Abstract
I report here a detailed protocol for seamless genome editing using the piggyBac transposon in human pluripotent stem cells (hPSCs). Recent advances in custom endonucleases have enabled us to routinely perform genome editing in hPSCs. Conventional approaches use the Cre/loxP system that leaves behind residual sequences in the targeted genome. I used the piggyBac transposon to seamlessly remove a drug selection cassette and demonstrated safe genetic correction of a mutation causing α-1 antitrypsin deficiency in patient-derived hPSCs. An alternative approach to using the piggyBac transposon to correct mutations involves using single-stranded oligonucleotides, which is a faster process to complete. However, this experimental procedure is rather complicated and it may be hard to achieve homozygous modifications. In contrast, using the piggyBac transposon with drug selection–based enrichment of genetic modifications, as described here, is simple and can yield multiple correctly targeted clones, including homozygotes. Although two rounds of genetic manipulation are required to achieve homozygote modifications, the entire process takes ∼3 months to complete.
Similar content being viewed by others


Introduction
Gene targeting via homologous recombination is a powerful method by which endogenous loci can be specifically modified in a predetermined manner, and it has been demonstrated in a wide range of organisms such as yeast and mice1,2. Almost the entire set of yeast genes was individually knocked out a decade ago3,4 and the international knockout mouse project has recently achieved more than 9,000 unique gene-targeting events in mouse embryonic stem cells (ESCs)5. The isolation of human ESCs, first reported in 1998 (ref. 6), has renewed attention to the stem cell biology field and highlighted the potential for therapeutic applications. In contrast to mouse ESCs, various technical difficulties have hampered genetic engineering–based research in human ESCs. One such difficulty was the inefficiency of single-cell cloning, which was caused by prominent apoptosis upon single-cell dissociation. Fortunately, this problem has been circumvented by the discovery of a Rho-associated protein kinase (ROCK) inhibitor (Y-27632), which suppresses apoptosis7. This, in turn, allows efficient single-cell cloning. Conventional gene targeting also seemed to be inefficient in human ESCs. This issue has only recently been solved with the development of double-strand break (DSB)-stimulated gene targeting8,9.
It has been known since the mid-1990s that DSBs can be repaired efficiently via one of the DSB repair pathways, namely homology-directed repair10. Homologous sequences (also known as the repair template or donor) can be located on the same chromosome adjacent to a DSB site, on a homologous chromosome or on an exogenously provided plasmid DNA (Fig. 1). By providing a predesigned repair donor (also referred to as a targeting vector) by means of plasmid transfection, DSB sites can be modified at will, resulting in targeted modification. Under optimized conditions, DSB-stimulated gene targeting can yield a targeting efficiency of 3–5% (ref. 11), which is several orders of magnitude higher than that obtained with conventional gene targeting (i.e., without DSBs). To harness the DSB repair machinery for genome modification, a method for creating site-specific DSBs at endogenous loci was required. A decade of technology development has resulted in two platforms: zinc finger nucleases (ZFNs)12,13 and transcription activator–like effector nucleases (TALENs)14,15. Both systems use custom synthesis of novel DNA-binding proteins16,17,18,19 that are fused to the FokI endonuclease domain20. Recently, an RNA-guided endonuclease has been discovered in the bacterial clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system21. It has been shown that the system is functional in mammalian cells22,23, and that it can stimulate homologous recombination in hPSCs24. In this system, construction of guide RNA–expressing vectors is simple, which is clearly advantageous for the targeting of novel sites. However, recent reports seem to show that the sequence specificity is not as high as expected25,26. Further technology development will be required before the CRISPR/Cas system can be applied to precise genome modification in hPSCs.

Two major mechanisms are involved in DSB repair: nonhomologous end joining (NHEJ) simply ligates broken ends and is associated with insertion or deletion (indel) mutations. Homology-directed repair (HDR) uses a repair donor (blue), which has sequence homology to the DSB site and results in gene conversion. The repair donor can be located on the same chromosome (i), on the homologous chromosome (ii) or on exogenous DNA (iii), i.e., plasmids. Missing sequences are retrieved from the repair donor and copied into the gap. Black circles represent centromeres.
Using these site-specific endonucleases, DSBs are readily introduced into cell lines such as HEK293 and K562 with high efficiency. However, hPSCs are less amenable to DSB induction, partly because of the inefficiency of transfection and a potential difference in the DNA repair response8. This makes it difficult to obtain correctly targeted hPSCs, which necessitates positive drug selection and screening. In addition, to restore the transcription unit of the targeted gene after gene conversion, the drug selection markers need to be removed from the correctly targeted clones. Removal of drug selection markers is typically performed by either the Cre/loxP or the Flp/FRT systems27. However, both systems leave behind a single loxP or FRT site after drug cassette excision (Fig. 2). Given that not all regulatory elements are known, the possibility that these small sequences may disturb some functional elements and adversely affect the phenotype of the cells cannot be discounted28. To resolve this issue, I developed a piggyBac transposon–based approach (Fig. 3), which allows precise and scarless modification of the genome; I have achieved homozygous correction of a point mutation causing α-1 antitrypsin deficiency in patient-derived induced pluripotent stem cells (iPSCs)29.

(a) Traceless genome modification using the piggyBac transposon. (i) The conventional technique using the Cre/loxP system leaves one loxP sequence after marker excision. Green arrowheads represent loxP sites. (ii) The piggyBac transposon carrying a drug selection marker is inserted into a genomic TTAA site. The intended modification (star) is present on one of the homology arms flanking the piggyBac transposon. This is followed by excision of the piggyBac transposon with PBase. Apart from the modified site, the genomic sequence remains the same. (b) The bipartite nonautonomous transposon system. The DNA transposon can be designed to carry any DNA sequence (gene of interest, GOI) flanked by terminal inverted repeats (blue and red arrowheads). The transposase is supplied in trans from a separate expression vector. (c) Traceless excision. A piggyBac transposon is integrated into its target TTAA site by the action of a piggyBac transposase (PBase). Upon integration, TTAA duplicates at both ends of the transposon. This is known as target-site duplication. When the PBase excises the transposon, the original sequence is restored. Most types of DNA transposon leave small insertions upon excision. This is known as a footprint.

Human iPSCs represent a potential source for generating unlimited quantities of cells for autologous transplantation. However, the application of iPSC-based therapies to genetic disorders requires the correction of disease-causing mutations in a manner that is fully compatible with their clinical use. The method described in this protocol meets this requirement. Moreover, the high efficiency of this genome modification method allows production of multiple lines of patient-specific corrected iPSCs. The ability to make precise genome modification in hPSCs (including both ESCs and iPSCs) not only will benefit cell-based therapies but also will be useful for in vitro disease modeling. Around 0.1% of the human genome varies in the population, and several thousands of single-nucleotide polymorphisms (SNPs) are present in protein-coding regions30. There are also copy number variations31 and SNPs in regulatory elements32, some of which have been implicated in inherited disorders. These natural variations will influence the differentiation potential of hPSCs33,34 and result in inappropriate interpretation of in vitro phenotypes when compared with 'control' hPSC lines that share little or no genetic variation. Precise genome modification in hPSCs allows us to generate isogenic controls that can be used as a point of reference when analyzing phenotypes of genetically modified cells.
Direct reprogramming of somatic cells into human iPSCs has now become increasingly common. It is now considerably easier to obtain hPSCs with genetic disease backgrounds. Moreover, the technology to introduce site-specific DSBs using ZFNs or TALENs has become more accessible. These recent advances allow researchers in a wide range of discipline to study disease etiology using genetically engineered hPSCs. It is hoped that this process will allow us to gain deeper insight into the pathophysiology of human diseases.
Comparison with other methods
The protocol described here uses plasmid DNA as the source of targeting vectors. As an alternative, single-stranded DNA oligonucleotides (ssDONs) can also be used19,35,36. These are typically 60–120 nt long and comprise a donor (substitution) sequence and homologous sequences of 30–60 nt on both sides. An advantage of ssDON-mediated modification is that only one round of genetic manipulation is required. In contrast, two rounds of genetic manipulation are required when using this protocol. However, the experimental procedure for ssDON-mediated modification is much more complicated. For example, cells need to be transfected with ZFN or TALEN expression vectors, ssDONs and a fluorescent marker. This marker is typically GFP, which allows successfully transfected cells to be sorted by flow cytometry. In addition, ssDON-mediated modification is quite inefficient, and thus a large number of colonies (200–400) generally have to be screened. Out of these, only a few will be correctly modified. In contrast, drug selection–based enrichment of genetic modifications allows for easier and simpler screening, which means that many fewer colonies need to be analyzed. This is especially true when only heterozygous targeting is required. The frequency of obtaining correctly targeted cells using plasmid DNA as the targeting vector is typically higher than 50%. Importantly, this high efficiency allows the isolation of homozygous targeted clones.
As described above, the Cre/loxP system is most commonly used to remove drug selection markers. Its high recombination efficiency enables us to isolate modified clones by simple subcloning36, whereas piggyBac-based excision of the marker gene requires negative selection. However, when used in combination with ZFNs/TALENs, the Cre/loxP system is less flexible than the piggyBac system. The loxP site has to be inserted into introns and its position needs to be at least 250 bp away from exons in order to avoid disturbing splicing elements37. Given that the gene conversion tract is short (up to 200–300 bp), the intended modification might be outside of the optimal conversion tract. In such cases, the frequency of obtaining cells with the correct modification will be impractically low. In contrast, this protocol uses an endogenous TTAA sequence for piggyBac insertion to achieve scarless modification. Theoretically, this allows genetic modification to be carried out at almost any site in the genome, as the average frequency of TTAA sites appearing in a genome is one in every 246 bp. However, it is known that the GC content of protein-coding regions is higher than that of the rest of the genome. It is therefore possible that, in certain loci, TTAA sites are less frequent than expected. In this case, TTAA sites can be created near the intended modification site by introducing subtle, single-nucleotide substitutions (e.g., TGAA > TTAA). The substitution should be carefully chosen so as to not disturb any regulatory elements, although the chances of this happening are much smaller than using loxP sequences.
Genomic integrity of modified hPSC lines is of great importance because genomic instability might cause unpredictable phenotypic changes. Both the Cre recombinase and the piggyBac transposase are not considered to cause genome instability. For example, constitutive expression of the piggyBac transposase is not tumorigenic and does not increase mortality in mice38. However, there are some differences between these two systems in cultured mammalian cells. Although a low-dose transient expression of Cre does not seem to cause genome instability in hESCs36, cytotoxicity and genotoxicity caused by prolonged expression of Cre is well documented39,40,41. In collaboration with Nancy Craig's laboratory, we developed a hyperactive version of the piggyBac transposase and extensively characterized its potential impact on genomic instability42. We found no evidence that this version of the piggyBac transposase increases genomic instability. Moreover, we isolated disease-corrected iPSC lines with no gross abnormalities29. Taken together, both systems can be used in genetic modification of hPSCs without causing marked genomic instability. Nevertheless, it is important to confirm by karyotyping that genomic integrity is maintained.
Limitation of the protocol
One remaining and unavoidable concern of the use of ZFNs and TALENs is off-target cleavage that may have unpredictable deleterious phenotypic consequences. A number of computational and experimental methods have been developed to determine sequence specificities and to predict off-target binding sites of ZFNs or TALENs43,44,45,46,47,48. Of these, two recent reports47,48 demonstrated more direct approaches to identifying off-target cleavage sites by ZFNs and showed that not all off-target cleavage sites could be predicted computationally. They, however, also found that all off-target cleavage events are far less frequent compared with on-target cleavage events, indicating that it is possible to obtain clones with no off-target cleavage. Given that TALENs can be designed to recognize longer sequences, off-target cleavage is probably much less frequent than with ZFNs. When designing ZFNs or TALENs, it is important to confirm that the target sequence is unique in the genome, to predict off-target sites computationally and to at least check that these off-target sites do not result in mutations. Further development of fast and easy methods for more comprehensive quantitative assessment of off-target cleavage is of great importance for safer use of custom endonucleases. At the moment, in addition to performing computational analysis, the most practical approach would be to generate multiple independent cell lines and confirm that all cell lines have the same phenotype.
Experimental design
There are two major components to this protocol: the design and construction of the targeting vector and the genetic modification of hPSCs. The construction of ZFNs or TALENs is not included in this protocol. Investigators would need to obtain custom ZFNs or TALENs from commercial providers or toolkits49,50,51.
The piggyBac transposon system. The piggyBac transposon is a moth-derived DNA transposon52, which shows highly efficient transposition in a wide range of organisms such as yeast and mammalian cells53,54,55. The DNA transposon systems consist of two components (Fig. 2b): a DNA element flanked by transposon-specific terminal inverted repeats and a transposase that catalyzes excision and integration. One of the unique characteristic of piggyBac is traceless excision56 (Fig. 2c). The piggyBac transposon integrates into a TTAA site and restores this original TTAA sequence when it is excised; no 'scars' are left behind. In addition, ∼50% of the excision events are not associated with re-integration back into the host genome, and hence the transposon can be permanently removed. This remarkable feature enables us to stably introduce a transgene into the genome by piggyBac transposition and subsequently remove the transgene with no alteration to the genome when it is no longer required. I had demonstrated the usefulness of the piggyBac system by generating transgene-free iPSCs from mouse embryonic fibroblasts (MEFs)57. We have also shown the correction of a disease-causing single-nucleotide mutation in iPSCs in our recent paper29. In addition, in collaboration with Nancy Craig's laboratory, we have developed a hyperactive piggyBac transposase that is approximately tenfold more efficient than the wild-type version42.
To achieve efficient transposon excision, the epigenetic status of the transposon is of great importance. The piggyBac transposon preferentially integrates into open chromatin regions, such as DNaseI–hypersensitive sites, and can be efficiently excised from these sites58. However, piggyBac excision from heterochromatic regions is far less efficient. When a genetic modification is conducted on non-expressed genes in hPSCs, the transposon is highly likely to succumb to gene silencing. These transposons then become extremely hard to remove. For successful remobilization of the inserted transposon, it is important to continuously culture targeted hPSC clones under drug selection and keep the transposon accessible to the transposase. Nevertheless, the achievable remobilization efficiency, followed by hyperactive transposase expression, is as high as 5% (ref. 42). To efficiently isolate clones whose transposons have been removed, I use a negative selection system with Herpes simplex virus–derived thymidine kinase (HSV-tk). I chose 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-5-iodouracil (FIAU) as a thymidine analog. Traditionally, gancyclovir has been more commonly used for HSV-tk–based negative selection in mouse ESCs. However, gancyclovir is toxic at the typically used concentration (2 μM) and often induces chromosomal instability. FIAU can be used at a ten-times-lower concentration (0.2 μM), and treatment with FIAU is not associated with genomic instability29. I would therefore recommend using FIAU for negative selection.
Design and construction of the targeting vector. An example of how to construct the targeting vector is illustrated in Figure 4. First, identify the closest TTAA site to the position where a genomic substitution is to be introduced. This TTAA site will be used for the insertion of a piggyBac transposon carrying a selection cassette, PGK-puroΔtk. The piggyBac transposon is, in turn, flanked by homology arms bearing the modification to be introduced. The distance between the intended genome modification site and the TTAA site directly affects how often the modification is incorporated into the genome. Thus far, we have measured incorporation frequency at the adeno-associated virus integration site 1 (AAVS1) locus in human iPSCs and found that 80% and 70% of the targeted clones carried the anticipated modification when these are 200 and 300 bp away from the TTAA site, respectively (K.U., unpublished results). As this success rate only applies to a single modification event, the frequency of obtaining homozygous mutants is considerably lower. In our experience, only about 10% of the targeted clones are homozygous at the serpin peptidase inhibitor, clade A (α-1 antiproteinase, antitrypsin), member 1 (SERPINA1) locus29. It is therefore vital to use the closest TTAA site to the intended modification site.

(a) Schematic of the design and construction of the targeting vector. In this example, endogenous A is to be modified to G. A TTAA site located within 300 or 200 bp is used to insert the piggyBac transposon. The length of the homology arms can be between 700 bp and 1 kb. Colored arrows indicate PCR primers (LA-Fw, LA-Rv, RA-Fw and RA-Rv), which are used to amplify the homology arms. Blue portions anneal to the genome. Green and red portions indicate restriction enzyme sites (A and B) and parts of the transposon sequences, respectively. These homology arms need to be digested by the appropriate restriction enzymes and ligated into the cloning vector together with the transposon fragment. The PGK-puroΔtk cassette should be cloned into the intermediate vector by gateway cloning. (b) Genetic correction at the SERPINA1 locus. A TTAA site is created by introducing silent mutations near the intended modification site. This TTAA site is used for the insertion of the piggyBac transposon. The genomic coordinates are based on the GRCh37. (c) Sequences of PCR primers adjacent to the transposon for the SERPINA1 locus modification. The transposon sequences are shown in red and the duplicated TTAA sites are shown in green. Genomic sequences are shown in black. An arrowhead indicates the intended modification (A→G).
Alternatively, an artificial TTAA sequence can be created near the modification site by introducing silent mutations. Note that the 4-bp TTAA sequence should not simply be inserted into the genome. Care must be taken to not disturb any known regulatory elements and to not cause either amino acid substitutions or frameshifts. Once the TTAA site, whether endogenous or artificially created, is chosen, the piggyBac transposon can be inserted. The nucleotide to be modified is present in one of the homology arms flanking the piggyBac transposon and can be edited at will. It is important to note here that the TTAA site must be duplicated on either side of the transposon (Fig. 4b). To clone the homology arms, PCR primers should extend from the piggyBac repeat into the genome (Fig. 4c). The two restriction sites (NsiI and BsiWI) should also be included in the primers, as these are used subsequently to ligate the homology arms and the piggyBac transposon together (Fig. 4c). The length of the homology arm does not seem to affect the targeting frequency substantially, and it is typically between 700 and 1,000 bp. Primers, however, should not be designed in repetitive regions.
Genetic modification of hPSCs. Although the protocol described here was only directly validated in human iPSCs, there is considerable evidence that the same approach is applicable to genetic modification in human ESCs8,9,59,60. Therefore, human ESCs and human iPSCs are collectively termed hPSCs in this protocol. A number of hPSC culture methods have been described61,62,63,64,65,66,67,68. I use a basic culture protocol: co-culture with MEF feeders in the KSR- and FGF-containing medium. To be able to perform drug selection, feeder cells with a drug-resistant gene are required. These feeder cells, however, may not be always available. Our first-choice drug is puromycin (1 μg ml−1), which can kill hPSCs efficiently. In contrast, wild-type MEF-feeder cells are not as sensitive to puromycin as hPSCs are, and this allows us to use wild-type MEFs as feeder cells during puromycin selection. In our original paper29, hPSCs were treated with puromycin at 1 μg ml−1 for 48 h from day 4 and 0.5 μg ml−1 thereafter. This selection scheme should allow sufficient wild-type feeder cells to survive and to support the growth of hPSCs. In the event that feeder cells progressively die and become too sparse, they can be supplemented as necessary.
In ZFN- or TALEN-assisted targeting, all plasmids are transfected in circular forms, which can considerably reduce random integration of the plasmids. If only heterozygous targeting is required, the number of resistant colonies to be analyzed can be as low as ten. Depending on the design of the targeting vector, however, the frequency of correct modification can be low. This is especially true when homozygous targeting is desired. Typically, at least 100 clones will need to be analyzed to identify homozygotes. The maintenance of hPSCs is labor intensive, and extra caution needs to be exercised when dealing with a large number of clones. For ease of handling, we analyze the genotype of the resistant colonies by PCR on the same day they are picked. For this purpose, each colony is divided into two halves with one half being used for PCR analysis and the other half being used for expansion (Fig. 5). This allows us to perform robust analyses quickly and substantially reduces the number of clones that need to be expanded.

These colonies have been under FIAU selection for 14 d. (a) A colony measuring 1 mm in size. Cut the colony into half with a pipette tip, scrape off the first half (1–3) and transfer the cell clump (arrowhead in 3) to a 96-well plate. This will be used for lysate-PCR and is the minimum amount of cells that will enable it—smaller colonies may fail lysate-PCR. Scrape the remaining of the colony, and transfer the cell clump (arrowhead in 5) to a 24-well feeder plate. (b,c) Examples of larger colonies, 1.5 mm (b) and 2 mm (c) in size. Transfer a similar-sized cell clump (the dotted area in b and c should be similar to the dotted area in a) into a 96-well plate for lysate-PCR. The remaining colony needs to be cut into several pieces before being transferred to a 24-well feeder plate. (d,e) Forty-eight hours after colony picking. Appropriate clump size generally results in extended hPSC colonies (d). Large clumps tend to result in the formation of embryoid bodies (EBs) (e). Outgrowth of pluripotent cells may occur underneath the EBs (dotted area in e) and can be used for subsequent expansion. These should not be disturbed until pluripotent cell growth is confirmed. Scale bars, 0.5 mm.
Materials
REAGENTS
-
Plasmids from the Sanger Institute Archives (http://www.sanger.ac.uk/form/Sanger_CloneRequests): pPB-R1R2_NeoPheS, pENTR-PGKpuroΔtk and pCMV-hyPBase (hyperactive mammalian-codon-optimized piggyBac transposase)
-
pBluescriptII KS+ (Agilent, cat. no. 212207)
-
Plasmids expressing ZFNs or TALENs: custom ZFNs or TALENs need to be obtained from commercial providers (e.g., Sigma-Aldrich, Cellectis) or be generated using toolkits49,50,51. See the Experimental design section for further information
-
Restriction enzymes: BsiWI (NEB, cat. no. R0553S), NsiI (NEB, cat. no. R0127S) and additional enzymes depending on choice
-
Genomic DNA from hPSC line of choice
-
Proteinase K, recombinant, PCR grade (Roche, cat. no. 03115879001)
-
Phusion hot start high-fidelity DNA polymerase (Thermo Scientific, cat. no. F-540S)
-
LongAmp Taq DNA polymerase (NEB, cat. no. M0323L)
-
PCR nucleotide mix (10 mM dNTPs; Roche, cat. no. 11814362001)
-
LigaFast rapid DNA ligation system (Promega, cat. no. M8225)
-
UltraPure agarose (Invitrogen, cat. no. 16500500)
-
DNA ladder, 1 kb (NEB, cat. no. N3232S)
-
Library efficiency DH5α competent cells (Invitrogen, cat. no. 18263012)
-
Gateway LR Clonase II Plus enzyme (Invitrogen, cat. no. 12538120)
-
QIAquick gel extraction kit (Qiagen, cat. no. 28704)
-
QIAquick PCR purification kit (Qiagen, cat. no. 28104)
-
QIAprep spin miniprep kit (Qiagen, cat. no. 27104)
-
Qiagen plasmid maxi kit (Qiagen, cat. no. 12163)
-
Yeast extract (Oxoid, cat. no. LP0021)
-
4-Chloro-Dl-phenylalanine (Sigma-Aldrich, cat. no. C6506-5G)
-
Agar (BD, cat. no. 214040)
-
Tris-EDTA (TE) buffer, pH 8.0 (Sigma-Aldrich, cat. no. 93283)
-
Sodium hydroxide (Sigma-Aldrich, cat. no. S8504)
-
Sodium chloride (Sigma-Aldrich, cat. no. S7653)
-
DMSO (Sigma-Aldrich, cat. no. D2650)
-
Glucose (VWR, cat. no. 101176K)
-
Kanamycin (Sigma-Aldrich, cat. no. K0129-20ml)
-
Ampicillin (Sigma-Aldrich, cat. no. A9518-5G)
-
Human pluripotent stem cell line(s) of choice
-
MEFs
-
Dulbecco's PBS without Ca and Mg (DPBS; PAA Laboratories, cat. no. H15-002)
-
DMEM, high glucose, GlutaMAX, pyruvate (Invitrogen, cat. no. 31966047)
-
Advanced DMEM/F12 (Invitrogen, cat. no. 12634)
-
KnockOut serum replacement (Invitrogen, cat. no. 10828-028)
-
MEM non-essential amino acids solution (NEAA; Invitrogen, cat. no. 11140050)
-
Gelatin, porcine (Sigma-Aldrich, cat. no. G1890)
-
2-Mercaptoethanol (Sigma-Aldrich, cat. no. M6250)
-
FBS (PAA Laboratories, cat. no. A15-101)
-
Culture-grade dH2O (Invitrogen, cat. no. 15230)
-
GlutaMAX (Invitrogen, cat. no. 35050038)
-
Basic fibroblast growth factor (bFGF; Invitrogen, cat. no. 13256029)
-
Penicillin-streptomycin (pen-strep; Gibco, cat. no. 15140122)
-
FIAU (Moravek, cat. no. M251)
-
Collagenase type IV (Invitrogen, cat. no. 17104-019)
-
Dispase (Gibco, cat. no. 17105-041)
-
Accutase (Millipore, cat. no. SCR005)
-
Y-27632 dihydrochloride (ROCK inhibitor; Sigma-Aldrich, cat. no. Y0503-1MG)
-
Mitomycin C (Sigma-Aldrich, cat. no. M4287-5X2MG)
-
Human stem cell Nucleofector kit 2 (Lonza, cat. no. VPH-5022)
EQUIPMENT
-
Inverted microscope
-
Cell culture centrifuge (Eppendorf, Centrifuge 5810)
-
Tissue culture plates: dishes, 10 cm (Corning, cat. no. 430167); plates, six well (Corning, cat. no. 3516); plates, 24 well (Corning, cat. no. 3524); round-bottom 96-well plate (Corning, cat. no. 3799)
-
Filter system, 0.2 μm: 250 ml (Corning, cat. no. 431096); 500 ml (Thermo Scientific, cat. no. 450-0020)
-
Filter cartridge, 0.2 μm (Nalgene, cat. no. 190-2520)
-
Microtube, 1.7 ml (Axygen, MCT-175-C-S)
-
Tissue culture tubes, 15 ml (BD, cat. no. 352097)
-
Tissue culture tubes, 50 ml (BD, cat. no. 352098)
-
Bacterial Petri dish (Sterilin, cat. no. 101R20)
-
P200 multichannel pipette (Axygen, cat. no. AP-12-200)
-
PCR thermal cycler (Life technologies, GeneAmp PCR system 7900)
-
Nucleofector 2b Device (Lonza)
REAGENT SETUP
Proteinase K, 10 mg ml−1
-
Prepare 10 mg ml−1 proteinase K by dissolving it in water. Divide the solution into aliquots and store it at −20 °C for up to 1 year.
Glucose, 20% (wt/vol)
-
Prepare 20% (wt/vol) glucose by dissolving it in water. Filter-sterilize it and store it at 4 °C for up to 6 months.
Mitomycin C, 1 mg ml−1
-
Prepare 1 mg ml−1 mitomycin C by injecting 2 ml of DPBS into the vial containing 2 mg of mitomycin C. Dissolve it. This may take up to 5 min by warming up with your hands. Aspirate the solution with a 5-ml syringe and filter-sterilize the solution with a 0.2-μm filter cartridge. This solution can be stored at 4 °C for 1 month.
Caution
Mitomycin C is harmful when it comes in contact with the skin. Wear gloves when handling it.
2-Mercaptoethanol, 0.1 M
-
Prepare 0.1 M 2-mercaptoethanol by adding 140 μl of 2-mercaptoethanol into 200 ml of DPBS. Filter-sterilize the solution. This solution can be stored at 4 °C for 6 months.
Caution
2-mercaptoethanol is harmful if inhaled, if it comes in contact with the skin and if swallowed. Wear gloves, safety glasses and work inside a chemical fume hood.
bFGF, 4 μg ml−1
-
Prepare bFGF in 0.5% (wt/vol) BSA-DPBS aliquots of 500 μl per tube. Store the aliquots at −20 °C for up to 6 months.
Y-27632, 1 mM stock solution
-
Prepare Y-27632 1 mM stock solution by dissolving 1 mg of Y-27632 powder in 3 ml of culture-grade water. Filter-sterilize aliquots of 500 μl per tube and store the aliquots at −20 °C for up to 6 months. This is a 100× concentrated stock solution for up to 6 months.
Gelatin, 0.1% (wt/vol)
-
Prepare 0.1% (wt/vol) gelatin by adding 0.5 mg of gelatin powder to 500 ml of water. Autoclave the solution. Store the solution at room temperature (15−25 °C ) for up to 6 months.
FIAU, 0.2 mM
-
Prepare 0.2 mM FIAU by adding 5 mg FIAU to 5 ml of water. Add 1 M NaOH gradually until the powder is completely dissolved. Adjust the solution to a total volume of 67 ml with water. Filter-sterilize the solution, and then aliquot and store it at −20 °C for up to 1 year.
YEG-CL agar
-
Combine 5 g of yeast extract, 5 g of NaCl, 2 g of 4-chloro-Dl-phenylalanine and 15 g of agar into 1 liter of water, mix thoroughly and autoclave the solution for 20 min at 121 °C. The 4-chloro-Dl-phenylalanine will not dissolve until autoclaving. When the temperature of the heated mixture has dropped below 55 °C, add 20 ml of 20% (wt/vol) glucose (to a final concentration of 0.4%) and ampicillin to a final concentration of 50 μg ml−1. Pour ∼25 ml of the agar per Petri dish.
MEF medium
-
Prepare MEF medium by adding 60 ml of FBS, 5 ml of NEAA and 5 ml of 0.1 M 2-mercaptoethanol into 500 ml of DMEM + GlutaMAX. The medium can be stored at 4 °C for 4 weeks.
Mitotically inactivated MEFs
-
Culture MEFs (at passage 3 or 4) in 15-cm dishes until they reach 100% confluence. Replace old medium with 15 ml of MEF medium containing 15 μg ml−1 mitomycin C, and then incubate the dishes at 37 °C for 2.5 h. Next, wash the cells twice with DPBS, add 15 ml of fresh MEF medium and incubate the cells overnight. On the following day, collect and count the inactivated MEFs. Re-plate the cells as indicated below for Gelatin/MEF-coated tissue culture plates or freeze them at –80 °C at desirable concentrations in MEF medium containing 10% (vol/vol) DMSO.
Caution
Mitomycin C is harmful when it comes in contact with the skin. Wear gloves when handling it.
Gelatin/MEF-coated tissue culture plates
-
Coat culture vessels with 0.1% (wt/vol) gelatin at room temperature for 10 min. Seed 0.8–1 × 106 mitotically inactivated MEFs per 10-cm dish (55 cm2). The feeder cells should be ∼60–70% confluent and used for hPSC culture within 3 d. Consider post-thawing viability when using frozen inactivated MEFs as feeders.
hPSC medium
-
Mix 380 ml of Advanced DMEM/F12, 100 ml of KnockOut serum replacement, 5 ml of GlutaMAX, 5 ml of NEAA, 5 ml of 2-mercaptoethanol, 5 ml of pen-strep and 500 μl of FGF2. The medium can be stored at 4 °C for 2 weeks.
Dispase stock solution
-
Dissolve dispase in Advanced DMEM/F12 at 1 mg ml−1. Filter-sterilize the solution and store it at 4 °C for up to 1 month.
Collagenase stock solution
-
Mix 400 ml of Advance DMEM/F12, 100 ml of KnockOut serum replacement, 5 ml of GlutaMAX, 5 ml of 2-mercaptoethanol and 500 mg of collagenase type IV. Place the solution at room temperature and wait until all the collagenase power has dissolved. Filter-sterilize the solution, divide it into 50-ml conical tubes and store aliquots at −20 °C for up to 6 months.
Collagenase/dispase (1:1) for passaging hPSCs
-
Defrost collagenase solution. Once thawed, this solution can be stored at 4 °C for 1 month. Mix dispase and collagenase at a ratio of 1:1 to obtain the volume required (1 ml for a well of six-well plate and 4 ml for a 10-cm dish). This solution can be stored at 4 °C for 2 weeks.
Procedure
Designing and constructing a targeting vector
Timing 14–21 d
-
1
Download the genomic sequence that contains at least 10 kb upstream and 10 kb downstream of the site to be modified, or download the entire target gene in a GenBank format from the Ensembl website (http://www.ensembl.org). This can be done via 'Export data' and by choosing the following GenBank options: 'Repeat features and Gene Information'. The downloaded GenBank file can be opened with various DNA construction software (e.g., Lasergene, VectorNTI and Gene Construction Kit). Gene annotation information should be incorporated.
-
2
Search for a TTAA sequence near the site to be modified. This TTAA site will be used for piggyBac transposon insertion.
Critical Step
As the distance between the modifying site and the TTAA site increases, the efficiency of incorporating the modified nucleotide(s) decreases. For heterozygous modifications, the distance can be up to 300 bp. For homozygous modifications, the distance should be no more than 200 bp. The general idea is to keep the distance as small as possible.
-
3
Design homology arms on either side of the TTAA site to be between 700 bp and 1 kb in length.
Critical Step
Avoid repetitive sequences at the 5′ and 3′ ends because PCR primer will have difficulties in specifically amplifying these regions. During subcloning, BsiWI and NsiI sites are used; the homology arms should therefore not contain these restriction sites.
-
4
Design PCR primers to amplify the left and right homology arms (Fig. 4). A forward primer for the left arm (LA-Fw) and a reverse primer for the right arm (RA-Rv) must include the appropriate restriction sites (labeled as A and B in Fig. 4a) at the 5′ end of the primers. In addition, design a reverse primer for the left arm (LA-Rv) and a forward primer for the right arm (RA-Fw), which must include parts of piggyBac transposon as illustrated in Figure 4c.
-
5
Amplify the left and right homology arms separately with a high-fidelity DNA polymerase using the genomic DNA from hPSC lines of choice as a template. Introduce the genetic modification (substitution) of choice into one of the arms. Set up the following PCR mixture. Use LA-Fw and LA-Rv for the amplification of the left homology arm and RA-Fw and RA-Rv for the right homology arm.
Table 5 -
6
Perform the PCR using the following conditions and run 5 μl of PCR products on a 1% (wt/vol) agarose gel.
Table 6 Critical Step
On average, there might be two SNPs in a total of 2-kb-long homology arms. This level of nucleotide difference between the homology arms and the hPSC genome does not affect modification efficiency. However, in terms of precise modification of the target nucleotide, it is highly recommended to use genomic DNA from the hPSC lines of choice as template DNA.
-
7
Purify the PCR products using a QIAquick PCR purification kit and elute into 30 μl of EB buffer (part of the kit).
-
8
Set up the following restriction digestions in a total volume of 50 μl. Incubate them for 2 h at the appropriate temperature. Note that BsiWI requires incubation at 55 °C; the reactions should first be incubated for 2 h at 37 °C, followed by an additional 2 h at 55 °C.
Table 7 -
9
Treatment of pBluescriptII KS+with alkaline phosphatase. Add the following reagents directly into the restriction digestion mixture and incubate the reaction mixture for 30 min at 37 °C.
Table 8 -
10
Run the digested DNA on a 1% (wt/vol) agarose gel and excise the desired bands. Digestion of the PCR products and pBluescriptII KS+ will generate single bands. Digestion of pPB-R1R2_NeoPheS will generate three bands (2.8, 1.8 and 1.1 kb). Excise the 2.8-kb fragment, which contains the piggyBac transposon. Purify all DNA fragments with a QIAquick gel extraction kit, and elute the fragments into 30 μl of EB buffer. Run 1 μl of each fragment on a 1% (wt/vol) agarose gel; estimate the DNA concentration by comparing the fragments with DNA ladders.
-
11
Set up the following ligation reaction and incubate the reaction mixture overnight at 16 °C.
Table 9 -
12
Transform competent bacterial cells (e.g., DH5α) with 5 μl of the ligation product (Step 11) according to the manufacturer's instructions. Plate the bacteria onto LB agar plate containing 50 μg ml−1 of kanamycin, and then incubate the plates at 37 °C overnight.
-
13
Pick four colonies and culture them in 1.5 ml of LB medium containing 50 μg ml−1 ampicillin at 37 °C in a shaking incubator overnight.
-
14
Isolate plasmid DNA from overnight cultures using a QIAprep spin miniprep kit, and then elute the plasmids into 50 μl of TE buffer. Verify the sequences of the PCR-generated homology arms by sequencing with the following primers: M13F97, PB3-P2, PB5-P2 and M13R84 (Table 1). If these primers do not cover the entire region of the homology arms, additional primers need to be designed within the homology arms to fill the sequencing gaps.
Table 1 Primer sequences. -
15
Exchange the NeoPheS cassette with the PGK-puroΔtk by Gateway cloning. To achieve controlled incubation temperature, it is recommended to set up the reaction in a PCR block.
Table 10 Vortex the Gateway ClonaseII PLUS enzyme mix briefly. Add to the sample 2 μl of LR ClonaseII PLUS enzyme mix, mix well by pipetting and incubate the mix at 25 °C overnight. Transform competent bacterial cells with 2 μl of the LR reaction and plate them onto YEG-CL agar containing 50 μg ml−1 of ampicillin. Incubate the plates at 37 °C overnight. Isolate plasmid DNA from the resistant colonies and verify the isolation by restriction digestion.
-
16
Prepare transfection-grade plasmids using a Qiagen plasmid maxi kit according to the manufacturer's instructions. Plasmid concentration should be higher than 1.0 μg μl−1.
Genetic modification of hPSC with ZFNs or TALENs
Timing 35 d
-
17
Passage hPSCs on MEF-feeder plates and grow them until they reach 70–80% confluence.
Critical Step
Keep cells in exponential growth for better transfection efficiency.
-
18
A day before transfection (next step), plate feeder cells onto five 10-cm plates. Electroporated hPSCs from Step 24 will be plated onto these plates.
-
19
On the day of transfection, replace old medium of hPSC culture with fresh hPSC medium containing 10 μM Y-27632 at least 4 h before transfection, and incubate the hPSCs at 37 °C.
-
20
Prepare the Amaxa human stem cell Nucleofector kit 2 by mixing 0.5 ml of supplement (provided with the kit) with 2.25 ml of the Nucleofection solution, and place it at room temperature. Prepare the plasmid cocktail (5 μg of ZFN/TALEN-left, 5 μg of ZFN/TALEN-right and 2 μg of the targeting vector) in a sterile microtube. Prepare plasmid cocktails for the control transfection as described in Table 2. Replace the medium in the 10-cm feeder plates (Step 18) with fresh hPSC medium containing 10 μM Y-27632, and then incubate these plates at 37 °C until Step 25.
Table 2 Controls for ZFN- or TALEN-mediated gene targeting (Step 20). -
21
Wash the hPSCs with DPBS, add Accutase (3 ml per 10 cm dish) and incubate the hPSCs at 37 °C for 5 min.
Critical Step
After 2–3 min of incubation, MEF feeder cells will start to detach much faster than hPSCs. When the feeder:hPSC ratio is high, it is recommended to gently swirl the culture dish and aspirate off the feeder cells. Thereafter, add fresh Accutase (3 ml per 10-cm dish) and incubate the plates at 37 °C until hPSCs start to dissociate. If the hPSCs are at a relatively higher density, feeder contamination will not affect the electroporation efficiency.
-
22
Add 3 ml of 10% (vol/vol) FBS in Advanced DMEM/F12 and disperse the hPSCs into a single-cell suspension by vigorous pipetting.
Critical Step
Mix the cells after adding 10% (vol/vol) FBS solution. Do not attempt to pipette the cells in only Accutase as this will cause cell lysis and high cell death. The presence of cell debris will substantially decrease electroporation efficiency.
-
23
Count the cells, transfer 2 × 106 cells into a 50-ml conical tube and centrifuge them at 200g for 3 min at room temperature.
-
24
Aspirate the supernatant and re-suspend the pellet in 100 μl of human stem cell Nucleofector kit 2. Combine this cell suspension with the plasmid cocktail (Step 20) and transfer the cell-DNA mixture to an electroporation cuvette (provided with the kit). Electroporate the cells using the Amaxa Nucleofector 2b device (program B-016).
-
25
Transfer the electroporated cells to a tube containing 900 μl of hPSC medium supplemented with 10 μM Y-27632 using a pipette (provided with the kit). Mix gently and plate 500 μl each onto two 10-cm feeder plates (Step 20). Incubate the plates at 37 °C. For the control transfection, plate 500 μl onto one 10-cm feeder plate and discard the remaining cells.
Critical Step
It is hard to predict how many colonies will be obtained per 10-cm dish after selection. Cells should therefore be plated reasonably sparsely. Thus far, we have not obtained too many resistant colonies after plating electroporated cells onto two 10-cm dishes. If too many resistant colonies form, it will be difficult to isolate single cell–derived colonies. Electroporation has to be repeated and the cells have to be plated onto more than two 10-cm dishes.
-
26
Analysis of transfection efficiency. Observe GFP expression under a fluorescence microscope and/or analyze it by flow cytometry. The percentage of GFP-positive cells should be higher than 30%. Viability of the cells should be higher than 50%.
-
27
For 3 d after transfection, replace the medium daily with hPSC medium supplemented with 10 μM Y-27632.
-
28
At day 4 after transfection, replace the medium with hPSC medium containing 1 μg ml−1 puromycin and 10 μM Y-27632.
-
29
From day 5, replace the medium with hPSC medium containing 0.5 μg ml−1 puromycin every other day for up to 14 d after transfection, if wild-type MEFs are used as feeders. Y-27632 can now be withdrawn. When puromycin-resistant feeders are available, keep the puromycin concentration at 1 μg ml−1. From day 10 onward, a daily medium change may be required, depending on the number of resistant colonies.
-
30
Pick resistant colonies 13–17 d after transfection. Plate out 24-well feeder plates 1 d before colony picking.
-
31
Pretreat resistant colonies with 10 μM Y-27632 for at least 4 h before colony picking.
-
32
Change the medium in the 24-well feeder plates to hPSC medium containing 10 μM Y-27632. In addition, prepare a round-bottom 96-well plate with 100 μl of DPBS in every well.
-
33
By using a P20 pipette set at 7 μl, cut colonies into halves. Scrape off one half and transfer it to a well in the round-bottom 96-well plate. Scrape off the other half of the colony, and transfer it to the 24-well feeder plate (Fig. 5).
-
34
When colony picking is done, incubate the 24-well plates at 37 °C. Do not disturb these cells for the next 48 h.
-
35
Suspend the cell clumps in the 96-well plates vigorously with a P200 multichannel pipette and break them down into the smallest pieces possible. Transfer the cells into 96-well PCR plates. Spin the 96-well plate at 200g for 3 min at room temperature.
-
36
Aspirate as much of the supernatant as possible without disturbing the cell pellets.
Pause point
The cell pellets can be stored at −20 °C for longer than a year.
-
37
Add 25 μl of water. There is no need to resuspend the pellets. Place the plate on a PCR machine and run the following program:
Table 11 -
38
Remove the plate from the PCR machine. Add 5 μl of 2 mg ml−1 proteinase K (freshly diluted in water from the stock solution) into each well (no need to resuspend). Place the plate back onto the PCR machine and run the following program:
Table 12 Critical Step
Conduct the following PCR genotyping (Steps 39–45) as soon as the proteinase K treatment is completed. Cell lysates can be stored at −20 °C for up to 7 d. Note that the frozen lysates may not maintain their quality even during this period. If PCR cannot be performed within 24 h, freeze and store the cell pellets at Step 36.
-
39
For genotyping, set up the following PCR mixture on ice. See Figure 6 for the primer combinations. For example, use primers F1, R1 and PB3-P2 (Table 1) to genotype the left arm.
Figure 6: PCR-based screening of targeted clones. 
(a) Schematic of gene targeting and primer design. Arrows represent primers used in genotyping. Arrowheads represent primers used to detect random integration of the targeting vector. Star represents the modified nucleotide. (b) PCR-based genotyping of alleles. Each arm is analyzed in a separate PCR reaction. (c) Possible genotypes of each banding pattern in b. (d–f) Examples of PCR. (d) The majority of colonies should show a simple pattern. (e) In some cases, deletions can be detected. (f) Heterogeneous colonies can be identified on the basis of the difference in intensity between the PCR bands of the original and targeted alleles.
Table 13 Critical Step
Perform genotyping for both left and right arms independently (Fig. 6a,b).
-
40
Perform the PCR using the following conditions:
Table 14 Critical Step
Heat the PCR block to 94 °C before placing the PCR plate on the PCR block.
-
41
Run the PCR products on a 1.5% (wt/vol) agarose gel. Analyze the results of the PCR genotyping carefully, as shown in Figure 6c,d.
Critical Step
Both left and right arms must have the correct genotype.
-
42
Set up the following PCR mixture on ice. This is to detect random integrations of the targeting vector. Use primers F3+R3 and F4+R4 (Fig. 6a).
Table 15 Critical Step
Perform PCR for both sides of the homology arm–plasmid junction individually (Fig. 6a). PCR products can be ∼300 bp. Handle the PCR reaction carefully to avoid plasmid DNA contamination.
-
43
Perform the PCR using the following conditions:
Table 16 -
44
Analyze PCR products on a 2% (wt/vol) agarose gel; choose the candidate clones with the desired genotype (Step 41) and with no random integration.
-
45
Amplify the region spanning the substitution site from the selected candidate clones using the transposon-specific primer and the outer primer (PB5-P2 (Table 1) and R2 in Fig. 6a), and then confirm the incorporation of the substitution by sequencing. When heterozygous targeting is desired, amplify the nontargeted allele using the 5′ and 3′ outer primers and confirm that the candidate clones do not have insertion or deletion mutations (indels).
-
46
At day 2 after picking colonies, replace the medium in wells containing positive clones seeded at Step 33. Y-27632 can now be withdrawn. Passage and freeze down the clones as soon as possible. Isolate genomic DNA from these clones, and then perform further genomic characterization by Southern blot analysis.
Critical Step
If you are planning to remove the transposon, keep selecting with puromycin during the expansion phase. This not only keeps the puroΔtk gene expressed but also helps to reduce background during tk-FIAU selection.

Transposon removal from hPSCs
Timing 35 d
-
47
Expand the correctly targeted hPSC clones grown under continuous puromycin selection.
-
48
A day before electroporation (next step), plate out two six-well feeder plates.
-
49
Electroporate 2 × 106 cells with 10 μg of pCMV-hyPBase as described above (Steps 19–24). To measure the background of the HSVtk-FIAU selection, a control transfection with GFP (10 μg) should be performed (Table 3). Cells transfected with GFP should be processed in parallel with the relevant experimental samples.
Table 3 Controls for piggyBac transposon excision (Step 49). -
50
Transfer the electroporated cells to 6 ml of hPSC medium containing 10 μM Y-27632 without puromycin using a pipette (provided with the kit) in a 50-ml conical tube. Mix the medium gently and aliquot 3 ml, 2 ml and 1 ml individually into each well of a six-well feeder plate. Top up the medium to 3 ml per well. Incubate the plate at 37 °C.
Critical Step
Once the transposon is removed, puromycin expression will be lost and these cells will become sensitive to puromycin. Puromycin should therefore be removed from the cell culture medium from this point onward.
-
51
On the following day, replace the medium with fresh hPSC medium supplemented with Y-27632.
-
52
At day 2 after transfection, passage cells that are 70–80% confluent. Wash the cells with 3 ml of DPBS. Add 0.5 ml of Accutase and incubate the cells at 37 °C for 3 min. Add 2.5 ml of hPSC medium, transfer the cells to a 15-ml conical tube and spin the tube at 200g for 3 min at room temperature. Aspirate the supernatant, re-suspend the pellets in 6 ml of hPSC supplemented with 10 μM Y-27632 and aliquot 3 ml, 2 ml and 1 ml individually into each well of a six-well feeder plate (split ratio is 1:2, 1:3 and 1:6, respectively). Top up the medium to 3 ml per well. Incubate the plate at 37 °C.
-
53
On the following day, replace the medium with hPSC medium supplemented with 10 μM Y-27632. Plate out three 10-cm feeder plates.
-
54
On the following day, seed the cells in 10-cm feeder plates. Wash the cells that are 70–80% confluent with DPBS. Add 0.5 ml of Accutase and incubate the cells at 37 °C for 3 min. Add 2.5 ml of hPSC medium, transfer the cells to a 15-ml conical tube and spin the tube at 200g for 3 min at room temperature. Aspirate the supernatant, re-suspend the pellets in 2 ml of hPSC medium and count the cells. Replace the medium of the 10-cm feeder plates (Step 53) with fresh hPSC medium supplemented with 10 μM Y-27632. Plate transposase- and GFP-transfected cells onto two and one 10-cm plates, respectively, at a density of 1 × 104 cells per plate.
-
55
On the following day, start FIAU selection. Change the medium to hPSC medium containing 200 nM FIAU and 10 μM Y-27632. Replace the medium every other day. At 2 d after the addition of FIAU, Y-27632 is no longer required. As colonies grow, a daily medium change may be required depending on the number of surviving colonies.
-
56
About 2 weeks later, pick colonies and prepare the cell lysates as described above (Steps 30–38).
-
57
Set up the following PCR mixture on ice. This step is for genotyping. See Figure 7a,b for the recommended primer combination.
Figure 7: PCR-based screening of transposon-excised clones. 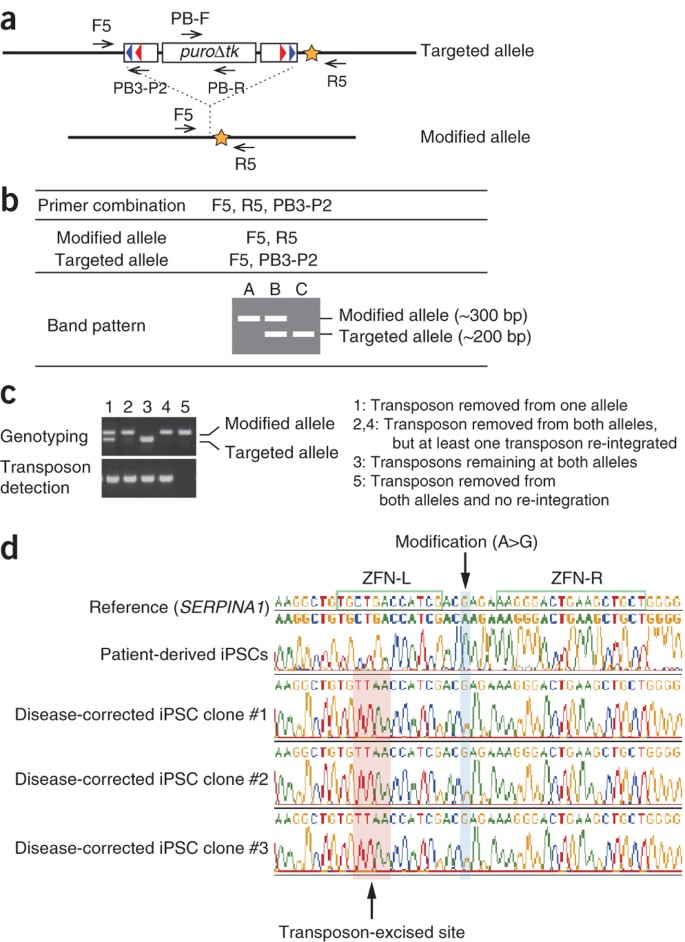
(a) Schematic of transposon excision and primer design. Arrows represent primers. PCR primers can be designed to amplify small amplicons (∼300–500 bp-long). Star represents the modified nucleotide. (b) PCR-based genotyping of alleles. (c) Examples of PCR results. Lane 5 shows an example of a clone where transposons have been removed from both alleles. Note that no re-integration events are observed. (d) Final confirmation of genetic modification by capillary sequencing. A genetic correction of disease-causing mutation associated with α-1 antitrypsin deficiency is shown here. The TTAA sequence should be observed at the transposon excision site. Modified nucleotides should also be detected. Adapted from ref. 29.
Table 17 -
58
Perform the PCR using the following conditions:
Table 18 -
59
Analyze PCR products on a 2% (wt/vol) agarose gel as shown in Figure 7b,c.
-
60
Set up the PCR mixture on ice. This step is to detect transposon re-integration (Fig. 7a).
Table 19 -
61
Perform the PCR using the following conditions:
Table 20 -
62
Analyze PCR products on a 2% (wt/vol) agarose gel. Choose the candidate clones without transposon re-integration and with the desired genotype (Fig. 7).
-
63
Sequence the PCR products from Step 58 using primers F5 and R5 to examine whether the transposon excision results in the expected TTAA sequence.
-
64
From the following day, change the medium daily in wells containing positive clones. Passage and expand the positive colonies, and freeze down the clones as soon as possible. Isolate genomic DNA and perform further genomic characterization.

Troubleshooting
Troubleshooting advice can be found in Table 4.
Timing
Steps 1–16, designing and constructing a targeting vector: 14–21 d
Steps 17–46, genetic modification of hPSCs with ZFNs or TALENs: 35 d
Steps 19 and 20, preparation before electroporation: 4.5 h
Steps 21–25, electroporation of hPSCs: 1 h
Steps 32 and 33, colony picking: 2 h per 96 colonies
Steps 35–38, lysate preparation, 2.5 h
Steps 39–45, PCR genotyping, 4 h
Step 46, expansion and cryopreservation of targeted hPSC clones, 20 d
Steps 47–64, transposon removal from hPSCs: 35 d
Anticipated results
The protocol described here was used to correct a single-nucleotide mutation causing α-1 antitrypsin deficiency in patient-derived iPSCs29. We achieved homozygous correction in iPSC lines from two independent patients. The protocol has also been used to modify several loci (AAVS1, BLM, 53BP1 and HPRT) in hPSCs. The numbers of resistant colonies and ratios between targeted and nontargeted clones vary depending on the loci and the cutting efficiency of individual ZFNs or TALENs. Thus far, modification has been always successful.
In our and others' studies, the background of HSV-tk–negative selection tends to be close to zero when the tk gene is inserted in expressed genes69. This figure is slightly higher in non-expressed genes29. Even if the target genes are expressed in hPSCs, there could be high background if puromycin selection is not applied during the expansion of targeted cells. The number of FIAU-resistant colonies from hyPBase- and GFP-transfected cells should be compared with each other. If no colonies are observed in the GFP-transfected cells, the transposon has been removed in most of the colonies. Even if there is background, removal will be successful as long as there are more colonies in the hyPBase-transfected cells compared with the GFP-transfected cells. However, a larger number of colonies (∼100 colonies) will need to be analyzed in this case.
PCR-based genotyping in ZFN- or TALEN-mediated gene targeting requires careful assessment of results. Figure 6 summarizes primer design for PCR genotyping (Fig. 6a), the expected band patterns (Fig. 6b) and the possible outcomes (Fig. 6c). A typical result is shown in Figure 6d. However, as ZFNs and TALENs introduce DSBs and these can also be repaired by nonhomologous end joining, indels are routinely observed. Small indels (∼20 bp) are the most frequent, and they are often not detectable by gel electrophoresis. Depending on the patterns of deletion, medium-size deletion (∼500 bp) can be detected by gel electrophoresis (Fig. 6e, lane 1). More rarely, large deletions (∼2 kb) can also occur, although these are not detectable because the primers' annealing sites are likely to be lost (ref. 29). As a result, compound heterozygous clones (comprising one targeted allele and one nontargeted allele with a small deletion) show the same pattern as heterozygous clones (Pattern B in Fig. 6c). Hemizygous clones (comprising only one targeted allele due to loss of the other allele) give the same pattern as homozygous clones (Pattern C in Fig. 6c). In addition, drug selection starts on day 4, which allows ZFNs or TALENs to be expressed and homologous recombination to occur. This sometimes results in heterogeneous colonies (Fig. 6f, lane 3). Although PCR genotyping is a rapid and convenient method for selecting candidate clones, further assessments of these clones by Southern blot analysis must be conducted before downstream genetic manipulation or phenotypic analysis.
In contrast, PCR analysis of transposon excision is fairly simple, as shown in Figure 7. Although the fidelity of transposon excision is ∼99%, there is a small chance that footprint mutations are introduced into the endogenous TTAA sites. It is therefore important to confirm the presence of the TTAA sequence at the excised site and the intended modification by direct sequencing of the PCR product (Fig. 7d).
References
Capecchi, M.R. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat. Rev. Genet. 6, 507–512 (2005).
Baudin, A., Ozier-Kalogeropoulos, O., Denouel, A., Lacroute, F. & Cullin, C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 21, 3329–3330 (1993).
Giaever, G. et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (2002).
Winzeler, E.A. et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 (1999).
Skarnes, W.C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011).
Thomson, J.A. et al. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998).
Watanabe, K. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681–686 (2007).
Hockemeyer, D. et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851–857 (2009).
Hockemeyer, D. et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 29, 731–734 (2011).
Jasin, M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 12, 224–228 (1996).
Porteus, M.H. & Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 300, 763 (2003).
Urnov, F.D. et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651 (2005).
Kim, Y.G. & Chandrasegaran, S. Chimeric restriction endonuclease. Proc. Natl. Acad. Sci. USA 91, 883–887 (1994).
Miller, J.C. et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143–148 (2011).
Boch, J. et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512 (2009).
Reyon, D. et al. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 30, 460–465 (2012).
Kim, Y. et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 31, 251–258 (2013).
Schmid-Burgk, J.L., Schmidt, T., Kaiser, V., Honing, K. & Hornung, V. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat. Biotechnol. 31, 76–81 (2013).
Ding, Q. et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 12, 238–251 (2013).
Doyon, Y. et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Methods 8, 74–79 (2011).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Ding, Q. et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394 (2013).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 10.1038/nbt.2623 (2013).
Hsu, P.D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 10.1038/nbt.2647 (2013).
van der Weyden, L., Adams, D.J. & Bradley, A. Tools for targeted manipulation of the mouse genome. Physiol. Genomics 11, 133–164 (2002).
Meier, I.D. et al. Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J. 24, 1714–1724 (2010).
Yusa, K. et al. Targeted gene correction of α-antitrypsin deficiency in induced pluripotent stem cells. Nature 478, 391–394 (2011).
Tennessen, J.A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012).
Conrad, D.F. et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010).
Maurano, M.T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Osafune, K. et al. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 26, 313–315 (2008).
Kajiwara, M. et al. Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 109, 12538–12543 (2012).
Chen, F. et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 8, 753–755 (2011).
Soldner, F. et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318–331 (2011).
Friedel, R.H., Wurst, W., Wefers, B. & Kuhn, R. Generating conditional knockout mice. Methods Mol. Biol. 693, 205–231 (2011).
Rad, R. et al. PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science 330, 1104–1107 (2010).
Loonstra, A. et al. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. USA 98, 9209–9214 (2001).
Silver, D.P. & Livingston, D.M. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol. Cell 8, 233–243 (2001).
Vallier, L., Alexander, M. & Pedersen, R. Conditional gene expression in human embryonic stem cells. Stem Cells 25, 1490–1497 (2007).
Yusa, K., Zhou, L., Li, M.A., Bradley, A. & Craig, N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 108, 1531–1536 (2011).
Meng, X., Thibodeau-Beganny, S., Jiang, T., Joung, J.K. & Wolfe, S.A. Profiling the DNA-binding specificities of engineered Cys2His2 zinc finger domains using a rapid cell-based method. Nucleic Acids Res. 35, e81 (2007).
Yanover, C. & Bradley, P. Extensive protein and DNA backbone sampling improves structure-based specificity prediction for C2H2 zinc fingers. Nucleic Acids Res. 39, 4564–4576 (2011).
Zykovich, A., Korf, I. & Segal, D.J. Bind-n-Seq: high-throughput analysis of in vitro protein-DNA interactions using massively parallel sequencing. Nucleic Acids Res. 37, e151 (2009).
Doyle, E.L. et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 40, W117–W122 (2012).
Pattanayak, V., Ramirez, C.L., Joung, J.K. & Liu, D.R. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods 8, 765–770 (2011).
Gabriel, R. et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 29, 816–823 (2011).
Maeder, M.L., Thibodeau-Beganny, S., Sander, J.D., Voytas, D.F. & Joung, J.K. Oligomerized pool engineering (OPEN): an 'open-source' protocol for making customized zinc-finger arrays. Nat. Protoc. 4, 1471–1501 (2009).
Sanjana, N.E. et al. A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 7, 171–192 (2012).
Cermak, T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82 (2011).
Cary, L.C. et al. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology 172, 156–169 (1989).
Mitra, R., Fain-Thornton, J. & Craig, N.L. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 27, 1097–1109 (2008).
Wu, S.C. et al. piggyBac is a flexible and highly active transposon as compared to Sleeping Beauty, Tol2, and Mos1 in mammalian cells. Proc. Natl. Acad. Sci. USA 103, 15008–15013 (2006).
Ding, S. et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122, 473–483 (2005).
Fraser, M.J., Ciszczon, T., Elick, T. & Bauser, C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol. Biol. 5, 141–151 (1996).
Yusa, K., Rad, R., Takeda, J. & Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nature Methods 6, 363–369 (2009).
Li, M.A. et al. The piggyBac transposon displays local and distant reintegration preferences and can cause mutations at noncanonical integration sites. Mol. Cell Biol. 33, 1317–1330 (2013).
Lacoste, A., Berenshteyn, F. & Brivanlou, A.H. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell 5, 332–342 (2009).
Rostovskaya, M. et al. Transposon-mediated BAC transgenesis in human ES cells. Nucleic Acids Res. 40, e150 (2012).
Beers, J. et al. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 7, 2029–2040 (2012).
Nandivada, H. et al. Fabrication of synthetic polymer coatings and their use in feeder-free culture of human embryonic stem cells. Nat. Protoc. 6, 1037–1043 (2011).
Braam, S.R. et al. Feeder-free culture of human embryonic stem cells in conditioned medium for efficient genetic modification. Nat. Protoc. 3, 1435–1443 (2008).
Lerou, P.H. et al. Derivation and maintenance of human embryonic stem cells from poor-quality in vitro fertilization embryos. Nat. Protoc. 3, 923–933 (2008).
Vallier, L. Serum-free and feeder-free culture conditions for human embryonic stem cells. Methods Mol. Biol. 690, 57–66 (2011).
Melkoumian, Z. et al. Synthetic peptide-acrylate surfaces for long-term self-renewal and cardiomyocyte differentiation of human embryonic stem cells. Nat. Biotechnol. 28, 606–610 (2010).
Rodin, S. et al. Long-term self-renewal of human pluripotent stem cells on human recombinant laminin-511. Nat. Biotechnol. 28, 611–615 (2010).
Villa-Diaz, L.G. et al. Synthetic polymer coatings for long-term growth of human embryonic stem cells. Nat. Biotechnol. 28, 581–583 (2010).
Schuldiner, M., Itskovitz-Eldor, J. & Benvenisty, N. Selective ablation of human embryonic stem cells expressing a 'suicide' gene. Stem Cells 21, 257–265 (2003).
Acknowledgements
I thank E.P. Tan for careful reading of the manuscript, and S.T. Rashid and members of the Allan Bradley laboratory (Sanger Institute, UK) and the Ludovic Vallier laboratory (University of Cambridge, UK) for discussion and for sharing their experience. This work was supported by Wellcome Trust (WT077187).
Author information
Authors and Affiliations
Contributions
K.Y. developed the protocol and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing financial interests.
Rights and permissions
About this article
Cite this article
Yusa, K. Seamless genome editing in human pluripotent stem cells using custom endonuclease–based gene targeting and the piggyBac transposon. Nat Protoc 8, 2061–2078 (2013). https://doi.org/10.1038/nprot.2013.126
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2013.126
This article is cited by
-
WNT signalling control by KDM5C during development affects cognition
Nature (2024)
-
CRISPR Del/Rei: a simple, flexible, and efficient pipeline for scarless genome editing
Scientific Reports (2022)
-
MAPRE2 mutations result in altered human cranial neural crest migration, underlying craniofacial malformations in CSC-KT syndrome
Scientific Reports (2021)
-
Non-coding cis-element of Period2 is essential for maintaining organismal circadian behaviour and body temperature rhythmicity
Nature Communications (2019)
-
Updated summary of genome editing technology in human cultured cells linked to human genetics studies
Journal of Human Genetics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
