
Abstract
Invasive and metastatic cells, as well as endothelial cells, must cross basement membranes (BMs) in order to disseminate or to form new blood vessels. The chemoinvasion assay using the reconstituted BM Matrigel in Boyden blind-well chambers is a very rapid, easy, inexpensive and flexible test that can be used to quantify the invasive potential of most cell types; it can be applied to detect the migratory activity associated with matrix degradation and can also be adapted to study the selective degrading activity on different matrix substrates. Transwell inserts can also be used. Once the optimal experimental conditions are empirically determined for specific cellular models, the chemoinvasion assay can be used for the screening of inhibitors of invasiveness and angiogenesis, or to select for invasive cellular populations. This protocol can be completed in 9 h.
Similar content being viewed by others
Introduction
Cell invasion of extracellular matrices is a program activated very early in ontogeny, already a fundamental process during implantation and embryonic development. Nevertheless, owing to its destructive potential on tissue integrity, cell invasion is tightly regulated and only a few cells, mostly leukocytes, are allowed to “wander” relatively undisturbed in the body under physiological conditions. Matrix remodeling represents a fundamental step both in physiological and pathological conditions: malignant tumor cells show a chronically activated phenotype resembling that found in their normal counterparts when tissue integrity is compromised.
Evaluation of cell migration and quantification of chemotactic potential of soluble mediators has been extensively investigated, although studies limited to cell migration have an intrinsic bias: in our body, cells do not move freely but they have to continuously cross matrix and/or cellular barriers. Models of invasion of stroma and BM are, therefore, better suited to mimic physiological and pathological conditions.
A comprehensive protocol of numerous variations of the chemoinvasion assay would be very lengthy. Here, we briefly focus on the crucial step in metastasis and angiogenesis: the invasion of BM. BM is a thin continuous sheet of extracellular matrix enveloping organs and represents “the barrier” to tumor cells; its degradation makes the difference between benign and malignant tumors, and patient's life or death. The original BM invasion test was performed using rough amnion BM stripped from placenta and fixed on plastic rings1. The chemoinvasion assay in Boyden blind-well chambers developed by us in 1987 (see ref. 2 and later modified3,4) has the advantage of using reconstituted BM extracted from the murine Engelbreth–Holm–Swarm sarcoma5, commonly known as and referred to here as “Matrigel”. Matrigel can simply be applied in its cold, liquid form over filters in the Boyden chamber chemotaxis assay, allowing the formation of a polymerized, ready-to-use matrix2. This assay and its numerous variations are still widely used for testing cells with different invasive abilities. Even though it was originally created to evaluate in vitro the metastatic potential of cancer cells, the chemoinvasion assay has been frequently used with endothelial cells as a model of endothelial activation and angiogenesis6,7. A highly flexible assay, it can be easily adapted for examination of the invasive potential of a wide variety of other cell types (i.e., myofibroblasts and phagocytes) and also for the investigation of other matrix barriers. In addition, it is adaptable and modular (i.e., it can be applied to the use of monolayers of cells grown on BM, mimicking physiological barriers).
In angiogenesis, the endothelial cells partially act similar to extravasating tumor cells, crossing vessel BMs with high efficiency. As the set of proteolytic enzymes (mainly matrix metalloproteinases (MMP) and urokinase plasminogen activator (uPa)) involved in BM digestion by endothelial and tumor cells is also largely shared, protease inhibitors could target cancer progression both by inhibiting metastatic and endothelial cell invasion. MMPs can be induced by oncogenes8,9 and by inflammation10,11,12 indicating their central role in metastasis and tumor progression through disruption of microenvironmental confinement.
Several growth factor tyrosine kinase receptors when stimulated by their ligands, or activated through transformation, induce tumor cell and endothelial cell motility and invasion. For example, vascular endothelial growth factor has been implicated in both lymph node metastasis and angiogenesis of esophageal squamous cell carcinoma13. Vascular endothelial growth factor, basic fibroblast growth factor, platelet-derived growth factor, epidermal growth factor, hepatocyte growth factor and other growth factors stimulate the motility and invasion of target cells, and are frequently used as chemoattractants in the chemoinvasion test. One important consideration for this test is that it allows comparative evaluation of basal random cell movements, shown by several cell lines. This “constitutive” activated status, mostly linked to the loss of cell–cell contact inhibition, or the use of growth factors in standard culture medium, can cause considerable background migration. Thus, in the chemoinvasion assay, the use of a control with no chemotactic stimuli is imperative to evaluate the true chemotactic potential of a specific molecule. However, when the test is applied to screening of inhibitors acting on pathways unrelated to the chemoattractant used to induce invasion, this control can be omitted.
The most frequent inaccuracy seen in the literature regarding the use of the chemoinvasion assay to measure the “pure” invasive components is the uncoupling of the chemoinvasion assay from a parallel chemotaxis assay, where no BM coating is applied. In fact, only contemporary evaluation of both phenomena can show the relative contribution of invasion to the total number of cells that “moved” to the lower part of the invasion chamber. The ratio between invaded and migrated cells in the absence of Matrigel or similar barrier is defined as the “invasive index”. This concept can be exemplified as follows: if you are comparing two cell lines to determine which one is more invasive and you only perform the chemoinvasion assay, you will never be able to say if the cells migrating more abundantly have actually invaded more (i.e., by activation of MMPs) or are simply more rapidly motile (they possess the same amount of active MMPs of their counterparts, but they are able to assemble microtubules or contractile filaments with higher efficiency). As a paradox, the comparison of chemotaxis and chemoinvasion assays can identify “slow” cells with very high invasive properties and “fast” cells with low invasive properties, inverting the results suggested by a chemoinvasion assay performed alone.
Many other factors can strongly affect chemoinvasion: the choice of the chemoattractant is crucial, as single growth factors frequently show a weak activity on target cells, whereas more complex “physiological” attractants, such as conditioned media from other cell sources, can represent optimal “cocktails” active on several cell models. Thus, if it is not necessary to test a specific molecule for chemotactic properties, but rather to find inhibitors of invasion, conditioned medium from normal murine or human fibroblasts would be the most appropriate efficacious attractant. This medium is particularly enriched in growth factors and matrix components, favoring both cell spreading on the chemotaxis filter and its migration through it.
The polycarbonate filters used for this test produced with different pore sizes must be chosen carefully: the cells under investigation should be able to pass through the pore size by actively “squeezing” through it, but should not passively fall through excessively large pores or get physically blocked by pores that are too small. For this reason, large epithelial and endothelial cells should be tested using filters with pores of 12 μm diameter. Many other types of cells do well with the standard 8 μm size, whereas leukocytes require 5 μm (monocytes, dendritic cells, lymphoblasts) or 3 μm (neutrophils, eosinophils) pore sizes. Very high backgrounds or no migration are respectively the effects of too large or too small pores.
Once the proper filter is identified, it is also possible to test several matrices: although chemoinvasion assay usually uses a full BM, Matrigel, for different purposes—that is, the test of inhibitors or pathways active only on a single MMP species—it is possible to create alternative barriers. Type IV collagen is suitable for testing MMP-2 and MMP-9 activity, whereas type I or III collagen will work for MMP-1 (see refs. 14,15). If the test is performed under sterile conditions, it can be used as a rapid procedure for selecting highly invasive and aggressive subclones of cells by isolation of the invaded cells from the lower filter surface by trypsinization, the selection is usually repeated several times.
The chemoinvasion assay can give affordable and reproducible results once established for the cell lines of interest. Cell and Matrigel concentrations, time of incubation and proper chemoattractants are to be defined for each cell type, although the standardized method described here will satisfy approximately 80% of cell types. Some compromise could be necessary when high and low invasive cells have to be compared, to avoid saturation or absence of invaded cells on filter. In an optimal test, you will find very low background (serum-free culture medium (SFM)-induced migration) and invasive cells being around 50% of the number of cells migrated by chemotaxis alone.
Occasionally, one finds that the invasive cells are more abundant than the migrated cells; this paradox is linked to two simultaneous phenomena: excessively low Matrigel quantities (weak or null barrier) and the haptotactic response of cells to some Matrigel components, typically laminin. In these conditions, a more stringent Matrigel concentration and the use of low-concentration Matrigel (5 μg per 50 μl) to coat chemotaxis filters will re-establish good protocol standards, even if a higher background migration can be expected.
Recently, readily available transwell-based chemoinvasion assays (http://www.bdbiosciences.com/discovery_labware/products/ display_product.php?keyID=128; http://www.chemicon.com/browse/productdetail.asp?ProductID=ECM550; http://www.trevigen.com/cultrexbme_cellinvasion.php) have had increasing success: this system is particularly suited for those laboratories not routinely using this technique, as it is accessible, disposable and well standardized. However, it is less flexible and quite expensive. One substantial advantage of this system is the sterility of the equipment. It is particularly suitable for living-cell selection. On the contrary, the fixed concentration of the Matrigel matrix used to cover porous filters can significantly increase test duration, which frequently creates difficulty in comparison of chemotaxis and chemoinvasion results when cells with low invasive potential are tested. Blind-well chemotaxis chambers are recyclable and, if are available in sufficient numbers, it is easy to process about 30–50 samples per day.
In this protocol, we describe the optimization of the original chemoinvasion method, based on chemotaxis blind wells (Fig. 1) derived from the Boyden chambers. This includes the manual coating of chemotaxis filters with an ideal Matrigel concentration2, a method that is so far the easiest, most economic, reliable and flexible, and is applicable to any cell type, providing rapid results.

The Matrigel-coated area (pink) must be larger than the surface of the lower chamber of the blind well containing the chemoattractants (orange).
Materials
Reagents
-
Viable, sub-confluent cells (usually 70% confluence is used) in tissue culture flasks.
Critical
Evaluate cell number before preparing the test: about 450,000 cells for each experimental point (in triplicate 150 × 3) should be calculated.
-
SFM without any growth factor
Critical
Use the same SFM (e.g., DMEM or RPMI) to prepare both chemoattractants and cell suspensions.
-
PBS and trypsin for cell harvesting
-
Chemotaxis inducer: usually a growth factor or a serum-free cell supernatant (in this protocol, we will refer to fibroblast-conditioned medium, which can be prepared as stocks by incubating subconfluent T75 flasks of fibroblasts with 8 ml of SFM for 24 h. After incubation, the culture supernatant is collected, centrifuged (at 800g for 5 min at room temperature (18–22 °C)) to eliminate cell debris, aliquoted in single 4 ml doses and conserved at −20 °C for up to 6 months)
-
13-mm-diameter polycarbonate chemotaxis filter (polyvinyl pyrrolidone-free) with suitable pore size: the 8 μm size usually works with most sarcoma and mesenchymal cells; 12 μm is suitable for epithelial and endothelial cells
Critical
Test filter lots for their ability to support cell attachment and migration per se: we note that some lots apparently do not support cell adhesion even in the presence of a matrix coating. Alternative sources of filters are Nucleopore, GE Osmonics' Labstore (http://www.osmolabstore.com/OsmoLabPage.dll?BuildPage&1&1&607) or Neuro Probe (http://www.neuroprobe.com/protocols/info_filters.html#Blind)
-
Gelatin (from porcine skin; Sigma)
-
Matrigel at a basal protein concentration of 10–14 mg ml−1 (BD Biosciences, http://www.bdbiosciences.com/discovery_labware/products/ display_product.php?keyID=230). Other alternatives with different commercial names are also available, for example, Sigma ECM gel (http://www.sigmaaldrich.com/catalog/search/ProductDetail?ProdNo=E1270&Brand=SIGMA) and ECMatrix by Chemicon (http://www.chemicon.com/browse/productdetail.asp?ProductID=ECM550). At any rate, each source and batch of matrix should be functionally tested
Critical
Matrigel should not contain polymerized flakes, which are an indication of poor preparation/conservation, compromising the real total protein quantification of your stock.
-
95% ethanol (ETOH v/v) for cell fixation
Caution
ETOH is an irritant and highly flammable.
-
Water
-
Toluidine blue 2% in distilled water for cell staining; dissolve with a stirrer and filter to eliminate debris. A wide variety of other cell staining systems can be used as an alternative
Equipment
-
Blind-well chemotaxis chambers (Fig. 1) (http://www.neuroprobe.com/products/blind.html): calculate three wells for each chemoinvasion experimental point and three wells for chemotaxis controls. In this protocol, we describe the use of wells with a lower volume of 200 μl (chemoattractant chamber) and an upper volume of 800 μl (loading cell chamber). However, for small samples, chambers with 25–50 μl lower and 200 μl upper well exist. Blind-well racks are available for organizing the chambers
-
Filters must be fixed and stained. We use custom-made metal filter boxes like the one shown in Figure 2, which allow bulk processing of separated filters. Alternatively, other methods can be used ranging from pinning the filters to wax carriers, or processing them in multiwell plates. A simple tea filter followed by individual separation of labeled floating filters at the final step could be an inexpensive alternative
Figure 2: These custom-made filter boxes are useful for singularly managing the filters for chemotaxis and chemoinvasion tests, but unfortunately are not commercially available and must be fabricated. 
A simple plastic or metal tea-filter could be used instead, with careful separation of the numbered filters in aqueous suspension at the end of the experiment.
-
Forceps with very thin tips to manage filters
-
Micropipettes with variable volume (a set of variable volume pipettes of 20, 200 and 1,000 μl is usually sufficient)
-
Filter paper
-
Thermostatic cell incubator with CO2
-
Plastic Petri dishes for cell culture, 15 ml polypropylene tubes
-
Four glass or plastic boxes (those used standardly for histological staining preparation)
-
A pen with ink resistant to ETOH

Procedure
Preparation of gelatin-coated filters for chemotaxis
Timing 2 h
-
1
Add 5 mg of gelatin to 1 liter of distilled H2O, warm it in a microwave oven at high power (750 W) for about 30 s; when approximately 60 °C is reached, gelatin will completely dissolve. Mix well.
-
2
Transfer the gelatin solution into a thermostatic bath at 98 °C.
-
3
Add filters to the metal filter box and immerse them in gelatin solution for about 1 h. If filter box is not available, filters can be left to freely float in the gelatin solution.
-
4
Transfer the filter box to an oven at 98 °C and leave until the filters are completely dry (usually less than 1 h). If a filter box is unavailable, single filters need to be picked using a forceps and put on an aluminum foil for oven desiccation.
-
5
Put filters in a Petri dish or in their original box.
Pause point
Gelatin-coated filters can be kept for 3–4 months at room temperature; so it is advisable to prepare large numbers in batches.
Critical Step
When testing endothelial cells, the gelatin coating is insufficient to attain good chemotaxis controls: in this case, filters should be coated manually with 50 μl of a 0.1 mg ml−1 solution (1% (v/v) acetic acid in water) of collagen type IV, available from the same sources as the Engelbreth–Holm–Swarm sarcoma matrix. The procedure is the same as described for Matrigel coating in Steps 7–11. These filters must be used within 3–4 days.
Polyvinyl pyrrolidone filters can be used as an alternative, by performing stripping in 5% acetic acid/water (v/v) solution at 60 °C before matrix coating; however, in our experience, the above method gives the best results.
-
6
Before using chemotaxis filters, mark them with numbers with an ETOH-resistant pen to distinguish each at the end of the test.
Preparation of Matrigel working aliquots
Timing 2 h
-
7
Thaw Matrigel stock solution in a water–ice bath.
Pause point
Frozen Matrigel stock can also be thawed overnight at 4 °C in a refrigerator, sparing much time.
Critical Step
Never force Matrigel to thaw in warm water or let it reach room temperature, as an irreversible polymerization often occurs.
-
8
Dilute Matrigel with cold (4 °C) SFM to a working concentration of 10 mg ml−1. Note that Matrigel is quite viscous; thus, care must be taken to thoroughly mix components after addition.
Pause point
Aliquots (100 μl) of Matrigel at 10 mg ml−1 can be placed in small tubes and stored at −20 °C for more than 1 year.
Preparation of Matrigel-coated filters
Timing 30 min
-
9
Prepare a tube with cold distilled water for further dilutions of Matrigel.
-
10
Collect the necessary polycarbonate filters (three for each experimental point). Polycarbonate filters have one rough and one shiny face. Place all filters with the rough face up in a Petri dish, ensuring they are not in contact with each other. Number each one on the rough face with a small consecutive number on the filter border using a pen with ETOH-resistant ink.
-
11
Make the first Matrigel dilution by adding 900 μl of cold water to the aliquot (1 mg ml−1). It is important to make Matrigel dilutions stepwise to the final concentration. The first time you test a cell line, the most appropriate Matrigel concentration should be established by performing a pilot test. Further dilute Matrigel 1:2, 1:3 and 1:4 in water, and manually coat each filter with 50 μl of this solution (filters will be coated with a layer of 25, 16.6 or 12.5 μg of Matrigel, respectively). This range is usually enough to find the optimal Matrigel barrier concentration (one that gives an approximately 50% reduction of cell migration as compared to chemotaxis controls). During filter coating, care must be taken to insure that the Matrigel solution is applied evenly over most of the surface of the filter, leaving a small rim (1 mm) uncoated to prevent leakage off the filter (see Fig. 1). Let filters air-dry completely at room temperature or at 37 °C (approximately 40 min). Then close the Petri dish.
Critical Step
Pay attention to electrostatic charges on the Petri dish; these can cause the filters to flip over or can induce capillary wicking of all the liquid Matrigel through the pores to the underside of the filter before it polymerizes.
Pause point
Store the Petri dish at 4 °C; the filters must be used within 48 h.
Preparation of cells
Timing 20 min
-
12
Rinse the tissue culture flask with PBS and harvest cells with trypsin. Remove trypsin by centrifugation, eventually with neutralization by addition of SFM with a small amount of serum.
-
13
Centrifuge the cell suspension to obtain a vital pellet (for most cells 5 min at 800g is sufficient). Discard the supernatant.
-
14
Resuspend the cell pellet in a few milliliters of SFM and count a small aliquot of cell suspension after staining with Trypan blue (0.04 % Trypan blue in PBS) to exclude dead cells.
Caution
Trypan blue is a possible cancer hazard: wear gloves.
-
15
Adjust the cell density to 150,000 cells per ml. According to the cell tested, this concentration can range from 100,000 to 300,000 cells per ml. The number of invaded cells is proportional to the number of initial cells plated in the upper well of the chambers. Use triplicates for each experimental point.
-
16
Every blind well should contain 800 μl of cell suspension; so the precise amount of cells needed is easily calculated, although it is always advisable to prepare the cell suspension in excess. If you are testing an inhibitor, unless it is active against the chemoattractant (i.e., growth factor antibody), it should be added to the cell suspension; so it is necessary to separate cells in the necessary aliquots.
Mounting blind wells
Timing 10 min
-
17
Prepare open blind wells on their racks.
-
18
Use a micropipette to collect 200 μl of chemoattractant and gently dispense it in the lower chamber of a blind well.
Critical Step
It is necessary to create a convex meniscus to avoid bubble formation; so it is advisable to increase the amount of chemoattractant by a few microliters (i.e., pipetting 205–210 μl per well).
-
19
Let Matrigel-coated filters detach from the Petri dish by pouring some SFM into the dish.
-
20
Take the appropriate filter by the edge with forceps, drip off residual SFM, and lower it into its location inside the blind well, paying great attention to let it slip in, so as not to create bubbles or trap air in the chemoattractant; this will block cell invasion.
-
21
Close the blind well by screwing the upper well on top of the lower and closing the filter between them.
-
22
Repeat this step until all wells are filled and closed.
-
23
Add 800 μl of cell suspension per well following your protocol.
-
24
When inhibitors are tested, they should be mixed with the cells, either at the time of experiment or with a preincubation time ranging from a few minutes to 2 h (some metabolic modulators—like retinoids—need a 24–48 h pretreatment during cell culture).
-
25
Incubate cells at 37 °C in a humidified atmosphere plus 5% CO2 for 6 h.
Critical Step
Although a 6-h incubation usually works for most cell types, the incubation period must be reduced in some models (4 h for exceptionally invasive cells, 90 min for monocytes, 30 min for neutrophils) or increased in some models (epithelial cells from an epidermis usually need an overnight incubation).
Stopping chemoinvasion
Timing 30 min
-
26
Prepare four histology glass boxes or similar vessels, containing, respectively, PBS (with Ca and Mg), 95% ETOH, tap water and toluidine blue solution, plus a large box for final washings.
-
27
Wash a filter box (the one depicted in Fig. 2) in PBS and open it.
-
28
Empty the upper compartment of blind well by rapidly inverting it and shaking the fluid away; alternatively, pipet the supernatant out, which can be stored for subsequent studies on the products of invading cells; twist off the cap and take the filter by the border with forceps, taking care not to strip or scratch it.
-
29
Clean the upper part of the filter on wet filter paper or dry paper towels to eliminate unmigrated cells. Cotton swabs can also be used, but one must be careful not to damage the migrated cells on the underside of the filter.
Critical Step
Pay attention to identify the right face of the filter looking at the number signed on it, as shiny and rough faces are not identifiable when the filter is wet.
-
30
Put the filter inside the filter box with the lower side up. This side contains the invaded cells.
-
31
Repeat until the filter box is filled; then close it.
-
32
Fix cells in 95% ETOH for 30 s.
-
33
Wash the cells in water.
-
34
Stain the cells in toluidine blue for 7–10 min.
-
35
Wash the filter box three times in a large volume of water.
-
36
Collect filters and put them on a glass slide with the unmigrated cells face down and let them dry completely.
Pause point
Filters can be left overnight in this condition.
-
37
Detach the filters from the glass slides with a rapid movement. This procedure will leave residual unmigrated cells stripped onto the glass slide.
-
38
Prepare new glass slides and put small drops of microscope immersion oil onto the slides. Place one filter onto each drop and close with a coverslip. Usually six filters in three staggered rows fit on a slide under a single coverslip.
-
39
Wash the blind wells with a mild, diluted detergent, rinse them thoroughly in tap water and, finally, in distilled water. Let them dry completely before closing.
Evaluation of cell invasion
Timing about 1 h for 12 filters
-
40
Count migrated (chemotaxis filters) and invaded (chemoinvasion filters) cells under a microscope at a magnification of × 160 to × 400 according to cell size and density (see Fig. 3). On each filter count at least 5–10 random fields (more fields per filter need to be counted if cells are not homogeneously distributed).
Figure 3: The appearance of migrated and invaded cells of different origin on filter surface. 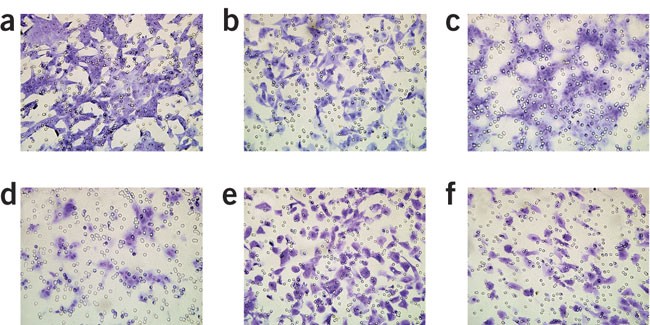
(a) Chemotaxis of mammary carcinoma cells, (b) the invasion of the same cell line. (c,d) Chemotaxis and invasion of endothelial cells. These cells usually do not scatter uniformly on the filter surface but form strings and clusters. (e,f) Chemotaxis and invasion of Kaposi's sarcoma cells. In all the panels, one can notice the smaller dimension of filter pores as compared to cells, which does not permit passive dropping of cells to the lower chamber.
-
41
Cell quantification can also be performed by evaluating the dye density of the scanned filter surface or by image analysis; fluorescent staining methods have also been developed by BD Biosciences (http://www.bdj.co.jp/labware/pdf/angiosympo.pdf). These methods, although more rapid, can generate errors if the invaded cell layer is partially damaged or irregular. Direct ocular counting so far has provided the best results, also allowing evaluation of the morphology of the invaded cells.
-
42
Average the values of migrated cells per field for each filter and use these values to calculate the mean and s.d. for the triplicate time point; so results can be shown as “number of cells/unit field”.
-
43
Calculate the invasive potential of a cell by the invasion index = no. of invaded cells/no. of migrated cells, or as percentage of invasive cells: no. of invaded cells/no. of migrated cells × 100. Results can be plotted as depicted in Figure 4.
Figure 4: An example of molecular correlates of invasive activity. 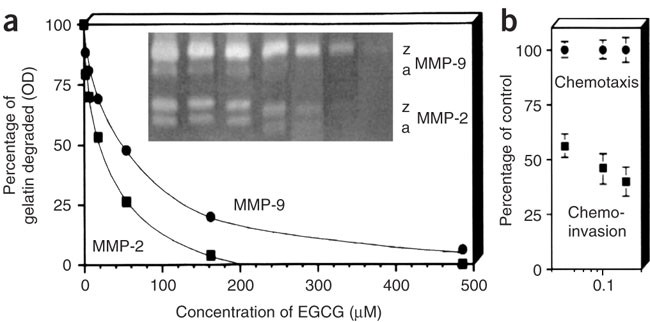
(a) Zymographic evidence of direct suppression of MMP by increasing concentrations of epigallocatechin-3-gallate (EGCG). Densitometric values of digestion bands were plotted for zymogen (z) and activated (a) forms of each gelatinase. (b) Cells invaded to the lower filter surface after 5 h on gelatin (chemotaxis) and Matrigel (chemoinvasion) coatings. EGCG dose dependently inhibits MMP activity and invasion but not chemotaxis. (Taken from ref. 20.)


Troubleshooting
Troubleshooting advice can be found in Table 1.
Anticipated results
Critical components of this assay are the measurement of background migration in the absence of a chemoattractant and migration in the absence of matrix barrier, that is, Matrigel coating.
Certain cells such as fibroblasts can be highly migratory but not invasive. Proper controls in addition to parallel chemotaxis assays include SFM alone in the lower chamber, which gives an index of random cell invasion (negative control), eventual positive controls with chemoattractants or inhibitors known to be active. “Checkerboard” assays where different concentrations of chemoattractants are placed in the upper and lower chambers, used in chemotaxis assays to assess chemokinesis as opposed to chemotaxis, can also be performed for invasion.
Invasion is time dependent. In the original paper2 we showed that the number of invaded cells is proportional to time of incubation in the blind-well chambers. This correlates well with the idea of invasion and metastasis being a matter of time also in the body. A mesenchymal–fibroblastic lineage generally needs 3–4 h to migrate and/or invade, whereas longer time periods are expected for endothelial and epithelial cells. Neutrophils are very fast, migrating and invading in less than an hour, as rapid invasion is part of their “duties”.
To assess the ideal conditions of invasion in a chemoattractant-stimulated sample, investigators should first perform some preliminary studies, testing various time durations and Matrigel concentrations. When compared with the transwell method, the original blind-well assay might seem more laborious; however, it is ideal for large-scale studies and for comparing migration and invasion with different coatings, as every transwell has one type of coating. The blind wells can be reused indefinitely; some of ours have been in use for over 10 years and are still in good condition. They can be purchased (in commercial wells, the bottom is acrylic, whereas the top is white acetate) or also custom cut in plastic (usually Teflon, which is easier to clean). Blind wells can also be made sterile and used for selection of cells; they fit in a 50 ml disposable centrifuge tube, which helps collecting the migrated cells to the bottom. We originally demonstrated this application for chemotaxis16 and other groups have used it to select invasive cells17. Invaded cells are cells selected for their intrinsic higher invasive phenotype, confirming that “metastatic seeds” or stem cells are already present in an original population, as recently postulated18,19.
Also, certain drugs can prevent invasion without affecting chemotaxis. A typical example of an experiment performed with HT-1080 fibrosarcoma cells is shown in Figure 4 (reprinted with permission of Nature group20). EGCG, an anti-invasive flavonoid inhibiting MMP-2 and MMP-9 activity, gives a dose-dependent block of tumor cell invasion, whereas it is essentially inactive on chemotaxis, showing selective effects. Our assay can be expected to be a good predictive preclinical test for anti-invasive anti-angiogenic drugs. A vast literature demonstrates that practically all anti-invasive inhibitors in trial, starting from the initial metalloproteinase blockers, do inhibit invasion in the Matrigel chemoinvasion assay. The test can therefore be used for selecting appropriate inhibitors of metastasis and/or angiogenesis before animal testing and after clinical trials.
The assay is compatible with the analysis of invaded cells, both morphologically and phenotypically, after selection. For example, we are currently performing microarray genotyping of selected breast cancer cells (unpublished results).
The assay can predict for high amounts of proteases in the supernatants of invading cells. Supernatants can be collected easily at the end of invasion and collected for zymography21. Also, stimulation of growth factor production during invasion can be assessed by ELISA.
Two major applications are envisaged for the chemoinvasion assay: the screening of potential pharmaceuticals and the identification of the capacity to induce invasion or “in vitro” angiogenic potential for a given factor. We and others have identified a number of new invasion inducers and angiogenic factors by applying this method, for example, the angiogenic activity of the HIV-Tat protein22. The angiogenic activity of Tat, confirmed by other groups, is associated to its capacity to activate the KDR tyrosine kinase22, involved in invasive migration of endothelial cells, and protease production.
References
Liotta, L.A., Lee, C.W. & Morakis, D.J. New method for preparing large surfaces of intact human basement membrane for tumor invasion studies. Cancer Lett. 11, 141–152 (1980).
Albini, A. et al. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 47, 3239–3245 (1987).
Albini, A. Tumor and endothelial cell invasion of basement membranes. The matrigel chemoinvasion assay as a tool for dissecting molecular mechanisms. Pathol. Oncol. Res. 4, 230–241 (1998).
Albini, A., Benelli, R., Noonan, D.M. & Brigati, C. The “chemoinvasion assay”: a tool to study tumor and endothelial cell invasion of basement membranes. Int. J. Dev. Biol. 48, 563–571 (2004).
Grant, D.S. et al. The basement-membrane-like matrix of the mouse EHS tumor: II. Immunohistochemical quantitation of six of its components. Am. J. Anat. 174, 387–398 (1985).
Thompson, E.W. et al. Differential regulation of growth and invasiveness of MCF-7 breast cancer cells by antiestrogens. Cancer Res. 48, 6764–6768 (1988).
Benelli, R. & Albini, A. In vitro models of angiogenesis: the use of Matrigel. Int. J. Biol. Markers 14, 243–246 (1999).
Baruch, R.R. et al. Altered matrix metalloproteinase expression associated with oncogene-mediated cellular transformation and metastasis formation. Cell Biol. Int. 25, 411–420 (2001).
Kuo, L., Chang, H.C., Leu, T.H., Maa, M.C. & Hung, W.C. Src oncogene activates MMP-2 expression via the ERK/Sp1 pathway. J. Cell Physiol. 207, 729–734 (2006).
Denkert, C. et al. Cytokine-suppressive anti-inflammatory drugs (CSAIDs) inhibit invasion and MMP-1 production of ovarian carcinoma cells. Cancer Lett. 195, 101–109 (2003).
Ishii, Y. et al. Induction of matrix metalloproteinase gene transcription by nitric oxide and mechanisms of MMP-1 gene induction in human melanoma cell lines. Int. J. Cancer 103, 161–168 (2003).
Melnikova, V.O., Mourad-Zeidan, A.A., Lev, D.C. & Bar-Eli, M. Platelet-activating factor mediates MMP-2 expression and activation via phosphorylation of cAMP-response element-binding protein and contributes to melanoma metastasis. J. Biol. Chem. 281, 2911–2922 (2006).
Mukherjee, T., Kumar, A., Mathur, M., Chattopadhyay, T.K. & Ralhan, R. Ets-1 and VEGF expression correlates with tumor angiogenesis, lymph node metastasis, and patient survival in esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 129, 430–436 (2003).
Guenzi, E. et al. The guanylate binding protein-1 GTPase controls the invasive and angiogenic capability of endothelial cells through inhibition of MMP-1 expression. EMBO J. 22, 3772–3782 (2003).
Rossello, A. et al. N-i-Propoxy-N-biphenylsulfonylaminobutylhydroxamic acids as potent and selective inhibitors of MMP-2 and MT1-MMP. Bioorg. Med. Chem. Lett. 15, 1321–1326 (2005).
Albini, A., Muller, P.K. & Parodi, S. A method to select cell populations with enhanced chemotactic activity. Biosci. Rep. 4, 311–318 (1984).
Hendrix, M.J., Seftor, E.A., Seftor, R.E. & Fidler, I.J. A simple quantitative assay for studying the invasive potential of high and low human metastatic variants. Cancer Lett. 38, 137–147 (1987).
Pfeffer, U., Noonan, D. & Albini, A. Re: microarray studies challenge theories of metastasis. J. Natl. Cancer Inst. 95, 829 (2003).
Pomeroy, S.L. et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415, 436–442 (2002).
Garbisa, S. et al. Tumor invasion: molecular shears blunted by green tea. Nat. Med. 5, 1216 (1999).
Leber, T.M. & Balkwill, F.R. Zymography: a single-step staining method for quantitation of proteolytic activity on substrate gels. Anal. Biochem. 249, 24–28 (1997).
Albini, A. et al. The angiogenesis induced by HIV-1 tat protein is mediated by the Flk-1/KDR receptor on vascular endothelial cells. Nat. Med. 2, 1371–1375 (1996).
Acknowledgements
This protocol has been supported by grants from the Compagnia di San Paolo, the AIRC (Associazione Italiana per la Ricerca sul Cancro), the MIUR Ricerca Finalizzata and ISS progetto Italia-USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Albini, A., Benelli, R. The chemoinvasion assay: a method to assess tumor and endothelial cell invasion and its modulation. Nat Protoc 2, 504–511 (2007). https://doi.org/10.1038/nprot.2006.466
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2006.466
This article is cited by
-
A Wnt-mediated phenotype switch along the epithelial–mesenchymal axis defines resistance and invasion downstream of ionising radiation in oral squamous cell carcinoma
British Journal of Cancer (2021)
-
A microcarrier-based spheroid 3D invasion assay to monitor dynamic cell movement in extracellular matrix
Biological Procedures Online (2020)
-
A Targeted Quantitative Proteomic Method Revealed a Substantial Reprogramming of Kinome during Melanoma Metastasis
Scientific Reports (2020)
-
Rapid Cancer Diagnosis and Early Prognosis of Metastatic Risk Based on Mechanical Invasiveness of Sampled Cells
Annals of Biomedical Engineering (2020)
-
Acetyl-L-Carnitine downregulates invasion (CXCR4/CXCL12, MMP-9) and angiogenesis (VEGF, CXCL8) pathways in prostate cancer cells: rationale for prevention and interception strategies
Journal of Experimental & Clinical Cancer Research (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
