiated in 10-cm culture dishes at 80% confluence overlaid with 3 ml PBS and the dish covered with plastic saran wrap to filter out any UVC. Following irradiation, the cells were incubated for the desired time in normal media (RPMI1640, 10% FBS, 100,000 U l−1 penicillin, 100 mg l−1 streptomycin sulphate, 2 mM glutamine and 200 nM 12-O-tetradecanoylphorbol-13-acetate (TPA)).
Chemotactic assays for Ccl8
The cDNA for mouse Ccl8 was amplified by RT–PCR and cloned into the pcDNA3.1 expression vector (Clontech). F5061 cells were transfected using FuGene HD (Roche). Empty pcDNA3.1 vector was transfected as control. In vitro chemotactic assay was performed on the RAW264.7 macrophage cell line in a Transwell system with 8-µm pore size (Corning). 1 × 105 cells were seeded in the top well, and the migrant cells were counted following haematoxylin staining. The experiment was done in duplicate. Five random microscopic fields from each replicate were counted for the number of migrant cells. For the in vivo chemotactic assay, 2.5 × 105 F5061-Ccl8 or F5061-vector cells were inoculated subcutaneously in syngeneic FVB/N mice. Skin samples from the area of the inoculation were harvested two days after inoculation, preserved in OCT compound, cryosectioned, and immunohistochemistry with anti-F4/80 antibody was performed to assess macrophage infiltration.
Tumorigenicity assay
F4/80+ macrophages were FACS-isolated from dorsal skins of pups 6 days after UVB irradiation at PD1. Macrophages were admixed with F5061 melanoma cells at a 1:5 ratio (0.5 × 105 macrophages: 2.5 × 105 F5061) and inoculated subcutaneously in syngeneic FVB/N mice (n = 10). Control mice were inoculated with 2.5 × 105 F5061 cells without macrophages (n = 10). Tumours were harvested after three weeks and were measured in three dimensions. Tumour volumes were calculated by the formula: V = length × width × depth × π × 1/6. For the tumorigenicity experiment with IFN-γ blockade, for each of the two groups with macrophages, four mice had 0.5 × 105 macrophages but another 6 mice from each group had 0.42 × 105 macrophages mixed with 2.5 × 105 F5061 cells. No differences in tumour growth were detected between the mice with these differing numbers of macrophages. All four groups contained ten mice, but two mice from the F5061 plus control-antibody group died prematurely and were taken out of the analysis.
TUNEL assay
The TUNEL assay was performed using TACS TdT Fluorescein kit (R&D Systems) following the manufacturer’s protocol.
Human and mouse melanoma tissue microarrays
The human melanoma tissue microarray was obtained from S. Hewitt at the National Cancer Institute/National Institutes of Health. Antigen retrieval was performed by microwave boiling in citrate buffer (pH 6). Immunohistochemistry with anti-CD68 and anti-IFN-γ antibodies (Supplementary Table 3) was performed serially with appropriate fluorescence-tagged secondary antibodies. The mouse melanoma tissue microarray has been generated by our laboratory and contains melanoma tissues from our HGF/SF transgenic mouse model, induced by a single neonatal UV radiation dose. Anti-F4/80 (macrophages), anti-CD3 (pan T cells) and anti-B220 (B cells) antibodies were used in conjunction with the Vectastain Elite ABC system (Vector Laboratories) following the manufacturer’s protocol.
Statistical analyses
All statistical analyses were performed using Graphpad Prism software. Two-tailed Student’s t-test and ANOVA with post-hoc Tukey analyses were performed as indicated to validate significant differences. Means ± s.e.m. are indicated for all statistical analyses. A P value of <0.05 was considered statistically significant.
Accession codes
Primary accessions
Gene Expression Omnibus
Data deposits
The microarray data have been deposited in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) under accessionnumber GSE25164.
References
Garibyan, L. & Fisher, D. E. How sunlight causes melanoma. Curr. Oncol. Rep. 12, 319–326 (2010)
Whiteman, D. C., Whiteman, C. A. & Green, A. C. Childhood sun exposure as a risk factor for melanoma: a systematic review of epidemiologic studies. Cancer Causes Control 12, 69–82 (2001)
Noonan, F. P. et al. Neonatal sunburn and melanoma in mice. Nature 413, 271–272 (2001)
De Fabo, E. C., Noonan, F. P., Fears, T. & Merlino, G. Ultraviolet B but not ultraviolet A radiation initiates melanoma. Cancer Res. 64, 6372–6376 (2004)
Nishimura, E. K. et al. Dominant role of the niche in melanocyte stem-cell fate determination. Nature 416, 854–860 (2002)
Walker, G. J. et al. Murine neonatal melanocytes exhibit a heightened proliferative response to ultraviolet radiation and migrate to the epidermal basal layer. J. Invest. Dermatol. 129, 184–193 (2009)
Schroder, K., Hertzog, P. J., Ravasi, T. & Hume, D. A. Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189 (2004)
Wolnicka-Glubisz, A. et al. Deficient inflammatory response to UV radiation in neonatal mice. J. Leukoc. Biol. 81, 1352–1361 (2007)
Darwich, L. et al. Secretion of interferon-γ by human macrophages demonstrated at the single-cell level after costimulation with interleukin (IL)-12 plus IL-18. Immunology 126, 386–393 (2009)
Li, D. et al. Rays and arrays: the transcriptional program in the response of human epidermal keratinocytes to UVB illumination. FASEB J. 15, 2533–2535 (2001)
Proost, P., Wuyts, A. & Van Damme, J. Human monocyte chemotactic proteins-2 and -3: structural and functional comparison with MCP-1. J. Leukoc. Biol. 59, 67–74 (1996)
DeNardo, D. G. et al. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 (2009)
Dunn, G. P., Koebel, C. M. & Schreiber, R. D. Interferons, immunity and cancer immunoediting. Nature Rev. Immunol. 6, 836–848 (2006)
He, Y. F. et al. Sustained low-level expression of interferon-γ promotes tumor development: potential insights in tumor prevention and tumor immunotherapy. Cancer Immunol. Immunother. 54, 891–897 (2005)
Aoki, H. & Moro, O. Upregulation of the IFN-γ-stimulated genes in the development of delayed pigmented spots on the dorsal skin of F1 mice of HR-1 x HR/De. J. Invest. Dermatol. 124, 1053–1061 (2005)
Hirobe, T. Histochemical survey of the distribution of the epidermal melanoblasts and melanocytes in the mouse during fetal and postnatal periods. Anat. Rec. 208, 589–594 (1984)
Wolnicka-Glubisz, A. & Noonan, F. P. Neonatal susceptibility to UV induced cutaneous malignant melanoma in a mouse model. Photochem. Photobiol. Sci. 5, 254–260 (2006)
Iliopoulos, D., Jaeger, S. A., Hirsch, H. A., Bulyk, M. L. & Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 39, 493–506 (2010)
Murphy, J., Summer, R., Wilson, A. A., Kotton, D. N. & Fine, A. The prolonged life-span of alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 38, 380–385 (2008)
Porter, G. A. et al. Significance of plasma cytokine levels in melanoma patients with histologically negative sentinel lymph nodes. Ann. Surg. Oncol. 8, 116–122 (2001)
Meyskens, F. L., Jr et al. Randomized trial of adjuvant human interferon gamma versus observation in high-risk cutaneous melanoma: a Southwest Oncology Group study. J. Natl. Cancer Inst. 87, 1710–1713 (1995)
Ascierto, P. A. & Kirkwood, J. M. Adjuvant therapy of melanoma with interferon: lessons of the past decade. J. Transl. Med. 6, 62 (2008)
Rebmann, V., Wagner, S. & Grosse-Wilde, H. HLA-G expression in malignant melanoma. Semin. Cancer Biol. 17, 422–429 (2007)
Derre, L. et al. Expression and release of HLA-E by melanoma cells and melanocytes: potential impact on the response of cytotoxic effector cells. J. Immunol. 177, 3100–3107 (2006)
Lee, N. et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl Acad. Sci. USA 95, 5199–5204 (1998)
Wischhusen, J., Waschbisch, A. & Wiendl, H. Immune-refractory cancers and their little helpers—an extended role for immunetolerogenic MHC molecules HLA-G and HLA-E? Semin. Cancer Biol. 17, 459–468 (2007)
Chen, Z., Koralov, S. B. & Kelsoe, G. Complement C4 inhibits systemic autoimmunity through a mechanism independent of complement receptors CR1 and CR2. J. Exp. Med. 192, 1339–1352 (2000)
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010)
Shah, K. V., Chien, A. J., Yee, C. & Moon, R. T. CTLA-4 is a direct target of Wnt/β-catenin signaling and is expressed in human melanoma tumors. J. Invest. Dermatol. 128, 2870–2879 (2008)
Wolnicka-Glubisz, A., King, W. & Noonan, F. P. SCA-1+ cells with an adipocyte phenotype in neonatal mouse skin. J. Invest. Dermatol. 125, 383–385 (2005)
Budd, P. S. & Jackson, I. J. Structure of the mouse tyrosinase-related protein-2/dopachrome tautomerase (Tyrp2/Dct) gene and sequence of two novel slaty alleles. Genomics 29, 35–43 (1995)
Urlinger, S. et al. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc. Natl Acad. Sci. USA 97, 7963–7968 (2000)
Tumbar, T. et al. Defining the epithelial stem cell niche in skin. Science 303, 359–363 (2004)
De Fabo, E. C., Noonan, F. P. & Frederick, J. E. Biologically effective doses of sunlight for immune suppression at various latitudes and their relationship to changes in stratospheric ozone. Photochem. Photobiol. 52, 811–817 (1990)
Serrano, M. A., Canada, J. & Moreno, J. C. Erythemal ultraviolet exposure of cyclists in Valencia, Spain. Photochem. Photobiol. 86, 716–721 (2010)
Team, R. D. C. R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, 2008)
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004)
Du, P., Kibbe, W. A. & Lin, S. M. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008)
Gentleman, R. Bioinformatics and Computational Biology Solutions using R and Bioconductor (Springer, 2005)
Cherwinski, H. M., Schumacher, J. H., Brown, K. D. & Mosmann, T. R. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J. Exp. Med. 166, 1229–1244 (1987)
Sheehan, K. C. et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 26, 804–819 (2006)
Goldszmid, R. S. et al. TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-γ production. J. Exp. Med. 204, 2591–2602 (2007)
Gramzinski, R. A. et al. Interleukin-12- and gamma interferon-dependent protection against malaria conferred by CpG oligodeoxynucleotide in mice. Infect. Immun. 69, 1643–1649 (2001)
Acknowledgements
We would like to thank the following individuals for their support: S. Yuspa for primary keratinocytes; C. Toniatti and H. Bujard for the rtTA2sM2 construct; V. Hearing for melan-c cell line; S. Hewitt for the human melanoma tissue microarray; M. Anver for immunohistochemical staining and production/analysis of mouse melanoma tissue microarray; K. Blas and E. Vega-Valle for technical help; N. Restifo and A. Hurwitz for suggestions and discussions. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. M.R.Z. was supported by a National Cancer Institute Director’s Innovation Career Development Award. E.C.D. and F.P.N. were supported by grants from the National Institutes of Health (awards CA53765 and CA92258), and the Melanoma Research Foundation.
Author information
Authors and Affiliations
Contributions
M.R.Z. designed and performed experiments, interpreted data and wrote the manuscript. S.D. performed statistical analysis of microarray data and generated heatmaps. F.P.N. interpreted data and reviewed the manuscript. C.G.-C. managed mouse colonies. T.S.H. performed flow cytometry and FACS. R.L.W. performed cDNA microarrays. L.F. produced Dct-rtTA transgenic mice. E.F. provided TRE-H2B–GFP mice. L.L. helped design interferon blockade experiments. H.A.Y. interpreted data and reviewed the manuscript. T.J.H. evaluated GFP expression in skin and reviewed the manuscript. H.A. evaluated embryonic expression of GFP and reviewed the manuscript. G.T. designed interferon blockade experiments, provided antibodies, and reviewed the manuscript. P.S.M. designed and performed analysis of microarray data and reviewed manuscript. E.C.D. designed, measured and performed UV irradiation experiments, supervised project, and reviewed manuscript. G.M. designed experiments, interpreted data, supervised the project and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-26 with legends and Supplementary Tables 1-3. (PDF 4164 kb)
Rights and permissions
About this article
Cite this article
Zaidi, M., Davis, S., Noonan, F. et al. Interferon-γ links ultraviolet radiation to melanomagenesis in mice. Nature 469, 548–553 (2011). https://doi.org/10.1038/nature09666
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature09666
This article is cited by
-
Sexual dimorphism in melanocyte stem cell behavior reveals combinational therapeutic strategies for cutaneous repigmentation
Nature Communications (2024)
-
Dedifferentiation maintains melanocyte stem cells in a dynamic niche
Nature (2023)
-
Interferon-gamma signaling promotes melanoma progression and metastasis
Oncogene (2023)
-
The journey from melanocytes to melanoma
Nature Reviews Cancer (2023)
-
Low doses of IFN-γ maintain self-renewal of leukemia stem cells in acute myeloid leukemia
Oncogene (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
