
Abstract
Osteoporosis and its consequence of fragility fracture impose a considerable demand on health-care services because fracture is associated with a series of adverse events, including re-fracture and mortality. One of the major priorities in osteoporosis care is the development of predictive models to identify individuals at high risk of fracture for early intervention and management. Existing predictive models include clinical factors and anthropometric characteristics but have not considered genetic variants in the prediction. Genome-wide association studies conducted in the past decade have identified several genetic variants relevant to fracture risk. These genetic variants are common in frequency but have very modest effect sizes. A remaining challenge is to use these genetic data to individualize fracture risk assessment on the basis of an individual's genetic risk profile. Empirical and simulation studies have shown that the usefulness of a single genetic variant for fracture risk assessment is very limited, but a profile of 50 genetic variants, each with odds ratio ranging from 1.02 to 1.15, could improve the accuracy of fracture prediction beyond that obtained by use of existing clinical risk factors. Thus, genetic profiling when integrated with existing risk assessment models could inform a more accurate prediction of fracture risk in an individual.
Key Points
-
Fragility fracture is common in the general population, and is associated with serious consequences, including mortality
-
BMD is the best predictor of fracture risk
-
Interindividual variation in fracture risk is partly determined by genetic factors
-
62 single nucleotide polymorphisms (SNPs) are associated with BMD in genome-wide association studies; among these SNPs, 18 are associated with fracture risk, of which only eight achieve genome-wide significance level
-
Any single SNP has little predictive value for fracture; however, genetic profiling of 50 SNPs could improve the accuracy of fracture prediction beyond that obtained by existing risk factors
Similar content being viewed by others
Introduction
One of the anticipated applications of the Human Genome Project is to use the genetic data obtained for early detection of predisposition to complex diseases. An enormous amount of genetic data has been generated since the completion of the Human Genome Project, and it is valid to assess the translational value of these data for the prediction of fracture risk. Fragility fracture offers an interesting case for genetic prediction because its susceptibility is determined by environmental and genetic factors, but existing predictive models have not made use of the genetic data. Yet, genetic data can potentially contribute to the individualized prevention, treatment and management of fracture.
Fragility fracture, the ultimate consequence of osteoporosis, is common in the elderly population and is associated with serious clinical consequences. From the age of 50 years, the residual lifetime risk of fracture is ∼50% in women and ∼30% in men.1 In women, the lifetime risk of hip fracture is actually equivalent to or higher than the risk of invasive breast cancer.1,2 In men, the lifetime risk of hip and vertebral fractures (17%) is comparable to the lifetime risk of being diagnosed with prostate cancer.2,3 With the rapid ageing of the global population, fracture will not only be a public health problem but also impose a great demand on medical services.
Fracture contributes to the loss of human life, which is not well known. Convincing evidence suggests that individuals with a pre-existing fracture have an increased risk of subsequent fracture4,5 and reduced life expectancy.6 Individuals with osteoporosis, fracture and recurrent fracture have increased mortality compared with those with only a single initial fracture, and patients with a single initial fracture have increased mortality compared with those without a fracture.6 Furthermore, numerous data accumulated during the past three decades have consistently shown that the relative risk of death in men with fracture (1.8-fold) is significantly greater than that in women (1.4-fold).4,6
An individualized predictive model
Several epidemiological studies during the past three decades have identified key risk factors contributing to the susceptibility to fracture. These risk factors include, advancing age, low BMD or low body weight, a personal history of fracture, a history of fall and prolonged use of corticosteroids.7,8,9 Treatment of individuals with low BMD and/or an existing fracture reduces their risk of fracture.10 Thus, The National Osteoporosis Foundation guidelines recommend treatment in the following clinical situations: women with T scores <–2 with no risk factors; women with T scores <–1.5 with one or more risk factors for fracture (including a prior fracture); and women with a prior vertebral or hip fracture.11
However, a problem exists with the initiation of treatment based on the dichotomization of BMD values into those that are osteoporotic versus nonosteoporotic. BMD is a continuous measurement and the relationship between BMD and fracture risk is continuous, such that no clear cut-off value exists to separate those who will sustain a fracture from those who will not. Thus, targeting individuals with a low BMD for treatment gives rise to the 'prevention paradox',12 which predicts that a large number of individuals who will have a fracture are in the nonosteoporotic group rather than in the high-risk osteoporotic group. Indeed, empirical data show that among individuals aged ≥60 years, 55% of women and 74% of men with fracture do not have osteoporosis (in other words, low BMD).13 As a result, treatment of individuals with osteoporosis can only reduce a modest proportion of the total numbers of fragility fractures in the general population.
The incomplete accounting for fracture incidence by BMD alone suggests that factors other than BMD have an important role in the determination of fracture risk. Indeed, fragility fracture is also a function of nonskeletal factors, such as fall propensity, which is affected by neuromuscular function, muscle strength and postural sway.14 The susceptibility to fracture is therefore a complex phenotype, in the sense that it is a constellation of bone strength and nonskeletal factors, and each of these factors might be determined by genetic factors.15 For example, an individual with osteoporosis and a history of fracture would be expected to have a greater risk of fracture than an individual with osteoporosis without a history of fracture. Furthermore, an individual with osteoporosis can have the same risk of fracture as an individual without osteoporosis if their genetic profiles are different.
Any assessment of fracture risk should, therefore, incorporate all relevant risk factors, which implies that the assessment of fracture risk should be individualized by using an individual's unique risk profile. The uniqueness of risk profile could be improved by including genetic factors. A logical step forward would be to construct multivariable risk prediction algorithms that combine genetic and nongenetic factors for the identification of individuals at high risk of fracture and to treat them appropriately.
Genetics of fracture susceptibility
That the susceptibility to osteoporosis has a genetic component is well established. In fact, BMD—the surrogate measure of osteoporosis and a major predictor of fracture risk—is among the most heritable traits in humans. The findings of several twin and family studies have consistently shown that genetic factors account for up to 80% of the interindividual variation in BMD.15,16 BMD varies with age, and the variation is also under genetic control.17 Moreover, quantitative ultrasound measurements of bone18 and bone turnover marker values19 are also highly heritable, with 40–65% of variance in these traits being due to genetic variation between individuals.
Evidence from family studies indicates that the risk of fragility fracture aggregates in families, but not in the strict Mendelian fashion. A key measure of genetic influence on a trait is the index of heritability, which is defined as the extent to which genetic individual differences contribute to individual differences in observed trait. Approximately 25–35% of the variance in the susceptibility to fracture is attributable to genetic factors.20,21 The index of heritability of fracture risk seems to be higher in younger individuals, and becomes progressively weaker in older individuals. In fact, among those aged ≥80 years, the genetic component of hip fracture risk was <3%.21 This finding suggests that hip fracture among the elderly is largely attributable to environmental factors.
A manifestation of this genetic influence is that women whose mothers had a hip fracture have a twofold increase in risk of hip fracture compared with women whose mothers had no hip fracture,22 possibly due to a family history of low BMD.23,24 Similar observations have been found in both elderly men and women.25 A meta-analysis found that a family history of hip fracture in parents is associated with a 50% increase in the risk of any fracture and 2.3-fold increase in the risk of hip fracture in their children.26 In summary, biometric studies have provided convergent evidence that the susceptibility to fracture is partly determined by genetic factors. An important implication of these findings is that genetic heterogeneity at least partly accounts for the phenotypic heterogeneity of osteoporosis and fracture.
In the light of knowledge that genetics influences the risk of fragility fracture, the search for genes that are either linked to or associated with fracture has been enthusiastically sought since the early 1990s. This search has been facilitated by the development of polymorphic DNA markers that can detect multiple forms of genes (genotypes) in chromosomal locations. A common epidemiologic approach to the search for genes is the candidate gene association study, in which specific DNA variant(s) are analysed in cases and unrelated controls. By use of this approach, polymorphisms in a number of genes have been implicated in fracture risk. These include polymorphisms in the genes that encode the vitamin D receptor, collagen type Iα1, osteocalcin,26 IL-1 receptor antagonist, calcium-sensing receptor, α2-HS-glycoprotein, osteopontin, osteonectin, oestrogen receptor α, IL-6, calcitonin receptor, collagen type Iα2, parathyroid hormone and transforming growth factor α1.27 However, findings from most candidate gene association studies have been poorly replicated, mainly owing to lack of statistical power28 and false-positive results.29
Even with technological advances in DNA analysis, the identification of specific genes associated with fracture risk has not been straightforward. The difficulty lies not only in the definition of phenotype but also in the analytic strategy. Fracture is a clinical event, and as a discrete trait it is an ultimate consequence of cumulative deterioration in bone strength and disturbances in bone remodelling, both of which are under strong genetic influence. Thus, it could be argued that fracture is a heterogeneous phenotype. The heterogeneity is not just in terms of clinical manifestations, but also in its risk factors. For instance, whereas fall is a major risk factor for hip fracture, it contributes little to the risk of vertebral fracture. This heterogeneity has major implications in the search for genes linked to osteoporosis. A 'fracture gene', or perhaps more accurately a gene that influences the risk of fracture, could be one that affects BMD, bone structure, or muscle strength that increase the likelihood of fall. Furthermore, a gene that is associated with hip fracture might not be predictive of vertebral fracture.
The case–control design has been widely used for the identification of genes associated with fracture. Although this design is very useful, it has a number of shortcomings. The selection of an appropriate control population can be a challenge, particularly for fractures that occurs mainly in later life. Any statistically significant association between a specific gene variant and fracture might not necessarily indicate a causative relationship because the association could have arisen from linkage disequilibrium and population stratification.30 Most candidate gene studies focused on a single gene; therefore, the P value threshold for declaring significance was commonly set at 0.05. Thus, if a study repeatedly examined 20 independent variants, it would be expected that one false-positive association would be obtained by chance alone. Not surprisingly, the decade in which candidate gene studies have blossomed has also been accompanied by increasing frustration with conflicting findings and false-positive results.28,31
Genome-wide association studies
Instead of focusing on a biologically plausible candidate gene, in genome-wide association studies (GWAS) the entire genome is scanned. Usually, hundreds of thousands of common single nucleotide polymorphisms (SNPs; minor allele frequency of common SNPs is >5%) are used to identify chromosomal regions harbouring genes that are likely to influence a trait. GWAS can be viewed as a large series of case–control candidate gene studies but performed in a single experiment on an array-based setting. GWAS are a hypothesis-free approach because they make no assumptions about the location and/or functional relevance of associated loci or their products.32 Moreover, inclusion of thousands of individuals from many settings and with validation samples means that GWAS have increased power to detect gene variants with modest effect size. Thus, the large sample size is a strength, but it can also be a weakness because the low effect size variants identified by GWAS might be of less clinical relevance. Nevertheless, GWAS have been highly successful in unravelling the genetic contribution to complex traits.33
The application of GWAS in osteoporosis has been slower than other fields. The earliest GWAS in osteoporosis examined the association between 71,000 genetic variants and BMD measured at different skeleton sites and found evidence of association for 40 SNPs. Several SNPs identified in this study were located in potential osteoporosis-associated genes, such as MTHFR, ESR1, LRP5, VDR and COL1A1,34 although none of these SNPs achieved the genome-wide significance threshold level (P <5 × 10−8),35 because the study was then considered to be underpowered. In another GWAS, 300,000 variants in an Icelandic population were screened and variants in the ZBTB40, ESR1, OPG and RANKL genes, and those in a novel region of 6p21, were found to be significantly associated with BMD at the genome-wide significance threshold level. In this study, some loci were also suggested to be associated with fracture risk, including variants in the 1p36 and 2p16 regions, and in OPG, LRP4, RANK and MHC genes. In the meantime, a mixed GWAS and candidate gene study in UK and Rotterdam cohorts found that variants in the TNFRSF11B and LRP5 genes were associated with BMD, whereas a variant in the LRP5 gene was also associated with fracture risk.36
To date, 12 GWAS have been conducted for bone density,37 and 62 SNPs have been identified to be associated with bone density at the genome-wide significance level. All GWAS conducted so far have been designed to identify loci associated with bone density, and fracture has not been considered a primary phenotype. However, among the 62 SNPs identified, eight SNPs were associated with fracture risk at the genome-wide significance level.37
Two meta-analyses of GWAS showed that variants in the ZBTB40, ESR1, LRP4, LRP5, TNFSF11, SOST and TNFRSF11A genes were associated with BMD,38,39 and that variants in LRP5, SOST and TNFSF11A were associated with fracture risk.39 Overall, results from GWAS and GWAS meta-analyses indicate that genes involved in the RANK–RANKL–OPG pathway (TNFRSF11B, TNFRSF11A and TNFSF11 genes), the Wnt–β-catenin pathway (LRP5, LRP4 and SOST genes), the oestrogen endocrine pathway (ESR1 gene) and the 1p36 region (ZBTB40 gene) were those strongly associated with osteoporosis. The latest GWAS meta-analysis, involving 81,949 cases and 102,444 controls, identified 56 loci that are associated with BMD, 13 of which were associated with fracture.40 Several of these loci or SNPs cluster within or near the location of genes that encode proteins involved in the RANK–RANKL–OPG system, mesenchymal stem cell differentiation, endochondral ossification and Wnt signalling pathways. To date, 32 SNPs have been identified to be statistically associated with fracture risk (Table 1). Ongoing studies will probably identify additional genetic variants relevant to fracture risk; however, a number of observations can be made from GWAS findings to date.
Notably, most genes associated with BMD (with the exception of LRP5) identified by candidate gene studies were not confirmed by GWAS. In other words, GWAS have discovered new genetic variants that were not previously suspected. The identification of new variants associated with BMD is clearly a success and a convincing demonstration of the power of GWAS. However, most regions identified in GWAS spanned 10–50 kb, which could harbour many genes; therefore, uncertainty remains concerning which genes are actually causal variants. Moreover, most of the variants identified by GWAS are located in the noncoding regions of the genome, and their functional relevance is yet to be determined.
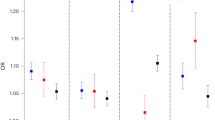
Effect sizes of individual genetic variants for both BMD and fracture risk are modest. Indeed, the median odds ratio of the 32 SNPs was 1.06 (1.2–1.15; Figure 1). In other words, the typical SNP increases the odds of fracture by about 5%, and the maximum increase by an SNP was only 15% (rs6696981). Interestingly, the relationship between odds ratios for fracture and allele frequency follows a U-shaped pattern (Figure 2), with the lowest odds ratios centred around SNPs with an allele frequency of 50%, and then increased in SNPs either below or above the 50% frequency. This finding is perhaps not surprising given that GWAS are based on the hypothesis of common variant–common disease, which predicts that complex diseases are caused by multiple common genetic variants, each with a modest effect size.

Red circles indicate the mean values. Abbreviation: SNPs, single nucleotide polymorphisms.

A U-shaped curve results from the lowest odds ratios being centred around SNPs with an allele frequency of 50%, and then increase in SNPs either below or above the 50% frequency. Loess refers to locally weighted scatterplot smoothing, a nonparametric regression technique. Abbreviation: SNPs, single nucleotide polymorphisms.
Translational use of genetic data
Statistical metrics
The genes listed in Table 1 that are associated with fracture risk are unlikely to represent the final list, as ongoing studies will probably identify more genetic variants relevant to the susceptibility to fracture. However, the listed genes provide an opportunity to examine the potential of using genetic profiling in the prediction of fracture. In the translational genetics of osteoporosis field, some current questions of interest include: how can we make use of the genetic data to predict an individual's risk of fracture?; can genetic variants alone identify individuals at high risk of fracture?; and can the genetic variants improve the prediction accuracy of fracture beyond that obtained with conventional clinical risk factors? Addressing these questions will help make advances towards the individualization of fracture risk prediction.
Despite this potential for individualized fracture risk prediction, the translation of genetic discoveries into clinical applications remains a major challenge. The issue is how to assess the usefulness of genes in fracture prediction and what metrics are suitable for the assessment. Simple measures of association, such as odds ratio, are not adequate.41 The usefulness of a genetic variant in terms of fracture risk prediction should be assessed in terms of discrimination, and more importantly, reclassification, as outlined below.
Discrimination measures how well a genetic variant can separate individuals who will have a fracture from those who will not.42 The primary metric of discrimination is the area under the receiver operating characteristic (ROC) curve (AUC), which can be interpreted as the probability that for a set of randomly selected pairs of fracture and nonfracture, the test result will be higher in patients that fracture than in individuals who do not fracture. In reality, AUC is a compromise between sensitivity and specificity, and is thus a global estimate of prognostic accuracy. As such, AUC is a rather insensitive measure of change.43 For example, a meaningful difference in prognostic value between two predictive models is not necessarily reflected by the AUC. Moreover, the AUC has no direct clinical meaning, and is therefore not helpful for clinical decision making.
A clinically meaningful metric of usefulness of a marker is risk reclassification.44 For a given threshold of risk, an individual can be classified as 'high risk' or 'low risk'. With additional risk factors (for example, a genetic variant) the individual might change risk category. Consider a predictive model with clinical risk factors, and a model with predictive factors plus genetic variants. If genetic variants are useful, then the addition of these genetic variants should result in an increased proportion of individuals with fracture being classified into the high-risk group than to the low-risk group; conversely, among individuals without a fracture, increased proportions would be classified into the low-risk group than the high-risk group. The net difference between the two proportions of re-classification is referred to as net reclassification improvement (NRI).45 Thus, when treatment decisions are based on risk threshold, the NRI can be helpful for making a clinical decision concerning an individual.
Utility of a single gene
The utility of a gene in predicting fracture is a function of several parameters. Both the 5-year incidence of fracture in the general population and the proposed or accepted level-of-risk threshold for treatment critically affects the NRI metric. Similarly, the frequency of a high-risk allele of the genetic variant in the general population, and the odds ratio of association between the genetic variant and fracture are important factors that affect the clinical usefulness.
Assuming that the 5-year incidence of fracture in the general population is 10% and that the risk threshold for treatment decision is 20% (in other words, individuals with a predicted risk >20% are considered 'high risk'), then it can be shown by simulation46 that a genetic variant with an odds ratio 1.1–1.4 is associated with an AUC of 0.52–0.55, and almost zero net reclassification improvement. In the above scenario, a genetic variant that confers an odds ratio of 3, a rare occurrence, could result in an AUC of 0.76 and NRI of 20%.
The relative risk of hip fracture for the COLIA1 risk genotype was 3.7.47 The AUC for a model including age and BMD was 0.83, but when COLIA1 genotype was added into the model, the AUC increased to 0.85,48 a very modest improvement. None of the genetic variants identified by GWAS achieved that magnitude of association (an OR >1.5). Collectively, these findings suggest that the contribution of any single gene to fracture risk prediction, no matter how large the effect size, is probably limited and would not be useful, particularly in the clinical setting. Actually, the predictive value of a single gene being very limited could be anticipated entirely from simple epidemiologic principles.49
Genetic profiling
Susceptibility to fracture is affected by multiple genetic variants, and any single genetic variant minimally contributes to the accuracy of fracture risk assessment. However, a lack of empirical data means it is unknown whether genetic profiling could enhance the predictive accuracy of fracture prediction. In the absence of data, our group have used methodology described by Pepe et al.46 to conduct simulation studies to test the usefulness of genetic profiling. The results are summarized in Table 2. For a given odds ratio, the discriminatory power and NRI increase proportionally to the number of genetic variants included in the simulation. With 50 genetic variants, each with an odds ratio of ∼1.1, the AUC value is expected to be 0.63; however, the NRI is ∼5%. However, genetic profiling with 50 genetic variants, each with an odds ratio of ∼1.3, the AUC is expected to be 0.80, and the NRI is 15%. Thus, it seems that genetic profiling alone can have useful prognostic value for fracture risk assessment.
Of note, the AUC value for genetic variants has an upper limit. Indeed, Wray et al.50 have elegantly shown that the AUC value for a genetic profiling is a function of disease prevalence and heritability of the disease. For example, for hip fracture, the heritability index is 30%,20,21 and if the 10-year incidence of fracture is 20%, the maximum AUC value is ∼0.80. On this theoretical basis, the estimated maximum discrimination of fracture by genes could not be >0.80.
In reality, genetic profiling only forms one component of fracture risk assessment, as clinical factors and anthropometric information are other important components. Indeed, existing models of fracture risk assessment, such as the Garvan Fracture Risk Calculator or nomogram51,52 and FRAX®,53 only include clinical risk factors. The prognostic performance of these models has been mixed, with AUCs ranging from 0.65 to 0.80, depending on type of fracture and the geographic population. The question remains whether including susceptibility genetic variants (in the form of genetic profiling) in risk assessment models could provide added value in terms of fracture risk prediction for an individual.
This question has been investigated by a partially simulated study, in which a set of 50 independent genetic variants (with allelic frequencies 0.01–0.60) were simulated so that odds ratios for fracture were 1.01–3.0.54 By adding simulated genetic profiling (in the form of a simple risk score) to the usual clinical risk factor model, the AUC increased from 0.77 to 0.88, with most of the improvement being in specificity, not sensitivity.54 These results suggest that genetic profiling could enhance the predictive accuracy of fracture prediction. However, this study was based on some rather optimistic assumptions, which would overestimate the contribution of genes to fracture prediction.54
Indeed, all SNPs identified by GWAS (Table 1) have a very modest association—albeit statistically significant—with fracture. Assuming that 50 SNPs have such magnitudes of association with fracture, the expected AUC value would be ∼0.72, which is significantly lower than that of the model with existing clinical risk factors (age, BMD, prior fracture and fall provide an AUC 0.77). However, when the 50 genetic variants are added to the model with the clinical risk factors, the AUC value increased to 0.83, and the NRI value was 21%. Thus, the integration of genetic variants (in the form of genetic profiling) could improve the accuracy of fracture prediction beyond that obtained with conventional clinical risk factors. More importantly, the incorporation of genetic profiling into the current prognostic models could significantly improve the 'correct' reclassification of fracture risk for an individual, and thus help improve treatment decisions.
Any improvement in predictive value with genetic profiling must be weighed against the cost. At present, the cost of genotyping 50 SNPs is relatively low compared with that of the analysis of bone turnover markers. However, as the technology improves, the genotyping cost is expected to reduce in the future. Thus, personal genetic profiling will probably become increasingly affordable. At the population level, the cost of genetic profiling for fracture risk should be considered in relation to the severity and costs associated with fracture, the predictive value of genetic profiling and available resources.
Problems with current models
GWAS are based on the assumptions of the common variant–common disease hypothesis. Under this assumption, multiple common genetic variants, each with small effect size, additively contribute to the risk of fracture. However, all genetic variants identified so far when considered in a multivariate model explain <5% of variance in BMD and fracture susceptibility. This fact is not consistent with the high heritability of BMD observed from twin and family studies, and is termed the missing heritability phenomenon.55 This phenomenon also raises the possibility that undiscovered rare variants exist with large effect sizes that could additionally contribute to the risk of fracture. This hypothesis is also known as rare variant–common disease. Individuals with a fracture can be hypothesized to carry many common genetic variants (identified by GWAS) and a number of rare variants that are unique to the individuals. This hybrid hypothesis could account for the missing heritability that persists after GWAS. The challenge is how to identify the rare variants. Current technology, such as next generation sequencing technology, which can sequence every base pair across a region of interest, could help unravel rare variants that have not been identified by GWAS.
Of note, all analytic models considered so far are somewhat simplistic. These models have assumed that genetic variants are inherited independently and that their effects on fracture are independent of each other (in other words, no interaction effect or epistasis). Although no gene–gene interaction effect has been identified, such an effect is likely to be identified in the future when enough data have been accumulated. At the individual level, gene–gene interaction predicts that two individuals can have a different fracture risk even if they share the same genotype at one locus. At the population level, epistasis suggests that heterogeneity and incomplete penetrance of fracture is expected. However, gene–gene interaction, which could partly explain the phenomenon of missing heritability, has been ignored in almost all studies to date.55,56
Of course, environmental factors, including hormones and behavioural factors, also contribute to fracture susceptibility.22,57,58,59 The risk of fracture is, hypothetically, a function of the interaction between the time-invariant genes and exposure to environmental factors. Consequently, the risk of fracture for an individual has to be considered not just in terms of gene–gene interactions but also in terms of gene–environment interactions. However, nearly no genetic analyses of osteoporosis have considered these interactions.
Towards individualized assessment
Medical practice is concerned with an individual, and each individual is unique, because no average individual exists in the population. In the absence of any risk factor and genotypes, all individuals in the population have the same risk of fracture and that risk is the population prevalence (in other words, the background risk or lifetime risk). In osteoporosis, this risk is relatively high (30% for men and 46% for women). With additional clinical information, individuals will have different risks. Once the individuals' genotypes are known, the risk of fracture will further differentiate among individuals, such that each individual has a unique risk. Individualized assessment of risk—or the prediction of risk for an individual given a risk profile—recognizes that uniqueness. The more risk factors that can be reliably measured and considered, the greater the likelihood that an individuals' uniqueness is defined.
Interest in individualized fracture risk assessment has been stimulated in the past decade from the advances in genetics, particularly GWAS. Genetic profiling can better define the uniqueness of an individual, and thus better predict the risk for the individual. Some major advantages derive from using genetic profiling as a prognostic factor of fracture risk. An individual's genetic profile is time-invariant; therefore, the risk of fracture for the individual can be predicted at young ages, well before the conventional risk factors become useful in any predictive sense. Although there is no 'genetic' therapy for individuals at high risk of fracture, the use of genetic variants could help segregate individuals at high risk from those with low risk of fracture, and help both with individual counselling about fracture prevention and bone health and targeting of treatment.
Individualized fracture risk assessment could have important implications in the optimization of treatment effectiveness. The effectiveness of an intervention is commonly expressed in terms of the number of patients needed to treat (NNT) to reduce one event. In several antifracture clinical trials, the NNT to reduce one vertebral fracture (compared with the untreated group) ranged from 8 to 83.60 For hip fracture, the NNT ranged from 91 to 250.60,61 The high variability of NNT is mainly attributable to the variation in baseline risks among the participants in the trials, which selected patients on the basis of BMD and/or a history of fracture. As the NNT is a function of baseline risk and absolute risk difference, selecting patients with high risk of fracture would reduce the NNT. With individualized risk assessment, by selecting individuals on the basis of their unique clinical and genetic risk profile, it is possible to increase the homogeneity of risks and hence optimize treatment effectiveness.
The application of individualized risk assessment must be in accordance with the stringent principle of evidence-based medicine. Until now, trials specifically testing the efficacy of individualized therapies chosen on the basis of multivariable risk-based assessments have not been carried out. Therefore, it is not clear whether treating individuals who are selected on the basis of their absolute risk will result in fracture reduction. However, a secondary analysis of a clinical trial suggested that treating women in the top quartile of fracture risk (average 10-year risk probability of 24%) resulted in a 23% reduction in fracture risk (HR 0.69, 0.53–0.90).62 These data are consistent with the hypothesis that treating individuals at high or moderate absolute risk could reasonably be expected to reduce fractures.
Conclusions
In conclusion, the assessment of individual fracture risk is currently based on conventional clinical risk factors. However, GWAS have identified several common genetic variants that are moderately associated with fracture risk. These genetic variants, despite their modest effect sizes, can yield predictive value beyond conventional clinical risk factors when integrated into an existing fracture risk assessment tool such as the Garvan Fracture Risk Calculator.51,52 These genetic variants, together with established clinical risk factors, could provide a useful index of the fracture risk of an individual. This individualized index will potentially help clinicians to tailor treatment to an individual and to make informed choices relating to lifestyle and preventive intervention. However, the incomplete discrimination and accuracy of prediction—probably related to the incomplete coverage of relevant variants and failure to take into account potential gene–gene and gene–environment interactions56—remains a major challenge. With rapid advances in DNA sequencing technology, more variants at a low frequency but with larger effect size will probably be identified. Advances in modelling approaches will refine the genetic profiling and enable a better assessment of fracture risk and individualized fracture prevention.
Review criteria
A search of original articles and meta-analysis published 1985–2012 indexed in PubMed and MEDLINE was carried out. The search terms were “fracture”, “osteoporosis”, “bone mineral density”, “genetics”, “heritability”, “genome-wide association study”, and “candidate gene association study”. All articles were English language, full-text papers. Abstracts were not considered. Owing to the size restriction of the Review, we could not include all of the references identified.
References
Nguyen, N. D., Ahlborg, H. G., Center, J. R., Eisman, J. A. & Nguyen, T. V. Residual lifetime risk of fractures in women and men. J. Bone Miner. Res. 22, 781–788 (2007).
Cummings, S. R., Black, D. M. & Rubin, S. M. Lifetime risks of hip, Colles', or vertebral fracture and coronary heart disease among white postmenopausal women. Arch. Intern. Med. 149, 2445–2448 (1989).
Shortt, N. L. & Robinson, C. M. Mortality after low-energy fractures in patients aged at least 45 years old. J. Orthop. Trauma 19, 396–400 (2005).
Bliuc, D. et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA 301, 513–521 (2009).
Center, J. R., Bliuc, D., Nguyen, T. V. & Eisman, J. A. Risk of subsequent fracture after low-trauma fracture in men and women. JAMA 297, 387–394 (2007).
Center, J. R., Nguyen, T. V., Schneider, D., Sambrook, P. N. & Eisman, J. A. Mortality after all major types of osteoporotic fracture in men and women: an observational study. Lancet 353, 878–882 (1999).
Johnell, O. et al. Predictive value of BMD for hip and other fractures. J. Bone Miner. Res. 20, 1185–1194 (2005).
Nguyen, N. D., Pongchaiyakul, C., Center, J. R., Eisman, J. A. & Nguyen, T. V. Identification of high-risk individuals for hip fracture: a 14-year prospective study. J. Bone Miner. Res. 20, 1921–1928 (2005).
Leslie, W. D., Lix, L. M., Tsang, J. F. & Caetano, P. A. Single-site vs multisite bone density measurement for fracture prediction. Arch. Intern. Med. 167, 1641–1647 (2007).
Delmas, P. D. Treatment of postmenopausal osteoporosis. Lancet 359, 2018–2026 (2002).
National Osteoporosis Foundation. Clinician's Guide to Prevention and Treatment of Osteoporosis [online], (2010).
Rose, G. Sick individuals and sick populations. Int. J. Epidemiol. 14, 427–432 (1985).
Nguyen, N. D., Eisman, J. A., Center, J. R. & Nguyen, T. V. Risk factors for fracture in nonosteoporotic men and women. J. Clin. Endocrinol. Metab. 92, 955–962 (2007).
Nguyen, T. et al. Prediction of osteoporotic fractures by postural instability and bone density. BMJ 307, 1111–1115 (1993).
Nguyen, T. V., Howard, G. M., Kelly, P. J. & Eisman, J. A. Bone mass, lean mass, and fat mass: same genes or same environments? Am. J. Epidemiol. 147, 3–16 (1998).
Pocock, N. A. et al. Genetic determinants of bone mass in adults. A twin study. J. Clin. Invest. 80, 706–710 (1987).
Makovey, J., Nguyen, T. V., Naganathan, V., Wark, J. D. & Sambrook, P. N. Genetic effects on bone loss in peri- and postmenopausal women: a longitudinal twin study. J. Bone Miner. Res. 22, 1773–1780 (2007).
Howard, G. M., Nguyen, T. V., Harris, M., Kelly, P. J. & Eisman, J. A. Genetic and environmental contributions to the association between quantitative ultrasound and bone mineral density measurements: a twin study. J. Bone Miner. Res. 13, 1318–1327 (1998).
Tokita, A. et al. Genetic influences on type I collagen synthesis and degradation: further evidence for genetic regulation of bone turnover. J. Clin. Endocrinol. Metab. 78, 1461–1466 (1994).
Deng, H. W. et al. Genetic determination of Colles' fracture and differential bone mass in women with and without Colles' fracture. J. Bone Miner. Res. 15, 1243–1252 (2000).
Michaëlsson, K., Melhus, H., Ferm, H., Ahlbom, A. & Pedersen, N. L. Genetic liability to fractures in the elderly. Arch. Intern. Med. 165, 1825–1830 (2005).
Cummings, S. R. et al. Risk factors for hip fracture in white women. Study of Osteoporotic Fractures Research Group. N. Engl. J. Med. 332, 767–773 (1995).
Seeman, E. et al. Reduced bone mass in daughters of women with osteoporosis. N. Engl. J. Med. 320, 554–558 (1989).
Seeman, E., Tsalamandris, C., Formica, C., Hopper, J. L. & McKay, J. Reduced femoral neck bone density in the daughters of women with hip fractures: the role of low peak bone density in the pathogenesis of osteoporosis. J. Bone Miner. Res. 9, 739–743 (1994).
Evans, R. A. et al. Bone mass is low in relatives of osteoporotic patients. Ann. Intern. Med. 109, 870–873 (1988).
Kanis, J. A. et al. A family history of fracture and fracture risk: a meta-analysis. Bone 35, 1029–1037 (2004).
Ralston, S. H. & Uitterlinden, A. G. Genetics of osteoporosis. Endocr. Rev. 31, 629–662 (2010).
Huang, Q. Y., Recker, R. R. & Deng, H. W. Searching for osteoporosis genes in the post-genome era: progress and challenges. Osteoporos. Int. 14, 701–715 (2003).
Nguyen, T. V. Pharmacogenetics of anti-resorptive therapy efficacy: a Bayesian interpretation. Osteoporos. Int. 16, 857–860 (2005).
Campbell, H. & Rudan, I. Interpretation of genetic association studies in complex disease. Pharmacogenomics J. 2, 349–360 (2002).
Ioannidis, J. P. A. Why most published research findings are false. PLoS Med. 2, e124 (2005).
Hirschhorn, J. N. & Daly, M. J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108 (2005).
Burton, P. R. et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Kiel, D. P. et al. Genome-wide association with bone mass and geometry in the Framingham Heart Study. BMC Med. Genet. 8 (Suppl. 1), 14 (2007).
Styrkarsdottir, U. et al. Multiple genetic loci for bone mineral density and fractures. N. Engl. J. Med. 358, 2355–2365 (2008).
Richards, J. B. et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet 371, 1505–1512 (2008).
Richards, J. B., Zheng, H. F. & Spector, T. D. Genetics of osteoporosis from genome-wide association studies: advances and challenges. Nat. Rev. Genet. 13, 576–588 (2012).
Rivadeneira, F. et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet. 41, 1199–1206 (2009).
Richards, J. B. et al. Collaborative meta-analysis: associations of 150 candidate genes with osteoporosis and osteoporotic fracture. Ann. Intern. Med. 151, 528–537 (2009).
Estrada, K. et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 44, 491–501 (2012).
Pepe, M. S., Janes, H., Longton, G., Leisenring, W. & Newcomb, P. Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am. J. Epidemiol. 159, 882–890 (2004).
Hanley, J. A. & McNeil, B. J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982).
Bolland, M. J., Grey, A. & Reid, I. R. Re: The calcium scare: what would Austin Bradford Hill have thought? Osteoporos. Int. 22, 3079–3080 (2011).
Pencina, M. J., D'Agostino, R. B. Sr, D'Agostino, R. B. Jr & Vasan, R. S. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat. Med. 27, 157–172 (2008).
Cook, N. R. Statistical evaluation of prognostic versus diagnostic models: beyond the ROC curve. Clin. Chem. 54, 17–23 (2008).
Pepe, M. S., Gu, J. W. & Morris, D. E. The potential of genes and other markers to inform about risk. Cancer Epidemiol. Biomarkers Prev. 19, 655–665.
Nguyen, T. V. et al. Contribution of the collagen I alpha1 and vitamin D receptor genes to the risk of hip fracture in elderly women. J. Clin. Endocrinol. Metab. 90, 6575–6579 (2005).
Tran, B. N., Nguyen, N. D., Center, J. R., Eisman, J. A. & Nguyen, T. V. Enhancement of absolute fracture risk prognosis with genetic marker: the collagen I α 1 gene. Calcif. Tissue Int. 85, 379–388 (2009).
Bacon, C. J. et al. Prevalent dietary supplement use in older New Zealand men. N. Z. Med. J. 124, 55–62 (2011).
Wray, N. R., Yang, J., Goddard, M. E. & Visscher, P. M. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet. 6, e1000864 (2010).
Nguyen, N. D., Frost, S. A., Center, J. R., Eisman, J. A. & Nguyen, T. V. Development of a nomogram for individualizing hip fracture risk in men and women. Osteoporos. Int. 18, 1109–1117 (2007).
Nguyen, N. D., Frost, S. A., Center, J. R., Eisman, J. A. & Nguyen, T. V. Development of prognostic nomograms for individualizing 5-year and 10-year fracture risks. Osteoporos. Int. 19, 1431–1444 (2008).
Kanis, J. A., Johnell, O., Oden, A., Johansson, H. & McCloskey, E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos. Int. 19, 385–397 (2008).
Tran, B. N. et al. Genetic profiling and individualized prognosis of fracture. J. Bone Miner. Res. 26, 414–419 (2011).
Bolland, M. J., Grey, A. & Reid, I. R. Authors' response to editorial. BMJ 342, d3520 (2011).
Zuk, O., Hechter, E., Sunyaev, S. R. & Lander, E. S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl Acad. Sci. USA 109, 1193–1198 (2012).
Center, J. R., Nguyen, T. V., Sambrook, P. N. & Eisman, J. A. Hormonal and biochemical parameters in the determination of osteoporosis in elderly men. J. Clin. Endocrinol. Metab. 84, 3626–3635 (1999).
Nguyen, T. V., Center, J. R., Sambrook, P. N. & Eisman, J. A. Risk factors for proximal humerus, forearm, and wrist fractures in elderly men and women: the Dubbo Osteoporosis Epidemiology Study. Am. J. Epidemiol. 153, 587–595 (2001).
Nguyen, T. V. et al. Lifestyle factors and bone density in the elderly: implications for osteoporosis prevention. J. Bone Miner. Res. 9, 1339–1346 (1994).
Delmas, P. D., Rizzoli, R., Cooper, C. & Reginster, J. Y. Treatment of patients with postmenopausal osteoporosis is worthwhile. The position of the International Osteoporosis Foundation. Osteoporos. Int. 16, 1–5 (2005).
Nguyen, N. D., Eisman, J. A. & Nguyen, T. V. Anti-hip fracture efficacy of bisphosphonates: a Bayesian analysis of clinical trials. J. Bone Miner. Res. 21, 340–349 (2006).
McCloskey, E. et al. Efficacy of clodronate on fracture risk in women selected by 10-year fracture probability. J. Bone Miner. Res. 22 (Suppl. 1), S131 (2007).
Author information
Authors and Affiliations
Contributions
Both authors contributed equally to researching data for the article, discussing the content, writing the manuscript, and reviewing and/or editing the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Nguyen, T., Eisman, J. Genetic profiling and individualized assessment of fracture risk. Nat Rev Endocrinol 9, 153–161 (2013). https://doi.org/10.1038/nrendo.2013.3
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrendo.2013.3
This article is cited by
-
Machine learning approaches for the prediction of bone mineral density by using genomic and phenotypic data of 5130 older men
Scientific Reports (2021)
-
Assessing the clinical utility of genetic profiling in fracture risk prediction: a decision curve analysis
Osteoporosis International (2021)
-
Machine Learning Approaches for Fracture Risk Assessment: A Comparative Analysis of Genomic and Phenotypic Data in 5130 Older Men
Calcified Tissue International (2020)
-
A profiling analysis of contributions of cigarette smoking, dietary calcium intakes, and physical activity to fragility fracture in the elderly
Scientific Reports (2018)
-
RANKL and OPG gene polymorphisms: associations with vertebral fractures and bone mineral density in premenopausal systemic lupus erythematosus
Osteoporosis International (2015)
