
Abstract
The involvement of collagen in bone biomineralization is commonly admitted, yet its role remains unclear. Here we show that type I collagen in vitro can initiate and orientate the growth of carbonated apatite mineral in the absence of any other vertebrate extracellular matrix molecules of calcifying tissues. We also show that the collagen matrix influences the structural characteristics on the atomic scale, and controls the size and the three-dimensional distribution of apatite at larger length scales. These results call into question recent consensus in the literature on the need for Ca-rich non-collagenous proteins for collagen mineralization to occur in vivo. Our model is based on a collagen/apatite self-assembly process that combines the ability to mimic the in vivo extracellular fluid with three major features inherent to living bone tissue, that is, high fibrillar density, monodispersed fibrils and long-range hierarchical organization.
Similar content being viewed by others

Main
Collagen I, a preponderant and ubiquitous protein in the human body, is the major organic component of the bone extracellular matrix (ECM). Collagen triple helices are secreted from the cell and assembled at the molecular level in a periodic staggered array into fibrils exhibiting a characteristic banding pattern. Different suprafibrillar arrangements of fibrils occur and coexist, forming dense structural hierarchies from the nanoscopic to the macroscopic length scales of bone1. Collagen combines with other bone ECM components among non-collagenous proteins (NCPs) and proteoglycans rich in acidic glycosaminoglycans. Furthermore, the organic matrix is strengthened by apatite mineral deposition. From the literature, there seem to be two means by which collagen mineralizes: interfibrillar2 and intrafibrillar3,4,5. Circulating fluids continuously bathe the calcified connective tissue over both collagen inter- and intrafibrillar spaces6.
Numerous studies focus on studying NCPs, as they are expected to play an important role in bone biomineralization7,8. The effects of NCPs on mineralization, in solution or cell culture systems, remain a matter of debate but have become clearer with the use of models developed with transgenic and knockout animals9. The results suggest that NCPs may be involved in different steps of bone mineralization, such as the formation of the putative amorphous calcium phosphate (ACP) precursor phase, apatite nucleation and crystal growth as well as inhibition7,8. Only a few examples point out the impact the surrounding matrix has on these proteins and how their confinement can result in unexpected activity10,11. The distribution and size of apatite crystals detected in the collagen bone disease, osteogenesis imperfecta12,13, highlight the necessity of having a hierarchically organized collagen matrix for mineral deposition. Also, both the cross-striated fibril and the three-dimensional (3D) suprafibrillar organization contribute to the high mechanical performance of the bone tissue14,15.
Although the structure–function relationship in bone is fundamental1, the long-range collagen/hydroxyapatite structure has been neglected in models proposed in vitro. Owing to the systematic use of low collagen concentrations in vitro, near 1 mg ml−1, only the level of the individual fibril has been reproduced1,16,17,18,19. These conditions lead to a large volume of extrafibrillar space (Fig. 1a), and neither the organization nor the density of the bone ECM (Fig. 1b) can be reproduced. Many studies are based on the examination of single mineralized collagen fibrils directly formed onto electron microscopy grids18,20. As previously addressed21,22, models based on isolated cross-striated fibrils (100–300 nm) or even tendon—which is close in density to bone but composed of uniaxial collagen fibrils—are limited because spatial information is lost. The use of such models in vitro to study bone biomineralization mechanisms partly explains why the investigations on protein activity have yielded contradictory findings, and limits the understanding of the role NCPs have on the interactions between the organic components and hydroxyapatite in bone23.

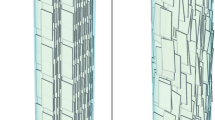
a–c, Stained thin-section TEM micrographs of a low-density collagen matrix (~5 mg ml−1) (a), a demineralized osteon in human compact bone (b) and a high-density collagen matrix (~250 mg ml−1) (c). The periodic rotation of collagen fibrils is illustrated in b and c by white bars, corresponding to fibrils positioned in the section plane, and dots, corresponding to fibrils normal to this plane. The green and orange colours illustrate the opposite extremities (N- and C-terminal) of the collagen fibrils. (Insets). The typical cross-striated pattern demonstrates the presence of collagen fibrils in both low- and high-concentration collagen matrices. In the low-concentration collagen matrix, collagen fibrils appear with no preferred orientation. In contrast, the pattern observed in the high-concentration collagen matrix—long-range helical organization—mimics the organization of the bone organic extracellular matrix, forming arced patterns in oblique sections52. Although the helical pitch (half-pitch=dotted red rectangle), the fibrillar density and the fibril diameter in c are similar to that in b, the lack of the other organic constituents present in the bone ECM may be the reason for the different aspect of the collagen. Schematic representations of the organization of collagen fibrils in a, (black rectangle) and in b,c in a 3D perspective are shown below (as indicated by the red arrows).
To address these shortcomings, bone-tissue-like dense collagen matrices are used. A limiting factor in working with very concentrated collagen solutions is their handling in vitro, which is difficult owing to their high viscosity. Above a certain concentration, collagen solutions are no longer fluids (Supplementary Fig. S1). Recently, a method was developed using compression to produce fibrillar collagen scaffolds at concentrations much higher than those proposed in the literature (reaching 170 mg ml−1), which is much closer to those found in living tissues24. Unfortunately, this process leads to the exclusion of a high amount of scaffold water, far from that found in living tissues, and the suprafibrillar network does not present in vivo-like preferred orientations. Inspired by a previous hypothesis25, another approach showed that similarities exist between the observed geometric organization of the fibrous collagen packing in vivo and the molecular order described in certain liquid crystals26. The 3D organization of collagen, similar to that described in human compact bone, has been reproduced in vitro by exploiting the capacity of collagen molecules to self-assemble spontaneously under high concentrations (at least 80 mg ml−1)26. The evidence of collagen liquid-crystalline phases in vivo has never been demonstrated, although collagen lyotropic properties in vitro are widely studied27. The stabilization of such a fluid cholesteric geometry of collagen molecules28 results in an organized and dense fibrillar matrix that was successfully, however poorly, mineralized29.
Interestingly, for all of the models cited above, that is, dilute and concentrated collagen conditions, the use of a calcium-binding polymer described as mimicking the role of non-collagenous acidic proteins was necessary to mineralize the intrafibrillar collagen spaces and induce the co-alignment between the c axis of the hydroxyapatite crystals and the long axis of the collagen fibril. From all of these models, a general consensus in the literature has recently risen on the necessity of a calcium-ion-binding polymer to mineralize the collagen in vivo and drive calcium phosphate amorphous precursors to ensure interactions between the collagen organic matrix and the apatite phase.
Here, we report on a bio-inspired mineralization of a tissue-like collagen matrix in the absence of calcium-binding polymer and non-collageneous proteins from the ECM of vertebrate calcifying tissues. We show that collagen is able to initiate and orientate the mineralization of carbonated hydroxyapatite (CHA) in vitro in a manner similar to that observed in bone, confirming a recent modelling study30. In addition, collagen influences the size and 3D distribution of apatite crystals. Unexpectedly, collagen has a further impact on the hydration environment and local structure of phosphate in apatite, which are both similar to those found in fresh untreated bone. Our collagen/apatite self-assembly method combined with mimicked extracellular fluid (ECF) provides insight into the complexities of collagen functions in bone, and could be expanded to other mineralization events by adding molecules from the ECM.
Collagen/apatite self-assembly and mimicked ECF
We set a model based on a continuous collagen injection with a dialysis process that recreates the dynamics of collagen fibrillogenesis after secretion by the cells31. Injection of an acidic collagen solution with a low concentration (~1 mg ml−1) into a permeable mould (dialysis membranes) organized type-I collagen molecules; the reverse dialysis performed against a concentrated polymer (poly(ethylene glycol) (PEG), ~300 mg ml−1) set the final high concentration of the collagen matrix. Because in bone, calcium, phosphate and carbonate ions are derived from the ECF and permeate the biological tissue continuously, the mineralization was performed by continuous injection of the carbonated apatite ion precursors with the collagen molecules. For this purpose, the initial acidic collagen and PEG polymer solutions were supplemented with calcium, phosphate and carbonate ions (Supplementary Fig. S2A). A calcium-to-phosphate (Ca/P) molar ratio of 3.33 together with a carbonate ion concentration of 33 mM is consistent in vitro with the formation of preferentially B-type CHA found in bone32. The carbonate substitution was checked based on Fourier transform infrared spectroscopy33 (Supplementary Fig. S3). The global ionic strength (165.9 mM) was kept constant throughout the process and was close to that described in physiological conditions. Neutralization through ammonia vapour led to the co-precipitation of collagen molecules into fibrils28 and CHA (Coll/CHA), as depicted in Supplementary Fig. S2A. Reproducing the condensed state of collagen triple helices and fibrils in vitro by the chemical method of pH control is a good alternative to enzymatic control, which fails at producing fibrils at high concentration28. Co-precipitation of calcium–phosphate salts and collagen molecules results in a matrix with a low mineralization degree29. To increase the mineral content, the matrix was kept at physiological temperature while continuously stirred in simulated body fluid (SBF; ref. 34; that is, a serum-like solution35) and replenished (Coll/CHA/SBF; Supplementary Fig. S2A). These conditions we will refer to as ‘dynamic’. The presence of mineral ions in the medium before and during fibrillogenesis mimics the circulating fluids in bone. Although this ECF is critical for a variety of physiological functions and biochemical reactions in the living bone tissue, it has not received, until now, deserved attention in models proposed in the literature.
Towards fundamental characteristics of collagen in bone
Transmission electron microscopy (TEM) investigations were performed on the Coll/CHA matrix. Observations of stained thin sections of the embedded matrix obtained from a highly concentrated collagen solution (Fig. 1c) show closely packed fibrils with a helicoidal organization that results from a continuously twisting orientation of collagen fibrils, similar to the ultrastructure of an osteon observed in decalcified human compact bone (Fig. 1b); in contrast, the matrix obtained from a dilute collagen solution (~5 mg ml−1) exhibits a random orientation of collagen fibrils (Fig. 1a). Tuning the injection rate of collagen molecules provides control over the solution viscoelasticity and the kinetics of self-assembly of the collagen molecule. This parameter leads to dense homogeneous matrices of fibrils that are monodisperse in diameter, with or without mineral (Supplementary Fig. S4), which is a main characteristic of biological tissues (Fig. 1b) and a notable structural property compared to previous studies on increased collagen concentration matrices (Supplementary Fig. S5; refs 29, 36). The presence of cross-striated fibrils (Fig. 1c, inset) reveals that fibrillogenesis in vitro was successfully induced within the Coll/CHA matrix, but because of stain precipitates and a probable low mineral content, crystals were hardly observed. On the basis of TEM observations, we show schematics of the fibrillar network in bone, and poorly concentrated and highly concentrated collagen matrices, that help visualize the density and the organization of collagen fibrils in a 3D perspective in these three environments (Fig. 1a–c). Neither compression nor harvesting was applied to the lyotropic collagen solutions during the process, implying that the resulting anisotropy is due to the inherent collagen properties induced by the experimental conditions at high protein concentration, and making this process easily reproducible. The final collagen concentration was determined by quantifying the amount of hydroxyproline by titration. The high collagen concentration (~250 mg ml−1) resulted in a half-cholesteric pitch (2–3 μm), close to that found in the femoral compact bone of adult humans. It can be assumed that the viscoelastic properties of collagen in bone are closer to those encountered in a high-concentration matrix than those obtained with a low-concentration matrix.
Towards fundamental characteristics of mineral in bone
Wide-angle X-ray diffraction (WAXD) studies were performed on freshly extracted hydrated bone from two-year-old sheep and compared with hydrated Coll/CHA and Coll/CHA/SBF matrices (Fig. 2a). The synthetic collagen matrix prepared without Ca2+, PO43− and CO32− (Coll) is shown as a reference in Fig. 2a, and exhibits the classical precipitated collagen diffraction profile14,37. One-dimensional radial averages of the WAXD patterns of both mineralized matrices exhibit the characteristic HA diffraction peaks (002) and the merged (211), (300), (202) reflections. X-ray diffraction performed on bone and Coll/CHA/SBF show similar patterns, with broad peaks attributed to small crystallite size and/or structural substitutions of carbonate ions for phosphate ions. Using Scherrer’s equation and the full-width at half-maximum (FWHM) of the corresponding peak (25.8°), the average crystallite size in the (002) direction (c axis) was determined to be 20 nm. Crystallites of this size are in the same range as those found in natural bone nanocrystals. The mineral content in Coll/CHA is low enough to observe the collagen pattern29, as found at very early stages of normal in vivo calcification37. The difference in intensity is consistent with the relative differences in the degrees of mineralization. Although the resulting organic-to-mineral weight ratio in the matrix (Coll/CHA) was quite low (~5%), a mineral content close to that published for mature bone6 was found after the immersion in SBF (~50%, Coll/CHA/SBF) (Supplementary Fig. S3).

a, 1D radial average of the WAXD patterns of the Coll matrix as control (purple line), and the Coll/CHA matrix before (green solid line) and after (green dashed line) immersion in SBF (Coll/CHA/SBF) and in sheep bone (blue line). Characteristic HA (002) and the merged (211), (300), (202) diffraction lines are seen in both mineralized matrices. The peak intensities in the Coll/CHA/SBF matrix reflect a higher degree of mineralization in this sample, similar to that in bone. b, Thin-section TEM micrograph of the unstained embedded Coll/CHA matrix shows equatorial (red arrow) and axial (white arrow) alignment of small apatite crystals, which illustrate the early and later stages of calcification, respectively. c, Higher mineral content in the collagen matrix reveals cholesteric domains, as seen on thin-section TEM micrographs of the unstained embedded Coll/CHA/SBF matrix. Features defined as Fig. 1. d, A spatial coexistence of other domains—aligned (al) and isotropic (is)—is also observed in this matrix. The mineral content is not homogeneously distributed within the matrix (c versus d).
To support X-ray diffraction data, further TEM observations were performed on thin sections of embedded matrices. Examining the crystal organization in bone by TEM is limited owing to the difficulty of thin-section preparation22 and artefacts from staining38. TEM was performed on unstained samples (that is, neither osmium nor uranyl acetate) because apatite and stain precipitates are sometimes indistinguishable from each other (Supplementary Fig. S6). The ability to observe apatite crystals without staining is possible because they are electron dense. TEM micrographs of the matrix with the lowest mineral content (Coll/CHA; Fig. 2b) show lateral (red arrows) and axial (white arrows) alignments of small apatite crystals with regards to the fibrils’ main axis. The lateral packing of crystals across the diameter of the collagen fibrils resembles the early stages of intrafibrillar mineralization, as described in the 2D channel model by Hodge and Petruska39; and the continuous apatite crystal deposition into both the intermolecular spaces between collagen molecules and the interfibrillar spaces of collagen fibrils illustrate the later stages of calcification. As no temporal sequence has been established here, the interfibrillar mineral formation in vitro could result from either a progression from inner to outer fibril portions or from sites of nucleation at the fibril surface2. The overall ultrastructure in Coll/CHA/SBF is revealed owing to a higher mineral content, which allows the observation of mineral in the inter- and intrafibrillar spaces using TEM techniques (Fig. 2c,d). Fibril arrays are observed in a variety of patterns—twisted plywood (Fig. 2c), parallel packing and non-oriented structures1 (Fig. 2d)—showing that cholesteric, locally aligned (al) and isotropic (is) domains of collagen molecules were present in the former acidic solution, before fibrillogenesis28. These domains are coexistent in the matrix, which is similar to the heterogeneous morphology of native bone tissue1,6. Concerning the collagen/apatite structural relationship, crystals seem to align with the fibrillar collagen network. The electron diffraction pattern (Fig. 3a, inset) obtained from the area where the fibrils lay parallel to the section, for example within the arced pattern (Fig. 2c), demonstrates that the apatite crystals are aligned with a preferred (002) orientation along the main axis of the collagen fibrils (Supplementary Fig. S7). This pattern is similar to that observed in mature sheep bone (Fig. 3b), although it is a newly formed mineral phase. The heterogeneity of collagen fibril mineralization inside the Coll/CHA/SBF matrix was further evidenced by CryoTEM observations. The banding pattern is easily observed although no stain was applied to the samples, indicating that crystals lie predominantly in the collagen gap region40 (Fig. 3c). However, in some parts it is difficult to see the banding (Fig. 3d,e). A single fibril can exhibit this heterogeneity (Fig. 3d). This disappearance of the fibril banding pattern may reflect the coexistence of low (Fig. 3c), intermediate (Fig. 3d), and high (Fig. 3e) degrees of fibril mineralization. Indeed, as previously described, because the cross-striated pattern is not observed in Fig. 3e, the mineral may occupy the entire collagen fibril20. However, the presence of a calcium-binding polymer (polyaspartic acid)17 in the solution was not needed in our model to achieve intrafibrillar mineralization and generate the collagen/apatite co-alignment. As there is no organic additive in the medium, collagen regulates the extent to which the crystals can develop. CryoTEM observations also show apatite crystals, isolated from the collagen matrix, that may result from sample preparation (Fig. 3c–e). The CHA particles have irregular edges and seem to be smaller than those found in the absence of a dense collagen network (Supplementary Fig. S8; ref. 29). In summary, the related results demonstrate that the 3D organization of the fibrillar collagen network controls the spatial distribution of apatite crystals. Collagen density and 3D organization, both at the molecular and fibrillar level, are key parameters to reproduce proper bone apatite mineralization in vitro.

a, Thin-section TEM micrograph of the unstained embedded Coll/CHA/SBF matrix. CHA is observed in parallel arrays. The corresponding electron diffraction pattern shows oriented CHA (002) diffraction arches demonstrating the co-alignment of the crystallite c axis and the fibrils’ long axes in the matrix. b, A similar pattern is observed for a thin-section TEM micrograph of unstained embedded fresh sheep bone. c–e, CryoTEM images of collagen fibrils in the Coll/CHA/SBF matrix may reflect different degrees of mineralization, low (c), intermediate (d) and high (e), supported by disappearance of the banding pattern. Apatite crystals isolated from the collagen matrix (arrows) appear with irregular edges and in the same size range as those found in bone6.
The local environment and hydration of apatite
To answer whether collagen may even affect the mineral structure in terms of substitution and crystallinity, 1H and 31P magic angle spinning (MAS) solid-state nuclear magnetic resonance (NMR) was used. Figure 4a shows quantitative 31P MAS NMR spectra of the hydrated Coll/CHA/SBF matrix, fresh sheep bone and CHA precipitated through the same process but in the absence of an organic matrix32. The NMR study was performed on fresh bone within 2 h after extraction (without any treatment) to prevent specimen alteration by chemical pre-treatment41. A single asymmetric resonance around 3 ppm, characteristic of phosphate ions in hydroxyapatite, is detected for each sample. The 31P MAS spectra of the hybrid matrix and the fresh bone are similar in terms of chemical shifts (3.2 ppm) and line width (FWHM = 510 Hz), whereas the spectrum of the pure CHA precipitated in the same conditions exhibits a resonance slightly shielded at 2.8 ppm, associated with a sharper line width (FWHM = 320 Hz). These differences in terms of line width can be attributed to a wider distribution of chemical shifts, because of either a higher atomic disorder or a higher degree of ion substitution in the apatitic phase. This result highlights the impact of the dense organic matrix on the mineral phase.

a, Quantitative 31P MAS spectra of the Coll/CHA/SBF matrix, fresh sheep bone and CHA precipitated in the absence of collagen (30° 31P pulse; recycle delay RD = 240 s; 1H SPINAL-64 decoupling during acquisition with 60 kHz of RF field). b, 1H–31P HetCor spectrum of the Coll/CHA/SBF matrix (RD = 3.5 s, contact time CT = 1 ms, 232 transients for each 128 t1 increment) and extracted 31P slices at δ(1H) = 0 and 4.85 ppm. c, 1H–31P HetCor spectrum of fresh sheep bone (RD = 3.5 s, CT = 1 ms, 400 transients for each 128 t1 increment) and extracted 31P slices at δ(1H) = 0 and 4.85 ppm. The similarity between hybrid matrix and fresh bone NMR spectra provide evidence that the chemical environment of phosphate anions is very similar in both samples. In particular, the hydration environment around CHA platelets is similar because a strong correlation peak at δ(1H) = 4.85 ppm is observed for both samples.
To further investigate the mineral phase, 2D spectra of the Coll/CHA/SBF matrix (Fig. 4b) and fresh bone (Fig. 4c), where 1H and 31P nuclei are spatially correlated through 1H–31P heteronuclear dipolar interaction (1H–31P HetCor), were recorded. We also evidence similar spectroscopic signatures for both samples. In particular, we can distinguish a correlation peak between the phosphate site from hydroxyapatite (3.2 ppm) and the hydroxyl anions (0 ppm). In agreement with XRD observations, we did not observe the presence of transition precursor phases described for HA (OCP; ref. 42) or contaminant phases (brushite, calcite). Furthermore, an intense correlation peak between the phosphate resonance and a proton resonance at 4.85 ppm is observed in both cases, which corresponds to water according to its chemical shift. As the HetCor experiment is based on a cross-polarization experiment, the latter signal cannot correspond to free water but instead to rigid molecules such as adsorbed water onto the crystal surface. A previous work43 has already reported the presence of such ‘rigid’ water molecules around HA nanocrystals in bovine cortical bone with a lower intensity of the characteristic correlation peak due to partial dehydration, probably induced by the cryogenic grinding procedure used in their study. A careful analysis of the 31P projections at δ(1H) = 0 ppm and 4.85 ppm (Fig. 4b) reveals that the resonance of the phosphate ions in the hydrated environment has a line width significantly broader (FWHM = 680 Hz) than the resonance coming from the apatitic phase (FWHM = 260 Hz). Once again, the Coll/CHA/SBF matrix possesses similar features to those observed in fresh sheep bone (FWHM = 690 Hz versus 290 Hz). The Gaussian line shape of the broad signal is characteristic of a distribution of 31P chemical shifts due to a variety of chemical environments in the hydrated domain, described as a putative amorphous calcium phosphate layer around synthetic44 and biological45,46 apatite platelets. As a result, the chemical environment of the different types of phosphate species, as well as the hydrated environment of the CHA platelets, are unambiguously similar in fresh bone and in the synthetic matrix, highlighting the impact of the collagen confinement on apatite formation at the atomic scale (Supplementary Fig. S9).
Collagen contribution in the bone mineralization process
To help identify the physico-chemical factors that are involved, and resolve their contribution in the bone mineralization process, changes in the experimental settings were performed as follows.
As a first control experiment, we undertook the mineralization of a matrix prepared without mineral ions and under ‘dynamic conditions’ in SBF (Coll(SBF); Fig. 5, Supplementary Fig. S2B). Abundant spherulitic crystals were observed on the surface (Fig. 5b) as well as within the most superficial layer of the collagen matrix (Fig. 5c,d) using electron microscopy (EM) techniques. WAXD (Fig. 5e) and 31P MAS NMR experiments (Supplementary Fig. S10) confirmed that the mineral phase was hydroxyapatite. Hence, diffusion of apatite ion precursors into the matrix seems limited. A possible explanation is that the addition of the matrix into SBF strongly modifies the solution composition and induces local supersaturation favouring apatite formation over the diffusion of apatite ion precursors into the organic matrix. In contrast, the Coll/CHA ionic composition may be closer to physiological conditions.

a, Disk-shaped matrix used for characterization. b,c, Examination of the surface (B) and fractured surface of the interior of the matrix (C) by SEM shows the presence of a thick mineral layer on the matrix’s surface (~15 μm, arrow) (b) and reveals that spherulitic crystals are also found within the most superficial layer of the matrix (c). d, Examination by TEM shows the polydispersity of the nanoparticle size and their random distribution inside the matrix at this scale. e, One-dimensional radial average of the WAXD pattern corresponding to the interior of the matrix shows the characteristic HA diffraction peaks—(002) and the merged (211), (300), (202) reflections—together with the classical precipitated collagen diffraction profile (see Fig. 2, Coll). A characteristic collagen diffraction signal seen at 5.7°, corresponds to the lateral packing of the collagen molecules with a period of 1.5 nm (refs 53, 54). The signal from collagen fibrils is strong, compared with that of HA, which points to a low mineralization degree of the matrix29,37. The presence of a potential amorphous calcium phosphate can be masked by the high intensity of the collagen signal. f, An EDX profile of the matrix confirms the presence of calcium and phosphate, and the low Ca/P ratio (~1.4±0.1) may indicate, together with HA crystals, the presence of amorphous calcium phosphate55, as confirmed by the 31P MAS NMR spectrum (Supplementary Fig. S10).
A matrix was prepared with a collagen concentration gradient ranging from ~20 to 100 mg ml−1. CHA ion precursors were continuously injected during the procedure but the resulting matrix was not kept in SBF afterwards (Fig. 6). According to scanning electron microscopy (SEM) observations and energy-dispersive X-ray spectroscopy (EDX) analysis, spherulitic CHA crystals were observed in the poorly concentrated region of the collagen matrix. In contrast, in the most condensed part of the collagen matrix, which corresponds to concentrations above 80 mg ml−1 (that is, the liquid crystal phase transition), a uniform mineralized coating was observed. Interestingly, previous work showed that spherulitic (non-carbonated) hydroxyapatite crystals precipitate in dense fibrillar collagen matrices with an irregular shape (Supplementary Fig. S1; ref. 29). This discrepancy in behaviour may be due to size polydispersity of the collagen fibrils (Supplementary Fig. S5), generating a higher volume of interfibrillar space and/or, in contrast to in vivo conditions, to the fact that mineral ions were added to the matrix after gelation of the collagen triple helices.

a, The blue colour gradation from left to right indicates the increase of collagen concentration (~20–100 mg ml−1; light and dark blue circles, respectively) of the cylindrically shaped matrix. b, Examination of the poorly concentrated part of the collagen matrix by SEM shows spherulitic crystals located on and beside the collagen fibrils. Collagen fibrils appear with no preferred orientation, as described in Fig. 1a. The corresponding EDX profile of the matrix confirms the presence of calcium and phosphate. In the absence of calcium phosphate contaminant phases, the Ca/P ratio (~1.5±0.1) and the presence of HA crystals may indicate the existence of amorphous calcium phosphate or calcium-deficient HA (CDHA; ref. 55). c, In contrast, a smooth appearance of the sample is observed at concentrations above 80 mg ml−1, as previously observed29. Such uniform mineral coating indicates that the mineral aligns with the closely packed collagen network, as described in Fig. 1c. The corresponding EDX profiles of the matrix confirm the presence of calcium and phosphate ions, and the Ca/P ratio (~1,6±0.1) indicates the presence of CHA crystals as expected55. The schematic representations of the collagen fibrils/apatite organizations of b and c in a 3D perspective are shown below (as indicated by the light and dark blue arrows respectively). The crystals are illustrated in red.
It is worth mentioning that only the main inorganic ions present in blood serum compose the carbonated apatite ion precursors (Na+, Ca2+, Cl−, HCO3−, HPO42−; ref. 35), and that they are injected with the acidic collagen solution. However, this solution is more concentrated in calcium than the physiological fluid. Increasing the concentration of the main inorganic ions of the apatite phase (Ca2+, PO43−, CO32−) may have an impact on the resulting mineralization6,35,47. Thus, SBF was used instead of the highly concentrated CHA salt precursors to prepare the collagen scaffold (Coll/SBF/SBF; Supplementary Fig. S2C). According to EM studies (Supplementary Fig. S11), spherulitic crystals are found within the superficial layer of the matrix as well as on the matrix’s surface. This apatite distribution is similar to that found in the Coll(SBF) control, suggesting that the concentrations and localization of these ions within the matrix are critical for the mineral distribution. Although homogeneous nucleation is unusual in vivo, a homogeneous nucleation mechanism is suggested to occur in Coll(SBF) (Supplementary Fig. S2B and Fig. 4) and Coll/SBF (Supplementary Figs S2C and S11). Indeed, spherulitic apatite crystals are also obtained when precipitation is performed without collagen32. This is supported by the random spatial distribution and size polydispersity of the crystals located on the surface of the spherulites in both matrices (Fig. 4d and Supplementary Fig. S11D). It should be noted that heterogeneous nucleation may also be occurring, however only heterogeneous nucleation seems to occur within Coll/CHA (Figs 1 and 2 and Supplementary Fig. S2A). The role of collagen confinement on apatite size can also be noted by the size of spherulites within dense collagen matrices, which is significantly smaller than those without additives29.
To understand the differences in mineralizing behaviour, we investigated the global nature of the collagen surface charge. Collagen has a negative zeta potential of −12.2 (±0.7) mV that decreases with the mineralization performed with either SBF (−6 (±0.81) mV) or CHA (−5.4 (±0.54) mV). As the matrices had been washed before the zeta potential measurement, this difference is attributed to the presence of counter-cations on collagen. A preliminary X-ray photoelectron spectroscopy study of Coll, Coll/SBF and Coll/CHA matrices shows the characteristic doublet of Ca2p, with a Ca2p3/2 contribution at 347.3 ± 0.2 eV, in both mineralized matrices, confirming this assumption (Supplementary Fig. S12). The Ca2p/N1s ratio in Coll/CHA (0.09) was found to be higher than in Coll/SBF (0.05). All these results strengthen the fact that the concentration of calcium ions strongly influences the mineral distribution in collagen matrices, as previously proposed in the literature6.
Because many other factors are involved in vivo (such as partial CO2 pressure), it is difficult to determine the specific influence of the physico-chemical processes on mineralization. However, on the basis of the above models and previous ones based on a self-assembling system21, we propose that the process locally increases the concentration of the inorganic components (Ca2+, PO43−, CO32−) to a level of supersaturation that results in carbonated apatite precipitation. More specifically, according to Fig. 2b, a higher concentration in inorganic components during collagen assembly suggests that the local increase in ion concentration occurs in the gap regions described as first site of nucleation for apatite crystals in vivo3,4,5. We hypothesize that some specific NCPs of bone ECM play a role in the local ion concentration in vivo. These ions are critical for many physiological and biochemical functions and they must be maintained within a very limited range of concentration6. It seems reasonable to propose that such proteins are already localized within the collagen scaffold during fibrillogenesis, as was previously proposed for the Dentin Matrix Protein 1 (DMP-1; ref. 48). This questions the redundant role of a calcium-binding polymer in driving ions inside the collagen fibrils used in numerous in vitro studies17,18,19,20,29,49. After the first crystals begin to nucleate and grow within the Coll/CHA matrix, the crystal continues to grow and multiply with the ions provided by the SBF inside the matrix (Fig. 2b,c). Collagen may serve as a structural template controlling both the formation of the initial Ca–P nuclei and the subsequent crystal growth because the orientation of the carbonated apatite crystals in the collagen fibrils is the same as that observed in bone20.
The function of collagen in the mechanisms of bone biomineralization remains a source of debate23. Our results strongly suggest that collagen initiates and orientates the apatite crystal growth and is responsible for the size and distribution of apatite crystals in bone. Furthermore, collagen has an influence on the hydration environment and local structure of phosphate in apatite.
Highly dense, monodispersed fibrils and the 3D suprafibrillar organization of collagen may be key structural parameters in the bone ECM for apatite crystal co-alignment with the collagen fibrils at the nanometre scale, as well as the long-range organization described in bone—the ‘fibril array pattern’ level, according to the previously proposed terminology by Weiner and Wagner1.
In our model, collagen can sequestrate large quantities of calcium, phosphate and carbonate ions, which precipitate spontaneously in apatite without additives. The concentration of calcium ions strongly influences the distribution of apatite crystals within the collagen matrix. Hence, the Ca-rich proteins in the ECF may play an important role as inhibitors of apatite precipitation whereas those in bone ECM may concentrate the ions locally in the gap regions described as the first site of nucleation for apatite crystals. The idea that those proteins localize within the collagen scaffold before or/and during the fibrillogenesis maintains that they can also interfere in mediating the size of collagen fibrils in vivo, as previously proposed with DMP-1 (ref. 48), and in stabilizing a potential amorphous calcium phosphate, which is confirmed as a precursor phase in zebrafish fin rays50.
Our model contradicts the mechanism proposed for the in vitro polymer-induced liquid-precursor collagen mineralization process17, where Ca-binding additives are usually used with composite materials containing single fibrils or fibre bundles that have radial symmetry. This result affirms the importance of physico-chemical processes occurring in biomineralization, as usually discussed from a biological control point of view.
In the context of bone tissue engineering, autologous bone is still considered as the gold standard51. Previously, we have shown that the density and organization of the collagen organic matrix is a key point for material performance29. As the resulting material is composed of the two main components in bone, collagen and apatite, and exhibits a hierarchical structure that resembles the one found in natural bone, its behaviour as bone graft and its capacity to enhance bone growth are currently being tested in sheep for tissue repair. Therefore, this process provides original models to study fundamental questions on biomineralization and represents a good starting point for applications in bone tissue engineering and for the design of new implantable materials.
Methods
Full details of the matrix synthesis and sample characterization techniques used are presented in the Supplementary Information.
References
Weiner, S. & Wagner, H. D. The material bone: Structure mechanical function relations. Annu. Rev. Mater. Sci. 28, 271–298 (1998).
Landis, W. J. et al. Mineralization of collagen may occur on fibril surfaces: Evidence from conventional and high-voltage electron microscopy and three-dimensional imaging. J. Struct. Biol. 117, 24–35 (1996).
Jackson, S. F. The fine structure of developing bone in the embryonic fowl. Proc. R. Soc. Lond. B 146, 270–280 (1957).
Weiner, S. & Traub, W. Organization of hydroxyapatite crystals within collagen fibrils. FEBS Lett. 206, 262–266 (1986).
McEwen, B. F., Song, M. J. & Landis, W. J. Quantitative determination of the mineral distribution in different collagen zones of calcifying tendon using high voltage electron microscopic tomography. J. Comput. Assist. Microsc. 3, 201–210 (1991).
Glimcher, M. J. in Medical Mineralogy and Geochemistry Vol. 64 (eds Sahai, N. & Schoonen, M. A. A.) 223–282 (Reviews in Mineralogy & Geochemistry, 2006).
George, A. & Veis, A. Phosphorylated proteins and control over apatite nucleation, crystal growth, and inhibition. Chem. Rev. 108, 4670–4693 (2008).
Palmer, L. C., Newcomb, C. J., Kaltz, S. R., Spoerke, E. D. & Stupp, S. I. Biomimetic systems for hydroxyapatite mineralization inspired by bone and enamel. Chem. Rev. 108, 4754–4783 (2008).
Jahnen-Dechent, W., Heiss, A., Schäfer, C. & Ketteler, M. Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 108, 1494–1509 (2011).
Hunter, G. K. & Goldberg, H. A. Nucleation of hydroxyapatite by bone sialoprotein. Proc. Natl Acad. Sci. USA 90, 8562–8565 (1993).
Frenkel-Mullerad, H. & Avnir, D. Sol–gel materials as efficient enzyme protectors: Preserving the activity of phosphatases under extreme pH conditions. J. Am. Chem. Soc. 127, 8077–8081 (2005).
Vetter, U., Eanes, E. D., Kopp, J. B., Termine, J. D. & Robey, P. G. Changes in apatite crystal size in bones of patients with osteogenesis imperfecta. Calcif. Tissue Int. 49, 248–250 (1991).
Landis, W. J. The strength of a calcified tissue depends in part on the molecular-structure and organization of its constituent mineral crystals in their organic matrix. Bone 16, 533–544 (1995).
Fratzl, P. & Weinkamer, R. Nature’s hierarchical materials. Prog. Mater. Sci. 52, 1263–1334 (2007).
Seto, J., Gupta, H. S., Zaslansky, P., Wagner, H. D. & Fratzl, P. Tough lessons from bone: Extreme mechanical anisotropy at the mesoscale. Adv. Funct. Mater. 18, 1905–1911 (2008).
Rhee, S-H., Suetsugu, Y. & Tanaka, J. Biomimetic configurational arrays of hydroxyapatite nanocrystals on bio-organics. Biomaterials 22, 2843–2847 (2001).
Olszta, M. J. et al. Bone structure and formation: A new perspective. Mater. Sci. Eng. R 58, 77–116 (2007).
Deshpande, A. S. & Beniash, E. Bioinspired synthesis of mineralized collagen fibrils. Cryst. Growth Des. 8, 3084–3090 (2008).
Liu, Y. et al. Hierarchical and non-hierarchical mineralisation of collagen. Biomaterials 32, 1291–1300 (2011).
Nudelman, F. et al. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nature Mater. 9, 1004–1009 (2010).
Hartgerink, J. D., Beniash, E. & Stupp, S. I. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 294, 1684–1688 (2001).
Weiner, S., Traub, W. & Wagner, H. D. Lamellar bone: Structure-function relations. J. Struct. Biol. 126, 241–255 (1999).
Boskey, A. L. Biomineralization: Conflicts, challenges, and opportunities. J. Cell. Biochem. 72, 83–91 (1998).
Brown, R. A., Wiseman, M., Chuo, C. B., Cheema, U. & Nazhat, S. N. Ultrarapid engineering of biomimetic materials and tissues: Fabrication of nano- and microstructures by plastic compression. Adv. Funct. Mater. 15, 1762–1770 (2005).
Bouligand, Y. Twisted fibrous arrangements in biological materials and cholesteric mesophases. Tissue Cell 4, 189–190; 192-217 (1972).
Giraud-Guille, M-M. Liquid crystallinity in condensed type I collagen solutions: A clue to the packing of collagen in extracellular matrices. J. Mol. Biol. 224, 861–873 (1992).
Giraud-Guille, M. M., Mosser, G. & Belamie, E. Liquid crystallinity in collagen systems in vitro and in vivo. Curr. Opin. Colloid Interf. Sci. 13, 303–313 (2008).
Besseau, L. & Giraud-Guille, M-M. Stabilization of fluid cholesteric phases of collagen to ordered gelated matrices. J. Mol. Biol. 251, 197–202 (1995).
Nassif, N. et al. Self-assembled collagen-apatite matrix with bone-like hierarchy. Chem. Mater. 22, 3307–3309 (2010).
Silver, F. H. & Landis, W. J. Deposition of apatite in mineralizing vertebrate extracellular matrices: A model of possible nucleation sites on type I collagen. Connect. Tissue Res. 52, 242–254 (2011).
Wang, Y. et al. Controlled collagen assembly to build dense tissue-like materials for tissue engineering. Soft Matter 7, 9659–9664 (2011).
Nassif, N. et al. In vivo inspired conditions to synthesize biomimetic hydroxyapatite. Chem. Mater. 22, 3653–3663 (2010).
Mkukuma, L. D. et al. Effect of the proportion of organic material in bone on thermal decomposition of bone mineral: An investigation of a variety of bones from different species using thermogravimetric analysis coupled to mass spectrometry, high-temperature X-ray diffraction, and Fourier transform infrared spectroscopy. Calcif. Tissue Int. 75, 321–328 (2004).
Rámila, A. & Vallet-Regí, M. Static and dynamic in vitro study of a sol–gel glass bioactivity. Biomaterials 22, 2301–2306 (2001).
Bohner, M. & Lemaitre, J. Can bioactivity be tested in vitro with SBF solution? Biomaterials 30, 2175–2179 (2009).
Gobeaux, F. et al. Fibrillogenesis in dense collagen solutions: A physicochemical study. J. Mol. Biol. 376, 1509–1522 (2008).
Chen, J. et al. In vitro mineralization of collagen in demineralized fish bone. Macromol. Chem. Phys. 206, 43–51 (2005).
Landis, W. J. & Glimcher, M. J. Electron diffraction and electron probe microanalysis of the mineral phase of bone tissue prepared by anhydrous techniques. J. Ultrastruct. Res. 63, 188–223 (1978).
Hodge, A. & Petruska, J. in Aspects of Protein Structure (ed. Ramachandran, G. N.) 289–300 (Academic, 1963).
Traub, W., Arad, T. & Weiner, S. Three-dimensional ordered distribution of crystals in turkey tendon collagen fibers. Proc. Natl Acad. Sci. USA 86, 9822–9826 (1989).
Zhu, P. et al. Time-resolved dehydration-induced structural changes in an intact bovine cortical bone revealed by solid-state NMR spectroscopy. J. Am. Chem. Soc. 131, 17064–17065 (2009).
Tseng, Y-H., Mou, C-Y. & Chan, J. C. C. Solid-state NMR study of the transformation of octacalcium phosphate to hydroxyapatite: A mechanistic model for central dark line formation. J. Am. Chem. Soc. 128, 6909–6918 (2006).
Cho, G., Wu, Y. & Ackerman, J. L. Detection of hydroxyl ions in bone mineral by solid-state NMR spectroscopy. Science 300, 1123–1127 (2003).
Jäger, C., Welzel, T., Meyer-Zaika, W. & Epple, M. A solid-state NMR investigation of the structure of nanocrystalline hydroxyapatite. Magn. Reson. Chem. 44, 573–580 (2006).
Huang, S-J., Tsai, Y-L., Lee, Y-L., Lin, C-P. & Chan, J. C. C. Structural model of rat dentin revisited. Chem. Mater. 21, 2583–2585 (2009).
Wu, Y. et al. Nuclear magnetic resonance spin–spin relaxation of the crystals of bone, dental enamel, and synthetic hydroxyapatites. J. Bone Miner. Res. 17, 472–480 (2002).
Fantner, G. E. et al. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nature Mater. 4, 612–616 (2005).
He, G. & George, A. Dentin matrix protein 1 immobilized on type I collagen fibrils facilitates apatite deposition in vitro. J. Biol. Chem. 279, 11649–11656 (2004).
Bradt, J-H., Mertig, M., Teresiak, A. & Pompe, W. Biomimetic mineralization of collagen by combined fibril assembly and calcium phosphate formation. Chem. Mater. 11, 2694–2701 (1999).
Mahamid, J. et al. Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc. Natl Acad. Sci. USA 107, 6316–6321 (2010).
Bauer, T. W. & Muschler, G. F. Bone graft materials—an overview of the basic science. Clin. Orthop. Relat. R 371, 10–27 (2000).
Giraud-Guille, M. M. Twisted plywood architecture of collagen fibrils in human compact bone osteons. Calcif. Tissue Int. 42, 167–180 (1988).
Fratzl, P., Fratzl-Zelman, N. & Klaushofer, K. Collagen packing and mineralization. An X-ray scattering investigation of turkey leg tendon. Biophys. J. 64, 260–266 (1993).
Maxwell, C. A., Wess, T. J. & Kennedy, C. J. X-ray diffraction study into the effects of liming on the structure of collagen. Biomacromolecules 7, 2321–2326 (2006).
Dorozhkin, S. V. & Epple, M. Biological and medical significance of calcium phosphates. Angew. Chem. Int. Ed. 41, 3130–3146 (2002).
Acknowledgements
We dedicate this work to the memory of Y. Bouligand (1935–2011). This work was supported by the Agence Nationale de la Recherche (ANR) through the ANR-09-BLAN-0120-01 ‘NanoShap’ program and the DRITT-SAIC (UPMC). We thank G. Laurent and M. Selmane for technical assistance with the NMR spectrometer and WAXD experiments respectively; A. Anglo, C. Illoul and E. Jallot for preparation of TEM samples; S. Mann, J. Peron, F. Michaux and Ö. Sel for insightful discussions and critical suggestions; A. Galtayries for X-ray photoelectron spectroscopy experiments and IMM Recherche, especially L. Behr, for providing the fresh bone samples.
Author information
Authors and Affiliations
Contributions
Y.W., T.A., M.R., C.C., P.L., G.P-A. and N.N. performed the research; F.B. looked for financial support for the project; Y.W., T.A., A.V., G.P-A., F.B., M-M.G-G. and N.N. analysed data; Y.W., T.A., C.C., A.V., M-M.G-G. and N.N. wrote the paper; N.N. designed the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Information (PDF 2769 kb)
Rights and permissions
About this article
Cite this article
Wang, Y., Azaïs, T., Robin, M. et al. The predominant role of collagen in the nucleation, growth, structure and orientation of bone apatite. Nature Mater 11, 724–733 (2012). https://doi.org/10.1038/nmat3362
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmat3362
This article is cited by
-
Hierarchical helical carbon nanotube fibre as a bone-integrating anterior cruciate ligament replacement
Nature Nanotechnology (2023)
-
Cartilage calcification in osteoarthritis: mechanisms and clinical relevance
Nature Reviews Rheumatology (2023)
-
Solid-state NMR studies on the organic matrix of bone
Nano Research (2023)
-
Multiscale Effects of Collagen Damage in Cortical Bone and Dentin
JOM (2023)
-
Functionalized Coatings on Degradable Magnesium Alloys for Orthopedic Implants: A Review
Transactions of the Indian Institute of Metals (2023)
