
Abstract
Triple-negative breast cancer (TNBC) is an aggressive form of breast cancer. Despite response to chemotherapy, relapses are frequent and resistance to available treatments is often seen in the metastatic setting. Therefore, identification of new therapeutic targets is required. With this aim, we have profiled the activation status of 44 receptor tyrosine kinases (RTKs) and their major signaling pathways in patient-derived TNBC tumors. Frequent co-activation of several RTKs as well as the extracellular signal-regulated protein kinases 1 and 2 (Erk1/2) and mammalian target of rapamycin (mTOR) routes was found. Pharmacologic targeting of the activated kinases indicated that agents that attack the mTOR route are more potent and efficient antitumoral treatments than agents targeting RTKs. mTOR signals through two multiprotein complexes, mTORC1 and mTORC2. We used a genetic approach to explore the contribution of each of the two mTOR branches to the regulation of cell number of TNBC cells. RNA interference experiments indicated that mTORC1 predominated over mTORC2 in the control of TNBC cell proliferation. Moreover, RNA interference of mTOR had a superior antiproliferative action than separately acting on mTORC1 or mTORC2. To analyze the relevance of mTOR targeting in vivo, we used mice with TNBC. Treatment of these mice with BEZ235, a drug that targets mTOR, slowed tumor growth. Mechanistically, BEZ235 delayed cell cycle progression without affecting cell viability. Our results show that TNBCs are particularly sensitive to inhibition of the mTOR pathway, and indicate that mTOR targeting may be a more efficient anti-TNBC therapy than exclusively acting on the mTORC1 branch of the pathway. This is relevant as most mTOR inhibitors used in the clinic act on mTORC1. Collectively with the fact that BEZ235 synergized with drugs commonly used in the treatment of TNBC, our data support the clinical development of agents that act on mTOR as a therapy for this disease.
Similar content being viewed by others

Introduction
Triple-negative breast cancer (TNBC) accounts for 15% of all breast cancers. It is defined at an immunohistochemical level as the lack of detectable expression of hormone receptors, and no human epidermal growth factor receptor 2 (HER2) gene amplification.1, 2, 3 Therapy of TNBC is based on chemotherapy with taxanes, vinorelbine or platinum compounds, which are likely to be effective in this type of tumors because of their rapid proliferation rates and frequent derangements in DNA repair mechanisms.4 Unfortunately, relapses are frequent in the early stage, and resistance to the chemotherapeutic agents is often seen in the metastatic setting.4, 5, 6 These facts, together with the relatively poor knowledge of the molecular alterations present in TNBC, have stimulated much research into this field to identify aberrant signaling networks that may be pharmacologically attacked in this aggressive type of breast cancer.4
Receptor tyrosine kinases (RTKs) play a key role in cell proliferation, and their deregulation has been linked to the pathogenesis of various tumors,7, 8 including TNBC.3, 9, 10, 11 These and other expression studies, together with the clinical success of targeting RTKs in other breast tumors,12 have established the bases to test drugs targeting some of these receptors in TNBC. Unfortunately, these clinical studies targeting individual RTKs have shown limited efficacy.13, 14 A circumstance that may explain this limited effect is concomitant expression of activated forms of various RTKs.11 Therefore, the targeting of an individual RTK may be ineffective if other proto-oncogenic RTKs are functional.11 To overcome this situation, it would be desirable to identify the active RTKs in a particular tumor to design drug combinations that target the different active RTKs. Another strategy could be the use of drugs that attack signaling pathways shared by different RTKs. The pharmacological intervention on these convergent signaling nodes could be effective and likely less toxic than using a multikinase RTK inhibitory approach.15 Importantly, recent deep-sequencing studies in TNBC have identified frequent clonal mutations in PI3K and PTEN, two genes that participate in signaling downstream of RTKs.16
With the aim of developing novel therapies in TNBC, we have profiled the activation status of 44 different RTKs and their major signaling pathways in a set of tumors from patients with TNBC. We observed co-activation of various RTKs in single tumors, together with frequent activation of the PI3K/Akt/mTOR and the Ras/Raf/Erk1/2 signaling routes. Genetic as well as pharmacological studies showed that TNBC cells were particularly sensitive to inhibition of the mammalian target of rapamycin (mTOR) pathway, opening the possibility for the incorporation of agents that target this route for the treatment of TNBC.
Results
Profiling of active kinases defines mTOR as a relevant target in TNBC
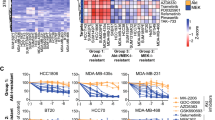
Although expression analyses are useful in describing the presence of kinases or their signaling intermediates, a more accurate assessment of their function is offered by evaluation of their activation status.7 The latter can be explored by analyzing tyrosine phosphorylation of RTKs, or phosphorylation of certain activating residues in signaling proteins.7 The activation status of 44 RTKs as well as of major RTK signaling pathways were analyzed in 26 TNBC tumors using antibody arrays. The clinical characteristics of this patient cohort are summarized in Supplementary Table 1. The most frequently activated kinases were the epidermal growth factor receptor (EGFR; 75% of patients), HER2 (33% of patients), HER4 (25% of patients), platelet-derived growth factor receptor-β (PDGFRβ; 71% of patients), Akt (88% of patients) and extracellular signal-regulated protein kinases 1 and 2 (Erk1/2; 53% of patients; Figure 1a). Concomitant activation of several RTKs in a single tumor was detected (see, for example, inset in Figure 1a, see also Supplementary Table 2).

(a) Expression of activated forms of RTKs and signaling proteins in tumor samples. The inset shows an array of active RTKs from one TNBC tumor. (b) Expression and activation of EGFR, HER2, HER4, PDGFRβ, AKT and Erk1/2 in seven TNBC cell lines. Activated forms of the RTKs were analyzed by immunoprecipitation with anti-receptor antibodies, followed by anti-PY blot. Detection of activated Akt and Erk1/2 was performed by western using antibodies that recognize residues phosphorylated in these proteins, which indicate active forms. (c) Effect of different kinase inhibitors on the MTT metabolization of four different TNBC cell lines. Cells were plated in 24-well dishes, and treated with the indicated concentrations of the drugs for 48 h. MTT uptake and metabolization was measured as described in the ‘Materials and methods’ section. Results are plotted as the mean±s.d.
To explore the relevance of these frequently activated RTKs and signaling pathways in TNBC, their level of activation was explored in seven TNBC cell lines. EGFR was expressed in all cell lines, and was tyrosine phosphorylated in five of them (Figure 1b). HER2 was detectable in five cell lines, and in those with higher levels (HCC1187 and HCC3153) resting tyrosine phosphorylation was evident. PDGFRβ was expressed in HS578T and HBL100, and was tyrosine phosphorylated. HER4 was undetectable in the cell lines analyzed. Akt was phosphorylated at serine 473 in all the cell lines, except in MDA-MB231. Detectable levels of dually phosphorylated Erk1/2 were observed in HS578T, HBL100 and BT549. MDA-MB231 and HCC70 cell lines showed higher phosphorylated Erk1/2 levels.
To define the importance of the activated RTKs or their signal transduction routes in TNBC, we explored the action of several inhibitory compounds, selected on the basis of being already approved or under clinical development in other tumor types. Lapatinib was used as an EGFR/HER2 dual inhibitor;17 dasatinib as a PDGFRβ inhibitor;18 sorafenib19 and selumetinib20 as Raf and MEK1 inhibitors, respectively; and BEZ235 as a PI3K/mTOR dual inhibitor.21 Dose-response experiments were performed in four TNBC cell lines, and estimation of the amount of cells analyzed by an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-based metabolization assay. As shown in Figure 1c, lapatinib, sorafenib and selumetinib had a rather modest effect on the MTT metabolization of the distinct TNBC cell lines tested. Dasatinib presented a mild inhibitory effect on these cell lines. The most effective and potent agent of those tested was BEZ235, which reached half-maximal inhibitory concentration values in the nanomolar range. Western blotting demonstrated the efficiency of these compounds in inhibiting the phosphorylation status of their targets or downstream signaling intermediates (Supplementary Figure 1).
mTORC1 and mTORC2 control the proliferation of TNBC cells
Given the substantial inhibitory action of BEZ235 on the proliferation of TNBC cells, and as this inhibitor targets mTOR, we decided to explore the importance of this pathway in TNBC. The mTOR kinase acts in concert with other proteins that constitute two multiprotein complexes, mTORC1 and mTORC2.22 mTORC1 includes mTOR, Raptor, PRAS40, mLST8/GβL and Deptor, whereas mTORC2 includes mTOR, Rictor, mLST8/GβL, Sin1, Protor and Deptor.
To investigate the role of mTOR and its two branches in the proliferation of TNBC, we performed knockdown of Raptor and Rictor as targets to decrease mTORC1 and mTORC2 signaling, as well as knockdown of mTOR as readout of the overall importance of this pathway in TNBC. For these experiments we used HBL100 and MDA-MB231 cell lines selected on the bases of their differential pAkt levels (Figure 1b). Western blotting of cell extracts from MDA-MB231 and HBL100 cells demonstrated the knockdown of these different components of the mTOR route (Figure 2a). mTOR knockdown exerted the largest inhibitory effect on the MTT metabolization values in both cell lines (72% inhibition in MDA-MB231 and 75% of inhibition in HBL100 with respect to the proliferation values of control cells, Figure 2b). Knockdown of Raptor had a higher inhibitory effect than the knockdown of Rictor in both cell lines. In the MDA-MB231 cell line the effect of the Raptor knockdown was very similar to the effect obtained by mTOR knockdown, indicating that mTORC1 is critical in channeling the cell growth signals transmitted through the mTOR pathway in this cell line. Similar data were generated using a distinct set of short hairpin RNAs (shRNAs; Supplementary Figure 2). Together, these data confirm the relevant role of mTOR in sustaining cell proliferation in these TNBC cell lines, and indicate a predominant role of mTORC1 over mTORC2 on this biological property.

(a) Knockdown of different mTORC components in HBL100 and MDA-MB231. The amount of the different proteins was analyzed by western blot. (b) Effect of the knockdown of mTOR, Raptor and Rictor on the proliferation of HBL100 and MDA-MB231. Cells were infected with viruses that included shRNAs for mTOR, Raptor or Rictor, selected with puromycin for 48 h, and MTT metabolization was measured at the indicated times. Results are plotted as the mean±s.d. of an experiment that was repeated twice with similar results.
mTOR inhibition provokes cell cycle arrest of TNBC cells
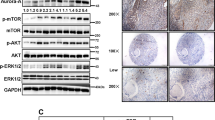
To confirm that BEZ235 acted on the mTOR route and to establish a dose that would efficiently block mTOR, we analyzed the phosphorylation status of pS6 and p4E-BP1 as readouts of mTORC1 activity, as well as pAkt as readout of mTORC2. BEZ235 decreased the resting phosphorylation status of pS6 and p4E-BP1 in a dose-dependent manner (Figure 3a). In MDA-MB231 cells, which did not present resting pAkt, treatment with BEZ235 provoked its phosphorylation at concentrations of BEZ235 between 10 and 50 nM. At higher doses of BEZ235 the pAkt signal decreased, disappearing at 250 nM (Figure 3a). In HBL100 cells, doses of BEZ235 of 10 and 25 nM caused a small but reproducible increase in pAkt with respect to the resting levels. At higher doses, BEZ235 decreased pAkt levels with complete disappearance of the signal at 250 nM. As this latter concentration of BEZ235 completely blocked phosphorylation of mTORC1 and mTORC2 readout proteins, we selected that dose for the following studies on the mechanism of action of the drug. It has been previously reported that treatment of HER2-overexpressing breast cancer cells with BEZ235 results in activation of mitogen-activated protein kinase signaling.23 However, in three different TNBC cell lines we did not observe such an effect (Supplementary Figure 3).

(a) Effect of BEZ235 on mTORC1 and mTORC2 readout proteins. Cells were treated with the indicated doses of BEZ235 for 24 h, and cell lysates analyzed by western blot using antibodies to the indicated proteins. Calnexin was used as a loading control. The asterisk indicates a non-specific band. (b) Effect of BEZ235 on the cell cycle of MDA-MB231 and HBL100. Cells were treated with BEZ235 for 24 h, fixed and permeabilized and stained with propidium iodide before analysis in a cytometer. The histograms represent the percentage of cells in the different phases of the cell cycle. (c) Effect of BEZ235 on apoptosis. Cells were treated with BEZ235 for 48 h, and then stained with Annexin V/FITC and propidium iodide. Stained cells were analyzed by flow cytometry. The percentage of Annexin V-positive or Annexin V-negative cells is shown.
We explored whether the inhibitory action of the compound on TNBC cells was due to inhibition of proliferation, stimulation of cell death or both. Treatment with BEZ235 increased the number of cells in the G0/G1 phases of the cell cycle (increase in G0/G1 of 32% and 11.5% in MDA-MB231 and HBL100, respectively, Figure 3b). BEZ235 did not increase Annexin V-positive cells, suggestive of failure of this drug to cause death (Figure 3c).
To define the action of BEZ235 on the cell cycle we used treatments that block cell cycle progression, followed by release from these blockers in the absence or presence of BEZ235. Nocodazole blocked cells in the M phase of the cell cycle, as indicated by the accumulation of cyclin B or by phosphorylation of the mitotic checkpoint protein BUBR1 (Figure 4a). After nocodazole washout, cells were released from mitosis and entered into G1. The biochemical (for example, BUBR1 dephosphorylation, Figure 4a) and cytometric profile (Figure 4b) at 3 h after release from nocodazole indicated that treatment with BEZ235 did not substantially affect exit of the TNBC cells from mitosis. In untreated samples, a substantial number of cells transited from G1 to S and G2/M, especially at 16 h after release (Figure 4b). However, in BEZ235-treated cells, the 2N population represented a substantial proportion (Figure 4b), suggesting that the drug slowed movement of the cells through G1 and S. The wide G1/S region in the propidium iodide staining also indicated that in HBL100 cells BEZ235 could not fully block G1 to S progression (Figure 4b, at T16, T18 and T21 h after release). In HBL100 cells, increases in cyclin D1, cyclin D3, cyclin A or cyclin B, which mark progression of cells through G1, S and G2/M, were inhibited by BEZ235 (Figure 4a). Western blotting of pS6 and pAkt confirmed that the drug inhibited their phosphorylation along the time course of the experiment (Figure 4a). Analogous experiments performed in MDA-MB231 generated data also compatible with the concept that BEZ235 slowed movement of cells through the cell cycle (Supplementary Figure 4). Synchronization of HBL100 at late G1 by a double thymidine block24 confirmed that BEZ235 slowed movement of cells at the G1/S transition (Supplementary Figure 5).

Effect of BEZ235 on the cell cycle. HBL100 cells were synchronized in mitosis by treatment with nocodazole for 14 h. Nocodazole was then washed out, and cells were placed in growth media in the absence or presence of BEZ235 (250 nM). Cells were harvested at the indicated times, and analyzed biochemically (a) and cytometrically (b). The asterisks indicate nonspecific bands.
Antitumoral action of BEZ235 in mice with epithelially deleted Brca1 and p53
To investigate the in vivo effect of BEZ235 on TNBC tumors we used a conditional mouse model in which deletion of Brca1 and p53 can be created by crossing to animals with K14/cre, which eliminates the Brca1 and p53 genes in epithelia, including breast epithelia.25 Female mice from this strain develop mammary tumors with characteristics of TNBC.25 We confirmed by western blot that HER2 as well as progesterone and estrogen receptors were undetectable or very low in these tumors (Figure 5a). We also failed to detect dually phosphorylated Erk1/2. In contrast, EGFR, PDGFRβ, pS6 and pAkt were expressed in these tumors (Figures 5a and b). Western analyses of the tumors from these mice showed that treatment with BEZ235 inhibited pAkt and pS6 levels (Figure 5b). BEZ235 significantly delayed tumor growth (70% inhibition of tumor growth after 21 days of treatment, P=0.022, Figure 5c).

(a) Expression of different proteins in tumors from two mice with epithelially deleted Brca1/p53. (b) Effect of BEZ235 on the activation status of Akt and S6. Tumors from mice treated or not with BEZ235 were excised, homogenized and cell extracts analyzed by western blot for the expression of activated forms of Akt and S6. GAPDH blots were performed as loading controls. (c) Effect of treatment with BEZ235 on tumor growth in mice with epithelially deleted Brca1/p53. Mice were randomly assigned to two groups and treated with BEZ235 following the schedule detailed under ‘Materials and methods’.
BEZ235 synergizes with standard of care drugs
As most antitumoral therapies are based on drug combinations, we explored whether combining BEZ235 with drugs used in the therapy of TNBC augmented their effect. To this end, three TNBC cell lines were treated with different doses of BEZ235, taxotere and carboplatin. Double and triple combinations were performed, and the results were analyzed by the Chou–Talalay algorithm.26 Double and triple combinations of BEZ235 with these drugs were found to be synergistic in HBL100 and HS578T cells (Figure 6a and Supplementary Figure 6A). In MDA-MB231, these double and triple combinations were additive. As BEZ235 affected cell cycle progression, it is possible that simultaneous administration of BEZ235 with chemotherapy might alter the efficacy of the latter. To test such a possibility, experiments in which BEZ235 and chemotherapy were added simultaneously or sequentially were performed. As shown in Figure 6b, sequential addition of BEZ235 and chemotherapeutic agents did not show antitumoral benefit as compared with the agents added together. Analogous results were also observed in the HS578T cell line (Supplementary Figure 6B).

(a) Effect of the combination of BEZ235 and standard of care drugs. Cell lines were treated with the indicated concentrations of the drugs, either in double or triple combinations, and their MTT values were measured. Combination indexes for the different drug combinations were obtained using the CalcuSyn program and plotted. (b) Effect of sequential versus concomitant administration of the drug combinations on MTT metabolization rates. Cells were plated, and 24 h later treated with the individual drugs (BEZ235, 7.5 nM; carboplatin, 100 μM; or taxotere, 0.5 nM), the first drug in the case of sequential combinations, or the drug combinations (B+C, or B+T) in the case of concomitant administration. For the sequential treatments, the second drug was added 15 h after the first drug. MTT metabolization was analyzed in all cases at 72 h. The results represent the mean±s.d. of quadruplicates of an experiment that was repeated twice. (c, d) Action of the drug combinations on the cell cycle (c) and apoptosis (d). Drugs were added for 48 h (BEZ235, 7.5 nM; carboplatin, 250 μM; or taxotere, 0.5 nM), and analyzed as described above. (e) In vivo antitumoral action of the drug combinations. Mice were injected with MDA-MB231 cells, and when tumors reached 200 mm3 mice were treated with the different drugs either alone or in combination, with the schedules indicated under ‘Materials and methods’. Data represent the mean±s.d. Statistical analyses were performed on the measurements obtained at the end of the experiment (day 24 of treatment) using the SPSS software v19, with the following results: Control vs BEZ235: mean volume=1.12 vs 0.53 cm3, difference=0.59 cm3, 95% confidence interval=0.77–0.41, P=0.000; Control vs TXT: mean volume=1.12 vs 0.50 cm3, difference=0.62 cm3, 95% confidence interval=0.81–0.44, P=0.000; Control vs carboplatin: mean volume=1.12 vs 0.84 cm3, difference=0.28 cm3, 95% confidence interval=0.47–0.083, P=0.009; BEZ235 vs BEZ235+TXT: mean volume=0.53 vs 0.27 cm3, difference=0.25 cm3, 95% confidence interval=0.37–0.13, P=0.000; BEZ235 vs BEZ235+carboplatin: mean volume=0.53 vs 0.32 cm3, difference=0.21 cm3, 95% confidence interval=0.34–0.079, P=0.004; TXT vs BEZ235+TXT: mean volume=0.50 vs 0.27 cm3, difference=0.22 cm3, 95% confidence interval=0.34–0.10 P=0.001; carboplatin vs BEZ235+carboplatin: mean volume=0.84 vs 0.32 cm3, difference=0.52 cm3, 95% confidence interval=0.68–0.36 P=0.000.
The action of the double combinations of BEZ235 with chemotherapy on cell cycle and cell viability was explored. Addition of carboplatin to MDA-MB231 and HBL100 cells caused accumulation of cells in the G2/M phases, and strongly decreased the presence of cells in the G1 phase (Figure 6c). Concomitant addition of BEZ235 and carboplatin augmented the proportion of cells in the S phase, in line with a potential effect of BEZ235 in slowing progression along G1 and S phases of the cell cycle. Taxotere had a profound effect on cell cycle profiles in both MDA-MB231 and HBL100, and addition of BEZ235 did not appreciably alter such profile. Annexin V staining, used to assess the effect of the drugs on apoptotic cell death, indicated that a combination of BEZ235 with carboplatin slightly increased the amount of Annexin V-positive cells in HBL100 (Figure 6d). In contrast, the level of cell death assessed by this technique did not change when BEZ235 was combined with taxotere.
Combinations of BEZ235 with taxotere or carboplatin were also analyzed in vivo. For these experiments, MDA-MB231 cells were injected in nude mice, and when tumors reached 200 mm3, mice were treated with the different drugs, either alone or in combination. Treatment with BEZ235 delayed tumor growth, consistent with the data obtained in the Brca1 and p53 mice. The combination of BEZ235 with carboplatin or taxotere had a higher antitumoral effect than individual treatments (Figure 6e).
Discussion
The dismal prognosis of TNBC, as well as the lack of new treatment options to fight the disease, has stimulated research in this pathology with the purpose of identifying novel treatments. In line with this, and given the importance of kinases in breast and other tumors, we investigated the level of activation of 44 different RTKs as well as several signaling intermediates in tumors from patients with TNBC. As the tumor samples and the cell lines analyzed frequently had co-activation of various RTKs, it is not unexpected that blocking only one of them may have a rather marginal effect on cell proliferation. These data may explain the relatively poor clinical activity of agents that target these receptors,14 and open the important question of how to design efficient therapies to overcome escape to antitumoral therapies because of proliferation signals originating at different RTKs. Several strategies can be contemplated, such as the concomitant administration of drugs that target different RTKs or the use of wide-spectrum multikinase inhibitors. Although some preclinical studies support these strategies,11, 27 some clinical studies with the multikinase inhibitor sunitinib have shown disappointing results.28 Another strategy could be the use of agents that target common signaling routes that funnel proliferative signals originated at distinct RTKs.9, 29 In support of the latter are the results obtained with the dual PI3K/mTOR inhibitor BEZ235. This drug was more potent and effective than agents that target active RTKs in TNBC, suggesting that targeting PI3K/mTOR could be important in TNBC. This idea was evaluated using a genetic approach. Knockdown of mTOR profoundly compromised the proliferation of the two TNBC cell lines tested. The knockdown of Raptor had a major inhibitory effect than Rictor knockdown on the proliferation of TNBC cells. Together, those results confirm the important role of the mTOR kinase routes in the control of proliferation of TNBC cells, and also suggest that signals channeled through the mTORC1 route are more important to sustain proliferation than those sent through mTORC2. This conclusion raises the possibility of using drugs that inhibit mTORC1, such as rapamycin or analogs, for the therapy of TNBC. However, several circumstances should be considered. First, our data indicate that attack to the mTORC1 pathway by rapalogs causes an increase in pAkt, indicative of upregulation of mTORC2, as already reported in other cellular models.30, 31 As our data with the knockdown experiments indicate that mTORC2 also facilitates proliferation of TNBC cells, the upregulation of mTORC2 by rapalogs could reduce effectiveness of these compounds. This concept, already reported in other cellular models and confirmed by us in TNBC cells, should be taken into account by clinicians evaluating rapalogs in different oncologic disorders. Furthermore, as activation of PI3K/Akt has been linked to resistance to therapies used in other breast cancer subtypes,32, 33 upregulation of pAkt by rapalogs may contribute to selection of tumoral clones resistant to standard therapies used in the treatment of TNBC. Moreover, it is important to mention that in the human samples, phosphorylation of S6, a surrogate marker of mTORC1 activity, was not as frequently detected as in cell lines. Therefore, strategies to target the mTOR signaling network should consider acting on mTOR, rather than just on the mTORC1 branch of the pathway.31, 34 This conclusion is highly relevant as most mTOR inhibitors already approved or in clinical trials target mTORC1.
The mechanistic studies on the antitumoral effect of BEZ235 not only provide insights into how the drug acts, but may also have therapeutic relevance. Of note, low doses of BEZ235 reduced pS6 but increased pAkt. As mTORC1 may inhibit mTORC2 signaling, it is therefore possible that inhibition of mTORC1 at low BEZ235 concentrations may explain the increase in pAkt observed when using these low BEZ235 concentrations.30 These findings also indicate the existence of distinct thresholds for mTORC1 and mTORC2 inhibition by BEZ235, as formerly reported in ovarian cancer cells.35 The antitumoral effect of BEZ235 in TNBC was mainly caused by slowing cell cycle progression, without any evident effect on apoptosis. The latter conclusion is particularly important, as reports in different cell types,36, 37 including a heterogeneous group of breast cancer cell lines,38 showed that the drug caused activation of caspases and PARP (poly (ADP-ribose) polymerase) cleavage, indicative of apoptotic cell death.39 However, BEZ235 failed to increase the number of Annexin V-positive cells in TNBC cell lines. Interestingly, although BEZ235 decreased tumor growth in mice, it did not cause shrinkage of the tumors, in line with a cytostatic rather than a cytotoxic effect of the drug also in vivo. The blockade-release synchronization experiments indicated that BEZ235 slows cell cycle progression. The results obtained indicated that progression of cells through G1 and S phases of the cell cycle was profoundly compromised by BEZ235. Biochemical experiments suggested that increases in the expression of various cyclins involved in cell cycle progression was inhibited in cells treated with BEZ235. Whether this is because of the role of mTOR on protein synthesis will require further investigation. These mechanistic studies suggest that eradication of the tumor will require combination of the effectiveness of BEZ235 as a cytostatic agent with drugs that provoke cell death. Importantly, BEZ235 synergized with other drugs used in the treatment of TNBC, and those combinations, may cause cell death. In fact, in HBL100 cells the combination of carboplatin with BEZ235 augmented the apoptotic action of the former. In addition to this proapoptotic potentiation, this drug combination also affected cell cycle profiles when compared with the individual treatments. The fact that the drug combination increased the proportion of cells in the S phase indicates that passage through this cell cycle phase is compromised by the drug combination, and might contribute to the increased antitumoral action of this drug combination when compared with the individual treatments. The mechanism of potentiation of the action of taxotere by BEZ235 is less clear. The combination of both agents did not cause substantial apoptosis. Taxotere provoked profound changes of the cell cycle profiles in both MDA-MB231 and HBL100 cells, and these profiles were unaffected by addition of BEZ235. We suspect that the profound derangement of the cell cycle profile caused by taxotere impedes adequate interpretation of a potential additive action of the BEZ235+taxotere combination on the cell cycle. Importantly, the in vivo experiments confirmed the major antitumoral action of the combined treatments when compared with the individual treatments, opening the possibility of evaluating such combinations for the therapy of TNBCs.
In summary, we show that co-activation of several RTKs is frequent in TNBC, as it is activation of downstream signaling pathways, especially the PI3K/Akt/mTOR route. Results obtained using in vitro and in vivo preclinical models indicate that targeting of mTOR is an effective antitumoral treatment in TNBC. The strategy of targeting this key pathway used by several proto-oncogenic RTKs may not only result in higher clinical activity, but could also represent a less toxic alternative to the use of a cocktail of RTK inhibitors. Finding a common target may in addition facilitate the management of TNBC, especially considering the genomic diversity of this disease. These conclusions, together with the fact that BEZ235 synergized with other drugs used in the treatment of TNBC, represent a starting point for the clinical development of mTOR inhibitors for the therapy of this disease.
Materials and methods
Reagents and antibodies
Cell culture media and fetal bovine serum were purchased from Invitrogen (Gaithersburg, MD, USA). Protein A-Sepharose was from GE Healthcare Life Sciences (Piscataway, NJ, USA). Puromycin and MTT were from Sigma Chemical (Madrid, Spain). Annexin V/fluorescein isothiocyanate (FITC) was purchased from BD Biosciences (San Jose, CA, USA). The polyethylenimide reagent was from Polysciences, Inc. (Warrington, PA, USA). BEZ235, lapatinib, sorafenib and selumetinib were from LC Laboratories (Woburn, MA, USA). Other generic chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA), Roche Biochemicals (Hoffmann, Germany) or Merck (Darmstadt, Germany). The anti-GAPDH, anti-cyclin E, anti-Wee1, anti-pErk1/2, anti-Erk2, anti-pTyr, anti-EGFR, anti-HER2, anti-HER4 and anti-PDGFRβ antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-mTOR, anti-Raptor, anti-pS6 (S240/244), anti-S6, anti-p4E-BP1 (T37/46), anti-4E-BP1, anti-Akt and anti-pCDK1(Y15) antibodies were from Cell Signaling Technologies (Beverly, MA, USA). The anti-cyclin A, anti-cyclin B, anti-cyclin D1, anti-cyclin D3 and anti-BUBR1 were purchased from BD Biosciences. The anti-ER and anti-PR were from Master Diagnóstica (Granada, Spain). The anti-Rictor was from Bethyl Laboratories (Montgomery, TX, USA). Horseradish peroxidase conjugates of anti-rabbit and anti-mouse immunoglobulin G were from Bio-Rad Laboratories (Hercules, CA, USA). The anti-calnexin antibody was from Stressgen Biotechnologies Corporation (British Columbia, Canada). The anti-pAkt (Serine 473) antibody has been described previously.40
Cell culture and infection with lentivirus
All cell lines were cultured at 37 °C in a humidified atmosphere in the presence of 5% CO2 and 95% air. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) or in RPMI medium containing high glucose concentration (4500 mg/l) and antibiotics (penicillin at 100 mU/ml and streptomycin at 100 μg/ml) and supplemented with 10% fetal bovine serum. Cell lines were provided by Drs J Losada and A Balmain (originally from Dr JW Gray’s Laboratory, who in turn obtained them from the ATCC or from collection development in the laboratories of Drs S Ethier and A Gazdar to avoid errors occurring when obtained through ‘second-hand’ sources).41
The lentiviral vectors containing shRNA for Raptor, Rictor and mTOR42 were obtained from Addgene (Cambridge, MA, USA). The following sequences were used:
mTOR_shRNA_1 sense:
5′-CCGGCCGCATTGTCTCTATCAAGTTCTTCCTGTCAAACTTGATAGAGACAATGCGGTTTTTG-3′.
mTOR_shRNA_1 antisense:
5′-AATTCAAAAACCGCATTGTCTCTATCAAGTTTGACAGGAAGAACTTGATAGAGACAATGCGG-3′.
Raptor_shRNA_1 sense:
5′-CCGGGGCTAGTCTGTTTCGAAATTTCTTCCTGTCAAAATTTCGAAACAGACTAGCCTTTTTG-3′.
Raptor_shRNA_1 antisense:
5′-AATTCAAAAAGGCTAGTCTGTTTCGAAATTTTGACAGGAAGAAATTTCGAAACAGACTAGCC-3′.
Rictor_shRNA_1 sense:
5′-CGGGCAGCCTTGAACTGTTTAACTTCCTgTCATTAAACAGTTCAAGGCTGCTTTTTG-3′.
Rictor_shRNA_1 antisense:
5′-AATTCAAAAAGCAGCCTTGAACTGTTTAATGACAGGAAGTTAAACAGTTCAAGGCTGC-3′.
mTOR_shRNA_2 sense:
5′-CCGGTTCAGCGTCCCTACCTTCTTCTCTCGAGAGAAGAAGGTAGGGACGCTGATTTTTG-3′.
mTOR_shRNA_2 antisense:
5′-AATTCAAAAATCAGCGTCCCTACCTTCTTCTCTCGAGAGAAGAAGGTAGGGACGCTGAA-3′.
Raptor_shRNA_2 sense:
5′-CCGGAGGGCCCTGCTACTCGCTTTTCTCGAGAAAAGCGAGTAGCAGGGCCCTTTTTTG-3′.
Raptor_shRNA_2 antisense:
5′-AATTCAAAAAAGGGCCCTGCTACTCGCTTTTCTCGAGAAAAGCGAGTAGCAGGGCCC-3′.
Rictor_shRNA_2 sense:
5′-CCGGTACTTGTGAAGAATCGTATCTTCTCGAGAAGATACGATTCTTCACAAGTTTTTTG-3′.
Rictor_shRNA_2 antisense:
5′-AATTCAAAAAACTTGTGAAGAATCGTATCTTCTCGAGAAGATACGATTCTTCACAAGTA-3′.
Preparation of lentiviral vectors was performed as described previously.30
Antibody arrays, immunoprecipitation and western blotting
We used two different commercial arrays, the human phospho-RTK array kit (R&D Systems, Abingdon, UK) and the PathScan RTK Signaling Antibody Array Kit (Cell Signaling). Quantitation of the different RTKs in the human phospho-RTK array kit was performed using the ImageJ 1.44 software (National Institute of Health, Bethesda, MD, USA), and quantitation of the different cell signaling intermediates was performed using the Odyssey V3.0 program (LI-COR, Lincoln, NE, USA). The procedure for immunoprecipitation and western blotting has been previously detailed.43
Cell proliferation, cell cycle and apoptosis assays
Cells were plated in 24-well plates at 10 000–20 000 cells/well and cultured overnight in DMEM or RPMI+10% fetal bovine serum. The next day, medium was replaced with DMEM or RPMI containing the different drugs. Cell proliferation was analyzed by an MTT-based assay as described previously.44 Unless otherwise indicated, the results are presented as the mean±s.d. of quadruplicates of a representative experiment that was repeated at least three times. To determine whether the combinations of BEZ235 with taxotere or carboplatin were synergistic, additive or antagonistic, we used the CalcuSyn v2.0 software program (Biosoft, Ferguson, MO, USA)45 as described previously.46 Results are plotted as the mean±s.d. values of quadruplicates from two experiments.
To analyze the effect of BEZ235 on the different phases of cell cycle, cells were synchronized in G1/S or in mitosis. Mitotic cells were obtained by treatment with nocodazole (275 nM during 14 h).24 The mitotic cells were collected by mechanical shake-off, plated and treated with BEZ235 for the indicated times. To enrich cells in G1/S, cells were treated with thymidine (2 mM during 14 h) as described previously.24 Then, the cells were treated with BEZ235 at different times. DNA content and cell cycle analyses were performed by using a FACScalibur flow cytometer and the CellQuest software (BD Biosciences).
For apoptosis analyses, cells were incubated with trypsin-EDTA, washed twice with cold phosphate-buffered saline and resuspended in binding buffer (10 mM HEPES free acid, pH 7.4, 140 mM NaCl and 2.5 mM CaCl2). Cells were incubated for 15 min in the dark with Annexin V/FITC (BD Biosciences) and propidium iodide staining solution.
In vivo studies
Mice were manipulated at the animal facility following legal guidelines. The Brca1/p53 conditional mice were generously provided by Dr J Jonkers (Division of Molecular Pathology, The Netherlands Cancer Institute, Amsterdam, The Netherlands).25 This model has a somatic insertion of Brca1 and p53 conditional alleles that, when crossed to K14/cre mice, create mice with deletions of these genes in the epithelia, including breast epithelia. The mice developed breast and skin tumors after 5 months. When the tumors were measurable, the mice were randomized and assigned into two groups (with equal average tumor volumes (50 mm3) before initiation of treatments): vehicle (n=5) and BEZ235 (n=4). When the tumors reached a volume of 150 mm3, the mice were treated daily and orally with 30 mg/kg BEZ235 dissolved in 10% N-Methyl-2-pyrrolidone (Sigma-Aldrich) and 90% PEG300. Analyses of diameters and volumes of the tumors were calculated as reported previously.46 For biochemical analyses, tumor samples were obtained after killing of the animals by CO2 inhalation, and 8 h after being treated with BEZ235, and immediately frozen in liquid nitrogen. The tumors were processed as described previously.40
Female BALBc nu/nu mice (7 weeks old) were obtained from Charles River Laboratories (Wilmington, MA, USA). A total of 5 × 106 MDA-MB231 cells in 75 μl of DMEM+10% fetal bovine serum and 75 μl of Matrigel (BD Biosciences) were injected into the mammary fat pad. When tumors reached 200 mm3, animals were randomized into 6 groups (n=5 animals for each condition, each bearing two tumors), and treatments were initiated (BEZ235 (30 mg/kg daily, p.o.), Docetaxel Hospira (15 mg/kg, weekly, intraperitoneally) and carboplatin Teva (20 mg/kg, each 5 days, intraperitoneally)).
Statistical analyses
Comparisons of continuous variables between two groups for the mice tumor model experiments were performed using a two-sided Student’s t-test. At least two independent experiments were performed for the in vivo studies. Differences were considered to be statistically significant when P-values were <0.05. Statistical data are presented as the mean±s.d. All data were analyzed using the statistical software SPSS 19.0 (SPSS Inc., Chicago, IL, USA).
Patient samples and immunohistochemical staining
The TNBC samples of patients were obtained from the University Hospital of Salamanca, and the studies with human samples followed institutional Ethics guidelines. Fresh tissue samples from the primary tumors were embedded in OCT (Tissue-Tek, Aj Alphen aan den Rijn, The Netherlands), and then in isopentane. Samples were placed at −80 °C. Selection of the tumoral tissue was performed after excision of part of the tumor and staining with hematoxylin/eosin. Processing of the frozen samples was performed as described previously.40
Immunohistochemical staining (DAKO HercepTest Kit, Dako, Dako Diagnósticos, S.A. San Just Desvern (Barcelona), Spain) and fluorescence in situ hybridization (FISH; DAKO HER2 FISH pharmDx Kit) to measure amplification of HER2 were performed on paraffin-embedded tissue following international guidelines.47 Scoring criteria include the intensity and pattern of membrane staining, and ranged from 0 to 3, considering positive a score of 3.48 Hormonal receptor status was analyzed by immunohistochemical staining with anti-ER and anti-PR antibodies (Master Diagnóstica), and using the Bond Polymer Refine Detection Kit (Bond Max Autostainer, Leica Microsystems, Buffalo Grove, IL, USA). Staining was scored as described previously.49
References
Cleator S, Heller W, Coombes RC . Triple-negative breast cancer: therapeutic options. Lancet Oncol 2007; 8: 235–244.
Hudis CA, Gianni L . Triple-negative breast cancer: an unmet medical need. Oncologist 2011; 16 (Suppl 1): 1–11.
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res 2004; 10: 5367–5374.
Carey L, Winer E, Viale G, Cameron D, Gianni L . Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 2010; 7: 683–692.
Metzger-Filho O, Tutt A, de Azambuja E, Saini KS, Viale G, Loi S et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol 2012; 30: 1879–1887.
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13: 4429–4434.
Lemmon MA, Schlessinger J . Cell signaling by receptor tyrosine kinases. Cell 2010; 141: 1117–1134.
Hunter T . Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol 2009; 21: 140–146.
Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012; 149: 307–321.
Hochgrafe F, Zhang L, O’Toole SA, Browne BC, Pinese M, Porta Cubas A et al. Tyrosine phosphorylation profiling reveals the signaling network characteristics of basal breast cancer cells. Cancer Res 2010; 70: 9391–9401.
Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 2011; 144: 703–718.
Higgins MJ, Baselga J . Targeted therapies for breast cancer. J Clin Invest 2011; 121: 3797–3803.
Baselga J, Stemmer S, Pego A, Chan A, Goeminne J-C, Graas M-P et al. Cetuximab+cisplatin in estrogen receptor-negative, progesterone receptor-negative, HER2-negative (triple-negative) metastatic breast cancer: results of the randomized phase II BALI-1 Trial. San Antonio Breast Cancer Conference 2010, (abstract PD01-).
Cristofanilli M, Morandi P, Krishnamurthy S, Reuben JM, Lee BN, Francis D et al. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: clinical activity and biological correlations. Ann Oncol 2008; 19: 1713–1719.
Ocana A, Pandiella A, Siu LL, Tannock IF . Preclinical development of molecular-targeted agents for cancer. Nat Rev Clin Oncol 2011; 8: 200–209.
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012; 486: 395–399.
Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW et al. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002; 21: 6255–6263.
Chen Z, Lee FY, Bhalla KN, Wu J . Potent inhibition of platelet-derived growth factor-induced responses in vascular smooth muscle cells by BMS-354825 (dasatinib). Mol Pharmacol 2006; 69: 1527–1533.
Hilger RA, Kredke S, Hedley D, Moeller JG, Bauer RJ, Stellberg W et al. ERK1/2 phosphorylation: a biomarker analysis within a phase I study with the new Raf kinase inhibitor BAY43-9006. Int J Clin Pharmacol Ther 2002; 40: 567–568.
Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 2007; 13: 1576–1583.
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008; 7: 1851–1863.
Laplante M, Sabatini DM . mTOR signaling at a glance. J Cell Sci 2009; 122: 3589–3594.
Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011; 30: 2547–2557.
Diaz-Rodriguez E, Pandiella A . Multisite phosphorylation of Erk5 in mitosis. J Cell Sci 2010; 123: 3146–3156.
Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci USA 2007; 104: 12111–12116.
Chou TC, Talalay P . Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–1043.
Barrios CH, Liu MC, Lee SC, Vanlemmens L, Ferrero JM, Tabei T et al. Phase III randomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer. Breast Cancer Res Treat 2010; 121: 121–131.
Hoeflich KP, O'Brien C, Boyd Z, Cavet G, Guerrero S, Jung K et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res 2009; 15: 4649–4664.
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22: 159–168.
Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov 2011; 1: 248–259.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 2008; 68: 9221–9230.
Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010; 120: 2406–2413.
Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 2010; 16: 205–213.
Montero JC, Chen X, Ocana A, Pandiella A . Predominance of mTORC1 over mTORC2 in the regulation of proliferation of ovarian cancer cells: therapeutic implications. Mol Cancer Ther 2012; 11: 1342–1352.
Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R . The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp Cell Res 2009; 315: 485–497.
McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res 2009; 69: 5835–5842.
Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res 2010; 70: 8097–8107.
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008; 68: 8022–8030.
Montero JC, Seoane S, Ocana A, Pandiella A . P-Rex1 participates in Neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene 2011; 30: 1059–1071.
Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10: 515–527.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM . Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307: 1098–1101.
Diaz-Rodriguez E, Cabrera N, Esparis-Ogando A, Montero JC, Pandiella A . Cleavage of the TrkA neurotrophin receptor by multiple metalloproteases generates signalling-competent truncated forms. Eur J Neurosci 1999; 11: 1421–1430.
Yuste L, Montero JC, Esparis-Ogando A, Pandiella A . Activation of ErbB2 by overexpression or by transmembrane neuregulin results in differential signaling and sensitivity to herceptin. Cancer Res 2005; 65: 6801–6810.
Chou TC, Motzer RJ, Tong Y, Bosl GJ . Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to clinical protocol design. J Natl Cancer Inst 1994; 86: 1517–1524.
Seoane S, Montero JC, Ocana A, Pandiella A . Effect of multikinase inhibitors on caspase-independent cell death and DNA damage in HER2-overexpressing breast cancer cells. J Natl Cancer Inst 2010; 102: 1432–1446.
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 2007; 25: 118–145.
de Alava E, Ocana A, Abad M, Montero JC, Esparis-Ogando A, Rodriguez CA et al. Neuregulin expression modulates clinical response to trastuzumab in patients with metastatic breast cancer. J Clin Oncol 2007; 25: 2656–2663.
Allred DC, Harvey JM, Berardo M, Clark GM . Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol 1998; 11: 155–168.
Acknowledgements
This work was supported by grants from the Ministry of Science and Innovation of Spain (BFU2009–07728/BMC to AP, and PS09/00868 to AE-O). JCM and SS were supported by the Cancer Center Network Program from the ISCIII (RD06/0020/0041). The Cancer Research Institute, and the work carried out at AP and AE-O laboratories, receive support from the European Community through the regional development funding program (FEDER), and from the Fundación Ramón Areces. AO receives research support from the FISCAM (Grant PI2010/017) and from the ISCIII (Grant PS09/02144). We thank Dr J Jonkers (Division of Molecular Pathology, The Netherlands Cancer Institute, Amsterdam, The Netherlands) for the generous gift of the Brca1/p53 conditional mouse model.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Supplementary information
Rights and permissions
About this article
Cite this article
Montero, J., Esparís-Ogando, A., Re-Louhau, M. et al. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene 33, 148–156 (2014). https://doi.org/10.1038/onc.2012.572
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2012.572
Keywords
This article is cited by
-
Altered proTGFα/cleaved TGFα ratios offer new therapeutic strategies in renal carcinoma
Journal of Experimental & Clinical Cancer Research (2021)
-
Ancestry-associated transcriptomic profiles of breast cancer in patients of African, Arab, and European ancestry
npj Breast Cancer (2021)
-
Clinical, genetic and pharmacological data support targeting the MEK5/ERK5 module in lung cancer
npj Precision Oncology (2021)
-
Drug-encapsulated blend of PLGA-PEG microspheres: in vitro and in vivo study of the effects of localized/targeted drug delivery on the treatment of triple-negative breast cancer
Scientific Reports (2020)
-
Identification of a stemness-related gene panel associated with BET inhibition in triple negative breast cancer
Cellular Oncology (2020)
