
Abstract
Squamous-cell lung cancer is one of the most prevalent subtypes of lung cancer worldwide and its pathogenesis is closely linked with tobacco exposure. Unfortunately, squamous-cell lung cancer patients do not benefit from major advances in the development of targeted therapeutics such as epidermal growth factor receptor (EGFR) inhibitors or anaplastic lymphoma kinase (ALK) inhibitors that show exquisite activity in lung adenocarcinomas with EGFR mutations or echinoderm microtubule associated protein like-4 (EML4)-ALK fusions, respectively. Major efforts have been launched to characterize the genomes of squamous-cell lung cancers. Among the new results emanating from these efforts are amplifications of the fibroblast growth factor receptor 1 gene and mutations of the discoidin domain receptor 2 gene as potential novel targets for the treatment of squamous-cell lung cancer patients. Here, we provide a review on these discoveries and their implications for clinical trials in squamous-cell lung cancer assessing the value of novel therapeutics addressing these targets.
Similar content being viewed by others

Main
Major advances in molecular biology and high-throughput sequencing techniques have boosted cancer drug treatment strategies over the past decades.1, 2, 3, 4, 5, 6 The envisioned paradigm is the identification of genetic aberrations that are unique for defined subgroups of patients, and the development of drugs that specifically aim at oncogenic targets and their signaling pathways of these patients. In lung cancer, this development has led to the identification of oncogenic EGFR mutations that are present in up to 20% of adenocarcinoma patients7, 8, 9 and confer exquisite sensitivity to EGFR inhibitors (for example, gefitinib, erlotinib).10, 11, 12 Another example is the identification of EML4-ALK, the first fusion gene that is causally linked with the development of lung cancer.13 Encouraging first clinical studies show that in patients with EML4–ALK fusions overall response rates of up to 57% can be observed when treated with crizotinib.14
However, the known ‘druggable’ targets are primarily enriched in the subgroup of adenocarcinomas of never smokers.15 In addition, molecularly targeted therapeutics such as erlotinib, gefitinib and cetuximab, as well as conventional chemotherapeutics such as pemetrexed are poorly active in squamous-cell lung cancer patients.11, 16 Drugs such as bevacizumab or sorafenib that primarily inhibit vascular endothelial growth factor receptor (VEGFR) signaling are also not registered for the treatment of squamous-cell lung cancer patients due to increased risk of fatal hemorrhage.17, 18 Thus, the therapeutic repertoire for squamous-cell lung cancer patients is rather limited.
Several genomic studies tackled this unmet medical need by systematically characterizing the landscape of genetic lesions of squamous-cell lung cancer patients in order to define new therapeutically amenable targets. The first large-scale analysis of significant copy-number changes in squamous-cell lung cancer involving a cohort of 47 specimens revealed recurrent amplification of the 3q26.33 chromosomal locus harboring the transcription factor gene SOX2.19 Bass et al.19 were able to show that in SOX2-amplified lung cancer cells SOX2 functions as an oncogene and that its expression is required for proliferation and anchorage-independent growth. Further studies confirmed recurrent amplification of SOX2 in up to 20% of squamous-cell lung cancer patients and further substantiated its role as a lineage-survival oncogene in these tumors.20, 21 Unfortunately, the amplified SOX2 gene does not represent a chemically ‘druggable’ target and its amplification does not lead to the activation of pathways that are immediate and obvious drug targets. However, studies involving a SOX2-driven mouse tumor model allowed the identification of genes such as CyclinD1 to be specifically overexpressed in these tumors.22 Such findings may represent rationale for the study of therapeutics that inhibit cell cycle progression (for example, polo-like kinase 1 (PLK1) inhibitors, cyclin-dependent kinase (CDK) inhibitors) but also allow the exploration of ‘druggable’ genes that are synthetically lethal with SOX2-amplification in squamous-cell lung cancer.
In an independent study our group analyzed recurrent copy number alterations in a cohort of 155 squamous-cell lung cancer specimens that were collected as part of an international collaborative initiative, the Clinical Lung Cancer Genome Project (CLCGP). Using Affymetrix 6.0 SNP arrays (Affymetrix, Santa Clara, CA, USA) we were able to identify the receptor tyrosine kinase fibroblast growth factor receptor 1 (FGFR1) to be focally and significantly amplified in a subgroup of squamous-cell lung cancer patients.23 Two independent cohorts showed that (a) FGFR1 amplification is highly enriched in the squamous-cell histology and that (b) FGFR1 amplification can be detected by fluorescence in-situ hybridization analysis and that it is present in approximately 15–20% of such patients. On the basis of these findings we used a genetically characterized lung cancer cell line collection24, 25 to analyze the cytotoxic effects of the FGFR inhibitor PD173074 as a function of genetic alterations present in these cells in an unbiased fashion. We observed that the cell lines with the highest sensitivity to the compound were enriched for lines bearing FGFR1 amplifications. In some of these lines FGFR1 inhibition induced apoptosis.23 Consequently, when grafted subcutaneously into nude mice, FGFR1-amplified cells formed tumors that regressed after oral application of the FGFR inhibitor. Further studies of the relevant pathways involved in signaling of FGFR1-amplified cells revealed that the MAPK was primarily inhibited upon inhibition of FGFR1 (Figure 1). This is in line with previous reports that clearly show preferential activation of the MAPK pathway in lung cancer cell lines that were stimulated with the FGFR1 specific ligands FGF2 and FGF9.26 However, further studies will be needed to clarify the role of the PI3K-signaling and other oncogenic pathways in FGFR1-amplified lung cancer cells.

Intracellular signaling engaged by amplified FGFR1 and mutant DDR2 in squamous-cell lung cancer. Schematic presentation of signaling cascades that mediate anti-apoptotic and proliferative effects in either FGFR1-amplified (left panel) or DDR2-mutant (right panel) squamous-cell lung cancer cells. As depicted by the size of the arrows amplified FGFR1 primarily engages the MAPK but not the PI3K pathway to execute its oncogenic signaling. Mutant DDR2 is known to engage SRC in order to mediate its oncogenic signaling in squamous-cell lung cancer.
Finally, we showed that the viability of FGFR1-amplified cells is significantly reduced when expression of FGFR1 is silenced by short-hairpin RNAs and that the introduction of a resistance allele of the FGFR1 protein abrogates inhibitor-mediated cell death. These results identify FGFR1 as the driving oncogene in the 8p amplicon in squamous-cell lung cancer. Together, these observations introduce the first therapeutically amenable oncogene alteration in squamous-cell lung cancers.23, 27 An independent study recently confirmed our initial observation.28
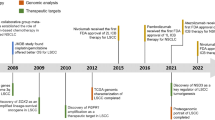
These findings add to the growing compendium of genetic alterations that can be found in the FGFR gene family across different cancer types. For instance, in subtypes of bladder cancer FGFR3 mutations are found in up to 50% of cases, FGFR2 is mutated in endometrial cancer and amplified in gastric cancer, and breast tumors frequently harbor amplifications of FGFR1 as well.29, 30, 31, 32 One of the challenges of personalized cancer medicine is to prospectively identify patients with such genetic aberrations as potent FGFR inhibitors have been developed and are currently undergoing clinical evaluation in genetically defined subgroups.33, 34, 35 FGFR1 amplifications can be detected in routine clinical setting by fluorescence in-situ hybridization. Initial experience at our own center showed that this assay is robust and accurate enough for clinical screening. Accordingly, prospective molecular screening of squamous-cell lung cancers for the presence of FGFR1 amplifications has been established at the cancer center of the University of Cologne. The goal of these efforts is to include patients with positive tumors into trials assessing the safety and efficacy of FGFR inhibitors.
In a recent study focusing on the identification of novel recurrent kinase mutations in squamous-cell lung cancer a group led by Matthew Meyerson analyzed three different squamous-cell lung cancer cohorts and could show that up to 4% of patients harbor mutations in the discoidin domain receptor 2 (DDR2) gene.36 Unlike most receptor tyrosine kinases the DDR receptor family signals in response to fibrillar collagens that have different specifities for the two known DDR receptors that are primarily expressed in either epithelial cells (DDR1) or interstitial cells (DDR2).37 The authors also found mutant DDR2 in two non-small-cell lung cancer cell lines that were growth-inhibited upon shRNA-mediated knockdown of DDR2. Furthermore, treatment with the FDA- and EMEA-approved src-abl inhibitor dasatinib, which has potent inhibitory activity on DDR2, effectively killed DDR2-mutant lung cancer cells. Furthermore, ectopic expression of mutant DDR2 led to growth factor-independent growth of Ba/F3 cells through activation of SRC signaling (Figure 1). Phosphorylation of wild-type DDR receptors and its downstream targets such as Ras largely depends on SRC activity37 but the importance of SRC signaling in DDR2-mutant cells remains to be clarified. The ectopic expression of mutant DDR2 also resulted in anchorage-independent growth of NIH3T3 in soft agar suggesting that the lung cancer-derived DDR2 mutations are oncogenic.36
The authors went on to show that DDR2-mutant cell lines form tumors in nude mice that regressed upon oral treatment with dasatinib. Finally, a squamous-cell lung cancer patient, who had responded to a combination treatment of dasatinib and the EGFR inhibitor erlotinib as part of a clinical trial,38 was found to bear a DDR2 mutation in the kinase domain. Unfortunately, it could not formally be shown that the mutation was actually somatic and the combination treatment makes it difficult to establish a causal relationship between the mutation and the response to the treatment. However, the tumor did not bear an EGFR mutation and the mutation in DDR2 (S768R) was in close proximity to one of the mutations recurrently found in the sequencing screen. These findings are therefore compatible with a dominant role of the DDR2 mutation in driving tumorigenesis in this patient and in rendering the tumor susceptible to treatment with dasatinib. In summary, these results provide evidence that mutations in DDR2 are important oncogenic events inducing therapeutically tractable oncogene dependency. As dasatinib is an FDA-approved drug these findings have immediate implications for the treatment of squamous-cell lung cancer patients and should rapidly translate into clinical reality.
Overall the discoveries of FGFR1-amplified and DDR2-mutant squamous-cell lung cancers give hope for a subgroup of patients that had previously been excluded from virtually all targeted cancer strategies as they significantly expand the list of druggable targets in squamous-cell lung cancer (Figure 2). An intriguing aspect is the fact that most of these patients were smokers and thus the predominant conception that only lung cancers of never smokers can be driven by monogenic ‘driver’ events such as EGFR mutations may not be correct. Large-scale genomics initiatives with a particular focus on squamous-cell lung cancer such as the TCGA (The Cancer Genome Atlas; http://cancergenome.nih.gov/) and the CLCGP (PI: Roman Thomas) efforts will clarify whether additional driver oncogenes are responsible for the growth of squamous-cell lung cancers and other histologically distinct subtypes such as small-cell lung cancers and others. Additionally, initiatives such as mycancergenome (http://www.mycancergenome.org/) have been launched with the goal of cataloging therapeutically tractable genome alterations in cancer. Such initiatives provide an important tool for the clinical community for making decisions based on the genomic landscape of cancers and are thus indispensable for translation of basic genomic discoveries into clinical practice.

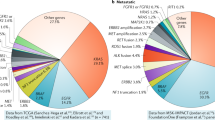
Overview of genetic aberrations that are present in squamous-cell lung cancer. A simplified overview of genetic aberrations found in squamous-cell lung cancer patients based on selected manuscripts discussed in the review and the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic/). The mutations are mutually exclusive except for PIK3CA mutations. amp, amplification; Mut, mutation.
If validated in clinical trials, analyses of FGFR1 amplifications and DDR2 mutations will become part of the routine molecular diagnostic workup of squamous-cell lung cancer. It will therefore be important to develop analytical approaches to address these needs in the near future. Lessons learned from the use of erlotinib and other EGFR inhibitors might help to streamline the endeavors of clinicians, pathologists and researchers alike and help to make these exciting findings a success story for the treatment of squamous-cell lung cancer patients.
References
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet 2007; 39: 347–351.
Thomas RK, Nickerson E, Simons JF, Janne PA, Tengs T, Yuza Y et al. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med 2006; 12: 852–855.
Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005; 309: 1728–1732.
Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I et al. Single-molecule DNA sequencing of a viral genome. Science 2008; 320: 106–109.
Lipson D, Raz T, Kieu A, Jones DR, Giladi E, Thayer E et al. Quantification of the yeast transcriptome by single-molecule sequencing. Nat Biotechnol 2009; 27: 652–658.
TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–1068.
Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I et al. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13306–13311.
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–1500.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–2139.
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009; 361: 958–967.
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–957.
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–2388.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448: 561–566.
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363: 1693–1703.
Sun Y, Ren Y, Fang Z, Li C, Fang R, Gao B et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol 2010; 28: 4616–4620.
Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet 2009; 373: 1525–1531.
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006; 355: 2542–2550.
Scagliotti G, Novello S, von Pawel J, Reck M, Pereira JR, Thomas M et al. Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol 2010; 28: 1835–1842.
Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 2009; 41: 1238–1242.
Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembele D et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One 2010; 5: e8960.
Yuan P, Kadara H, Behrens C, Tang X, Woods D, Solis LM et al. Sex determining region Y-Box 2 (SOX2) is a potential cell-lineage gene highly expressed in the pathogenesis of squamous cell carcinomas of the lung. PLoS One 2010; 5: e9112.
Lu Y, Futtner C, Rock JR, Xu X, Whitworth W, Hogan BL et al. Evidence that SOX2 overexpression is oncogenic in the lung. PLoS One 2010; 5: e11022.
Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2010; 2: 62ra93.
Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest 2009; 119: 1727–1740.
Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci USA 2009; 106: 18351–18356.
Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol 2009; 75: 196–207.
Turner NC, Seckl MJ . A therapeutic target for smoking-associated lung cancer. Sci Transl Med 2010; 2: 62ps56.
Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T et al. Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer. PLoS One 2011; 6: e20351.
Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet 1999; 23: 18–20.
Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA 2008; 105: 8713–8717.
Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res 2008; 68: 2340–2348.
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010; 70: 2085–2094.
Sarker D, Molife R, Evans TR, Hardie M, Marriott C, Butzberger-Zimmerli P et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin Cancer Res 2008; 14: 2075–2081.
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 2008; 68: 4774–4782.
Turner N, Grose R . Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2009; 10: 116–129.
Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discovery 2011; 1: 78–89.
Ikeda K, Wang LH, Torres R, Zhao H, Olaso E, Eng FJ et al. Discoidin domain receptor 2 interacts with Src and Shc following its activation by type I collagen. J Biol Chem 2002; 277: 19206–19212.
Haura EB, Tanvetyanon T, Chiappori A, Williams C, Simon G, Antonia S et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J Clin Oncol 2010; 28: 1387–1394.
Acknowledgements
RKT is supported by the German Ministry of Science and Education (BMBF) as part of the NGFNplus program (grant 01GS08100), by the Max Planck Society (M.I.F.A.NEUR8061 to RKT), by the Deutsche Forschungsgemeinschaft (DFG) through SFB (TP6), the Ministry for Innovation, Science, Research and Technology of the State of Nordrhein-Westfalen (MIWT, 4000- 12 09), by the Fritz-Thyssen-Stiftung (grant 10.08.2.175) and by an anonymous foundation. MLS is supported by an IASLC Lung Cancer Fellowship Award. RKT received consulting and lecture fees (Sanofi-Aventis, Merck, Roche, Boehringer Ingelheim, Astra-Zeneca, Atlas-Biolabs, Johnson&Johnson), as well as research support (AstraZeneca, Merck).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
There is potential conflict of interest.
Rights and permissions
About this article
Cite this article
Sos, M., Thomas, R. Genetic insight and therapeutic targets in squamous-cell lung cancer. Oncogene 31, 4811–4814 (2012). https://doi.org/10.1038/onc.2011.640
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2011.640
Keywords
This article is cited by
-
Non-small-cell lung cancer
Nature Reviews Disease Primers (2015)
