
Abstract
miR-374a has been reported to function as an oncogene during tumor pathogenesis. In this study, miR-374a is observed to reduce nasopharyngeal carcinoma (NPC) cell proliferation, migration, invasion, metastasis and cisplatin (DDP) resistance in vitro and in vivo. Mechanistic analyses indicate that miR-374a directly targets CCND1 to inactivate pPI3K/pAKT/c-JUN forming a negative feedback loop, as well as suppressing downstream signals related to cell cycle progression and epithelial−mesenchymal transition (EMT). Interestingly, we also observed that miR-374a direct targeting of CCND1 is modulated by tumor suppressor PDCD4 via suppressing pPI3K/pAKT/c-JUN signaling. In clinical specimens, miR-374a was positively and negatively correlated with expression of PDCD4 and CCND1, respectively. Our studies are the first to demonstrate that the miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop induced by PDCD4 supresses NPC cell growth, metastasis and chemotherapy resistance.
Similar content being viewed by others


Introduction
Previous studies have shown that miRNAs have major roles as either oncogenes or tumor suppressors during carcinogenesis.1, 2, 3, 4, 5, 6 Hence, understanding the expression profile and function of individual miRNAs will help to validate their use as diagnostic markers and also provide therapeutic targets for cancer treatment. miR-374a was first reported to be upregulated in primary small cell lung cancer compared to normal lung tissue.7 Further, Cai et al. observed that miR-374a activated WNT/β-catenin signaling by targeting WIF1, PTEN and WNT5A to promote metastasis in breast cancer.8 Silencing of miR-374a has been shown to inhibit cell migration and invasion in gefitinib-resistant NSCLC. In addition, high expression of miR-374a was associated with poor disease-free survival.9 The above mentioned studies suggest that miR-374a functions as an oncogene in certain tumors. Interestingly, other studies have reported conflicting results. In early-stage NSCLC, low expression of miR-374a was associated with poor patient survival.10 In breast cancer, the relative expression of miR-374a was lower in the center of low-invasive and metastatic tumors than that at the edges. In highly invasive and metastatic tumors, it was lower at the edges than that in the center, and lower in cancer tissues than in normal tissues.11 Taken together, the functions and molecular basis of miR-374a are complex and contradictory, thus more studies are needed to confirm its context-specific role.
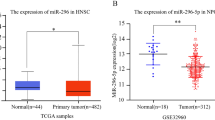
Nasopharyngeal carcinoma (NPC) is an Epstein–Barr virus (EBV)-associated tumor that originates from the nasopharynx and is one of the most common carcinomas in Southern China. It has a highly malignant phenotype and distinct geographical pattern characteristics. In recent studies, abnormal expression of miRNAs was broadly implicated in NPC pathogenesis.12, 13, 14, 15, 16, 17 However, the role of miR-374a in NPC still remains largely unknown.
Programmed cell death 4 ‘PDCD4’ is a classical tumor suppressor deregulated in various tumors including NPC.13, 18, 19 Our previous studies indicated that PDCD4 suppressed cell growth and survival by causing miR-184-mediated direct targeting of c-MYC and BCL-2 via inactivating PI3K/AKT/c-JUN signaling in NPC.13 These findings demonstrated the significance of PDCD4 in NPC pathogenesis. However, a more detailed role of PDCD4 in the suppression of cell growth and metastasis in NPC has not been elucidated.
In this study, we examined the relationship between miR-374a, CCND1 and PDCD4 in NPC and found an atypical miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop modulated by PDCD4. This pathway suppresses cell growth and metastasis, and enhances the chemotherapeutic sensitization of NPC cells to DDP. Together these results provide a novel mechanism for miR-374a as a tumor-suppressive miRNA in NPC.
Results
miR-374a suppresses NPC cell growth in vitro and in vivo
To further explore its biological role in NPC, miR-374a mimics or inhibitors were respectively introduced into EBV-negative SUNE1 and EBV-positive HONE1 NPC cell lines to induce or reduce the expression of miR-374a (Supplementary Figures S1a and b). Compared with negative controls, miR-374a mimics significantly inhibited cell growth from Day 2 in SUNE1 cells. In HONE1 cells, miR-374a mimics suppressed cell growth from Day 3 (Figure 1a). Further, we observed that miR-374a mimics retarded G1/S cell cycle transition (Figure 1b, Supplementary Figure S2). Inversely, knocking down miR-374a with its specific inhibitor significantly restored cell proliferation (Supplementary Figure S3a) and G1/S cell cycle transition (Supplementary Figure S3b). However, miR-374a did not induce cell apoptosis as measured by FACS analysis (Supplementary Figure S4).

miR-374a suppresses NPC cell growth in vitro and in vivo. (a) miR-374a mimics markedly inhibited cell proliferation in SUNE1 and HONE1 cells by MTT assay. (b) miR-374a mimics markedly inhibited G1 to S and G2 cell cycle transition in vitro. (c) Compared with negative control cells, tumorigenicity of miR-374a cells was markedly reduced in vivo. Original magnification, × 200; scale bar, 25 μm. Data are presented as mean±s.d. for three independent experiments (*P<0.05). Ctr, negative control cells; NC, negative control cells.
Subsequently, NPC cell proliferation was measured in vivo by innoculating Lv-miR-374a-GFP SUNE1 and HONE1 cells (Supplementary Figures S5a and b) into nude mice. The average xenograft tumor weight from both lines overexpressing miR-374a was significantly decreased compared with control tumors (Figure 1c). These above results suggest that miR-374a exerts a significant inhibitory effect on NPC cell growth.
miR-374a inhibits NPC cell metastasis in vitro and in vivo
Compared with negative controls, miR-374a reduced cell migration and invasion in Transwell assays (Figure 2a), wound-healing assays (Figure 2b, Supplementary Figure S6) and Boyden assays (Figure 2c). Subsequently, NPC cell metastasis was measured in vivo by innoculating Lv-miR-374a-GFP SUNE1 and HONE1 cells under liver capsule of mice. Notably, fluorescent image detection confirmed increased intra-hepatic dissemination and intestine metastasis established in the control group compared with NPC miR-374a cells (Figure 2d, Supplementary Figures S7a and b). Conversely, suppressing miR-374a with its specific inhibitor elevated cell migration (Supplementary Figure S8a) and invasion (Supplementary Figure S8b).

miR-374a significantly inhibits NPC cell migration, invasion and metastasis in vitro and in vivo. (a) miR-374a mimics suppressed cell migration in SUNE1 and HONE1 cells. Blank: SUNE1 or HONE1 parental cells. Original magnification, × 200; scale bar, 25 μm. (b) Wound-healing assay showing migration of SUNE1 and HONE1 cells transfected with miR-374a or negative control at 0 and 24 h. Bar graph shows size of wound at 0 and 24 h. Original magnification, × 100; scale bar, 100 μm. (c) miR-374a mimics suppressed cell invasion of SUNE1 and HONE1 cells. Blank means SUNE1 or HONE1 parental cells. Original magnification, × 200; scale bar, 25 μm. (d) SUNE1 and HONE1 cells with stable miR-374a expression and their respective control cells were inoculated under the liver capsules of nude mice. Fluorescent images are shown for four representative mouse models. Intra-hepatic dissemination and intestine metastasis predominantly appeared in the control mice compared with miR-374a cell-injected mouse models. Data are presented as mean±s.d. for three independent experiments (*P<0.05). Ctr, negative control cells.
miR-374a enhances DDP chemosensitivity in vitro and in vivo
Stably overexpressing miR-374a significantly enhanced NPC cell chemosensitivity to DDP. Inhibition rates after 48 h treatment with different concentrations of DDP were calculated before and after miR-374a transfection (Figure 3a). The IC50 of DDP in SUNE1 cells was reduced from 12.4 to 4.1 μM after miR-374a transfection. A similar IC50 reduction from 12.2 to 6.3 μm occurred in HONE1 cells. In vivo efficacy of DDP was evaluated in mice bearing abdominal tumors originating from miR-374a-overexpressing cells or their controls and a fitted curve was generated to determine the weight of each group (Figure 3b). Kaplan-Meier analysis was then used to examine effects on survival. Survival time of NC+DDP and miR-374a+NS groups was much longer than that of the untreated NC+NS group, but still shorter than miR-374a+DDP-treated group (Figure 3c) (P<0.001). There was no significant difference between the NC+DDP or miR-374a+NS groups. The average survival of mice in NC+NS, miR-374a+NS, NC+DDP and miR-374a+DDP groups were 28.3, 34.8, 35.2 and 47.0 days, respectively.

miR-374a enhances cisplatin chemosensitivity in vitro and in vivo. (a) miR-374a significantly elevated NPC cell sensitivity to DDP. (b) After 30 days of DDP or NS treatment, a fitted curve was generated for the bodyweight of each group. (c) Animals were divided into four groups: control cell group (NC)+Normal saline (NS), miR-374+NS, NC+DDP and miR-374+DDP (each group: N=10). Overall survival time ranging from low to high, was as follows: NC+NS<miR-374a+NS<NC+DDP<miR-374+DDP.
miR-374a suppresses PI3K/AKT, cell cycle and epithelial−mesenchymal transition (EMT) signals
To determine the molecular mechanism by which miR-374a functions as a tumor suppressor relevant to NPC, it was overexpressed in SUNE1 and HONE1 cells. We observed that miR-374a not only suppressed the levels of pPI3K (Tyr458), pAKT (Ser473) and c-JUN (Figure 4), but also significantly decreased expression of cell cycle-related genes including CCND1, c-MYC, E2F1 and pRB (Ser780) (Figure 4). Furthermore, mesenchymal-related genes including Snail and N-cadherin were also decreased concomitant with increased expression of epithelial-marker E-cadherin (Figure 4). However, levels of β-catenin (Figure 4), total PI3K and AKT (Supplementary Figure S7) were unchanged. Similarly, mitochondrial apoptosis-related proteins PDCD4, cleaved-CASP9 and cleaved-PARP (Supplementary Figure S9) were unaffected by ectopic miR-374a. Taken together, these data suggest that miR-374a suppresses PI3K/AKT, cell cycle signaling and EMT, but does not regulate apoptosis in NPC.

miR-374a modulates expression of cell cycle and EMT genes via inactivating PI3K/AKT pathway. miR-374a mimics suppressed the expression of pPI3K (Tyr458), pAKT (Ser473) and c-JUN, as well as cell cycle-related genes including c-MYC, CCND1, pRB (Ser780) and E2F1. Mesenchymal genes including Snail and N-cadherin were also reduced while E-cadherin expression was increased. No expression changes in β-catenin were observed in either SUNE1 or HONE1 NPC cells. β-actin served as a loading control.
CCND1 is a direct target of miR-374a which reverses suppression of cell growth, metastasis and DDP sensitivity
Through miRwalk20, 21 and RNAhybrid algorithms, CCND1 and c-MYC were predicted to be direct targets of miR-374a (Supplementary Figures S10a and b). Overexpressing or suppressing miR-374a respectively downregulated or elevated the protein expression of both CCND1 and c-MYC in SUNE1 and HONE1 cells (Figure 4 and Figure 5a). Luciferase reporter assays were used to determine whether miR-374a could directly target the 3′UTR (untranslated region) of CCND1 and c-MYC. Cotransfection miR-374a mimics with CCND1 3′UTR wt vector significantly decreased luciferase reporter activity (Figure 5b, lanes 2 and 3; P<0.001) while miR-374a inhibitor had the opposite effect (Figure 5b, lanes 4 and 5; P<0.001). These effects on luciferase activity were abrogated when cotransfected with mt vector (Figure 5b, lanes 7 and 8, P=0.7937; lanes 9 and 10, P=0.3804). However, no significant change in luciferase activity for c-MYC was observed when using miR-374a mimics compared with miR control (Figure 5c). Taken together, these results demonstrated that CCND1, but not c-MYC, is a direct target of miR-374a in NPC cells. Further, transiently transfecting CCND1 into miR-374a overexpressing NPC cells (Supplementary Figure S11a) not only significantly reduced miR-374a expression (Supplementary Figure S11b), but also enhanced cell growth (Supplementary Figure S11c), migration (Supplementary Figure S11d) and invasion (Supplementary Figure S11e), as well as reversed sensitizing effects to DDP (Supplementary Figure S11f). Moreover, miR-374a mimics or its specific inhibitor respectively suppressed or restored cell growth (Supplementary Figure S12a), migration (Supplementary Figure S12b) and invasion (Supplementary Figure S12c) through upregulating CCND1 (Supplementary Figure S12d). These indicate that CCND1 can overcome the suppression of cell growth, metastasis and DDP chemoresistance induced by miR-374a.

miR-374a directly targets CCND1, but not c-MYC. (a) CCND1 and c-MYC expression were elevated after SUNE1 and HONE1 cells transfected with 100 nM miR-374a inhibitor. β-actin served as a loading control. (b) SUNE1 cells were cotransfected with miR-374a mimics, inhibitor or their corresponding control vectors and luciferase reporters carrying either the predicted miRNA target site in CCND1 3’UTR (wt) or its corresponding mutant (mt), or only transfected with wt or mt. (c) Luciferase reporter assays confirmed that miR-374a did not directly target c-MYC. Data are presented as mean±s.d. for three independent experiments (*P<0.05).
Suppression of CCND1 inhibits PI3K/AKT, cell cycle signaling and EMT as well as induces miR-374a
Subsequently, we found that expression of c-MYC, pPI3K (Tyr458), pAKT(Ser473), c-JUN, pRB (Ser780), E2F1, N-cadherin and Snail were significantly downregulated while E-cadherin was markedly increased (Supplementary Figure S13a) after CCND1 knockdown. This effect was consistent with changes observed after miR-374a overexpression. No alterations in β-catenin, PI3K and AKT expression were observed (Supplementary Figure S13a). Further, siRNA-mediated suppression of c-MYC downregulated expression of pPI3K (Tyr458) and pAKT (Ser473) in NPC cells (Supplementary Figure S13b). Total levels of PI3K and AKT (Supplementary Figure S13b) were unchanged. Finally, suppression of CCND1 induced miR-374a expression in NPC cells (Supplementary Figure S13c). These data suggest that CCND1 activates PI3K/AKT signaling through c-MYC through yet undetermined mechanisms.
PI3K/AKT pathway modulates miR-374a-CCND1 expression by modulating c-JUN
Next, we wondered whether PI3K/AKT signaling could regulate the expression of miR-374a. Interestingly, bioinformatics predictions indicated that the promoter region of human miR-374a contains three AP-1 (c-JUN) binding sites, thus we speculated that the PI3K/AKT pathway reduced miR-374a expression by modulating c-JUN. Inactivating PI3K phosphorylation reduced the expression of pAKT (Ser473) and c-JUN (Figure 6a) as well as stimulated the expression of miR-374a (Figure 6b). Further, knocking down c-JUN (Figure 6c) induced miR-374a expression (Figure 6d), which suggests that c-JUN is an upstream regulator of miR-374a. Subsequently, we used chromatin immunoprecipitation combined with qPCR analysis to determine whether c-JUN could bind the putative miR-374a promoter and reduce its expression (Figure 6e). A reduction of wild-type miR-374a promoter luciferase activity was observed upon upregulation of c-JUN in SUNE1 cells (P<0.05), and a similar effect was observed when c-JUN binding sites were mutated alone or in combination (P<0.05) (Figure 6f). Finally, suppression of c-JUN reduced the expression of CCND1 in NPC cells (Figure 6c).

PI3K/AKT pathway suppresses expression of miR-374a by modulating c-JUN. (a) Suppressing PI3K by its specific inhibitor Ly294002 (50 nM) reduced expression of pPI3K (Tyr458), pAKT (Ser473) and c-JUN in SUNE1 and HONE1 cells, but did not affect total PI3K or AKT levels. β-actin served as a loading control. (b) Ly294002 (50 nM) enhanced expression of miR-374a in SUNE1 and HONE1 cells. (c) Expression of c-JUN and CCND1 was examined in c-JUN siRNA-treated SUNE1 and HONE1 cells by western blot. β-actin served as a loading control. (d) Knocking down c-JUN by siRNA stimulated the expression of miR-374a in SUNE1 and HONE1 cells. (e) Cross-linked chromatin from mock and pcDNA3.1-c-JUN-transfected SUNE1 cells was immunoprecipitated with anti-c-JUN antibody or normal rabbit IgG. AP-1 binding sites (labeled as P1, P2 and P3) on the immunoprecipitated DNA were determined by quantitative RT-PCR. Amplification of input chromatin (input) prior to immunoprecipitation served as a positive control for chromatin extraction and PCR amplification. Chromatin immunoprecipitation using a non-specific antibody (normal human IgG) served as a negative control. (f) Relative luciferase activity of the indicated promoter vectors in SUNE1 cells transfected with c-JUN plasmids. Data are presented as mean±s.d. for three independent experiments (*P<0.05).
PDCD4 inhibits NPC cell migration and invasion by mediating EMT signal
Our previous study demonstrated that PDCD4 blocks cell proliferation, cell cycle transition through the PI3K/AKT pathway, as well as induces NPC cell apoptosis.13 In this study, we confirmed that overexpression of PDCD4 notably reduced cell migration (Supplementary Figure S14a) and invasion (Supplementary Figure S14b) in 5−8 F cells. The opposite results were observed in SUNE1 and HONE1 cells with stable suppression of PDCD4 (Supplementary Figures S14c and d). These results suggest that PDCD4 exerts a significant inhibitory effect on metastasis.
Furthermore, PDCD4 overexpression reduced expression of EMT-related genes Snail and N-cadherin (Supplementary Figure S14e), as well as increased E-cadherin. The reverse effects were observed in SUNE1 and HONE1 cells with stable PDCD4 suppression (Supplementary Figure S14f). No alterations in β-catenin were observed. These results indicate that PDCD4 inhibits cell migration and invasion through EMT signaling.
miR-374a participates in PDCD4-mediated suppression of cell growth and metastasis
In our previous study, we investigated the effects of PDCD4 on miRNAomics in NPC.13 In this study we reanalyzed this data and interestingly found that miR-374a was markedly downregulated in shPDCD4 NPC cells (Supplementary Figure S15a). This finding was confirmed by real-time PCR after PDCD4 knockdown as well as the opposite effect after PDCD4 overexpression (Supplementary Figures S15b and c). We previously noted that PDCD4 regulated the PI3K/AKT pathway and inactivated c-JUN.13 Here we observed that c-JUN directly suppresses miR-374a expression. c-JUN binding to the miR-374a promoter was notably reduced in PDCD4-overexpressing 5−8 F NPC cells by a ChIP assay combined with qPCR analysis (Supplementary Figure S16). Furthermore, expression of miR-374a was upregulated in PDCD4-suppressed NPC cells after Ly294002 treatment (Supplementary Figure S17). Suppression of miR-374a partially restored CCND1 and pPI3K levels, while total PI3K or PDCD4 were not changed after c-JUN knockdown (Supplementary Figure S18). These data indicate that PDCD4 induces miR-374a expression through the PI3K/AKT/c-JUN pathway. Transiently transfecting miR-374a mimics into PDCD4-silenced NPC cells inhibited cell proliferation (Supplementary Figure S19a), migration (Supplementary Figure S19b) and invasion (Supplementary Figure S19c). These data indicate that miR-374a can overcome the effects on cell growth and metastasis induced by PDCD4 suppression.
The correlation between miR-374a, PDCD4 and CCND1 mRNA levels
We next measured the mRNA levels of miR-374a, PDCD4 and CCND1 in NPC specimens and nasopharyngeal tissues. Expression levels of miR-374a (Figure 7a,1) and PDCD4 (Figure 7a,3) were decreased in NPC specimens compared to NP tissues while CCND1 was significantly increased in cancer tissues (Figure 7a,2). As a result, miR-374a expression was negatively correlated with CCND1 expression (Figure 7b,1) (Supplementary Table S1) and positively associated with PDCD4 expression (Figure 7b,2) (Supplementary Table S2). In addition, CCND1 expression was negatively correlated with PDCD4 expression (Figure 7b,3) (Supplementary Table S3).

CCND1 and PDCD4 correlation with miR-374a expression and NPC prognosis. (a) Decreased expression of miR-374a (a1) and PDCD4 (a3) and increased expression of CCND1 (a2) in NPC specimens compared to nasopharyngeal tissues. (b) miR-374a expression was negatively correlated with expression of CCND1 mRNA (b1) and positively correlated with PDCD4 mRNA expression (b2) in NPC tissues. Furthermore, CCND1 mRNA expression was negatively correlated with PDCD4 mRNA expression (b3). (c) Representative cases from 149 primary NPC specimens were analyzed by immunohistochemical staining. Original magnification, × 400; scale bar, 25 μm. (d) Survival analysis showed cumulative overall survival time ranging from low to high, was as follows: high CCND1+low PDCD4<low CCND1+low PDCD4<high CCND1+high PDCD4<low CCND1+high PDCD4.
CCND1 expression is negatively correlated with PDCD4 expression, and predicts poor NPC patient prognosis
PDCD4 protein expression was examined in NPC tissues by immunohistochemistry (IHC) in our previous study.13 In this study, CCND1 exhibited nuclear and cytoplasmic expression patterns while statistical analysis confirmed it was negatively correlated with PDCD4 levels (Figure 7c). Survival analysis ranked cumulative overall survival time as: high CCND1+low PDCD4<low CCND1+low PDCD4<high CCND1+high PDCD4<low CCND1+high PDCD4 in NPC. Thus, high expression of CCND1 together with low PDCD4 expression was associated with poorer prognoses for NPC patients. Inversely, patients with low CCND1 and high PDCD4 expression had the longest survival times (P<0.001). Low PDCD4+ low CCND1 patients also had better survival than those with low PDCD4+high CCND1 (P=0.018) (Figure 7d).
Disscussion
miR-374a has been identified as an oncogene in breast cancer progression, yet its function in lung cancer pathogenesis is unclear.7, 8, 9, 10, 11 It is likely that miR-374a serves a complex role in carcinogenesis that is context dependent.
In this study, we observed that miR-374a repressed cell cycle progression, cell growth, migration, invasion and metastasis in vitro. These effects were also observed in vivo, but interestingly miR-374a did not affect apoptosis. We also found that miR-374a overexpression elevated NPC cell sensitivity to DDP and significantly increased survival time of tumor-bearing nude mice. Our data indicate that miR-374a acts as a tumor suppressor in NPC, different from Cai and Wang’s reports of miR-374a functioning as an oncogene in breast cancer and lung cancer.
It is well known that cell cycle transition and EMT are two key downstream effects of the PI3K/AKT pathway activation which lead to cell growth, metastasis and chemotherapy resistance.22, 23, 24, 25, 26, 27, 28, 29 In this study, miR-374a was observed to form a negative feedback loop via key oncogenic genes including CCND1, PI3K/AKT and c-JUN. This signaling cascade suppressed cell cycle transition and EMT thus reducing cell growth and metastasis as well as sensitizing NPC cells to DDP. These findings are different than a previous report of activation through the Wnt/b-catenin pathway.8
CCND1 is a classical oncogene which induces G1 to S cell cycle transition promoting cancer cell growth.30, 31 Recent studies have suggested a dual role of cyclin D1 in promoting or inhibiting cellular migration in breast cancer32 and prostate cancer.33 In our study, CCND1 was observed to be a direct target of miR-374a and reversed miR-374a-mediated suppression of cell growth, metastasis and DDP chemoresistance. This was accomplished by activation of PI3K/AKT pathway via c-MYC as well as downstream cell cycle and EMT signals.34, 35, 36 The detailed mechanisms of c-MYC activating pPI3K/AKT in NPC remain unclear. As a downstream target of pPI3K/AKT signaling, c-JUN was also observed to be downregulated after CCND1 knockdown in NPC cells, an effect consistent with miR-374a overexpression. Interestingly, we also observed increased or decreased expression of miR-374a in CCND1-suppressed NPC cells or CCND1-overexpressed miR-374a cells, respectively. These results support that CCND1 is a direct target of miR-374a which activates the PI3K/AKT/c-JUN pathway. This ultimately induces feedback signaling to suppress miR-374a expression in NPC.
Analysis of a region upstream to the miR-374a locus revealed multiple putative binding sites for c-JUN, a downstream regulator of PI3K/AKT signaling.12, 13 We observed that c-JUN suppressed miR-374a expression by directly binding to its promoter and further increasing CCND1 expression. Suppression of pPI3K/AKT-c-JUN signaling by a specific pPI3K inhibitor induced miR-374a expression. Taken together, these results suggest that the miR-374a can induce its own expression through CCND1-pPI3K/AKT-c-JUN feedback, which may contribute to NPC pathogenesis.
PDCD4 is a tumor suppressor predominantly expressed in the cytoplasm where it plays a significant role in inducing cell cycle arrest by blocking PI3K/AKT/c-JUN and downstream cell cycle factors CCND1 and c-MYC.13 In this study, PDCD4 repressed cell migration and invasion by modulating EMT pathways downstream of PI3K/AKT,34, 35 including suppressing Snail and N-cadherin while inducing E-cadherin expression. Taken together, PDCD4 suppresses NPC cell growth and metastasis by blocking PI3K/AKT-mediated cell cycle and EMT signaling, a role similar to that of miR-374a.
We observed that miR-374a was induced by PDCD4 after reanalyzing our miRNAomics data from a previous study.13 We confirmed that miR-374a as a downstream positive modulator of PDCD4 in NPC, though the precise molecular mechanisms of this interaction are still unclear.
In our previous investigation, PI3K/AKT/c-JUN mediated induction of CCND1 was suppressed by PDCD4.12, 13 In this study, the PI3K/AKT/c-JUN-miR-374a-CCND1 negative feedback loop suppressed cell growth and metastasis in NPC. We suspected that miR-374a is induced by PDCD4 by inactivating PI3K/AKT/c-JUN signaling. Indeed, miR-374a was significantly upregulated in PDCD4-suppressed NPC cells treated with a PI3K inhibitor. Further, suppression of miR-374a partially restored CCND1 and pPI3K levels, while total PI3K and PDCD4 were not significantly changed after c-JUN knockdown. In addition, PDCD4 suppressed the binding of c-JUN to the miR-374a promoter. miR-374a ovexpression restored inhibitory effects on cell growth, migration and invasion mediated by PDCD4 knockdown. Taken together, the miR-374a-CCND1-PI3K/AKT/c-JUN feedback loop is modulated by PDCD4 and participates in NPC pathogenesis.
Consistent with their known roles in NPC, we observed reduced expression of miR-374a and PDCD4 but elevated CCND1 mRNA expression in clinical NPC tissues. miR-374a was positively correlated with PDCD4 mRNA expression yet negatively correlated with CCND1. CCND1 was also observed to be negatively correlated with PDCD4 mRNA expression. These relationships were also consistent at the protein level. NPC patients with CCND1 overexpression and reduced PDCD4 protein levels had the shortest overall survival times while those with increased PDCD4 together and decreased CCND1 expression had the longest overall survival times. Our study underscores the significance of the PDCD4-miR-374a-CCND1 axis in mediating NPC pathogenesis.
As summarized in Figure 8, the miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop stimulated by PDCD4 suppresses cell growth and metastasis, and enhances NPC sensitization to DDP. This finding is consistent with the increasing appreciation that microRNAs form complex regulatory networks whose perturbation can underlie numerous pathologies.37 Therefore, the cellular context is important for understanding the specific role of microRNAs. It is tempting to speculate that once induced by PDCD4, the miR-374a-mediated feedback loop allows NPC cells to become less autonomous. This would reduce NPC cell proliferation and metastasis, and would explain the robust inhibitory effect on cell growth and metastasis we have observed.

The miR-374a-CCND1-pPI3K/AKT-c-JUN negative feedback loop induced by PDCD4 suppresses cell growth and metastasis and sensitizes NPC cells to DDP by blocking cell cycle and EMT signals.
Materials and methods
Cell culture
Two NPC cell lines (SUNE1 and 5−8 F) were obtained from Cancer Center of Sun Yat-sen University, Guangzhou, China. EBV-positive NPC cell line HONE1 was kindly provided by Professor George SW Tsao from the University of Hong Kong.15 shPDCD4-SUNE1, shPDCD4-HONE1 and PDCD4-overexpressed 5−8 F cells were maintained by Cancer Research Institute of Southern Medical University.13 All cell lines were confirmed to be free of mycoplasma contamination and cultured in RPMI-1640 with 10% fetal bovine serum in a humidified chamber with 5% CO2 at 37 °C.
Collection of primary NPC and non-cancerous nasopharynx specimens and Ethics Statement
Samples analyzed included 70 fresh NPC tissues, 20 fresh nasopharynx tissues and 149 paraffin-embedded NPC tissues obtained at the time of diagnosis before any therapy from People’s Hospital in Zhongshan City (Guangdong, China). Clinical processes were approved from the Ethics Committees of People’s Hospital of Zhongshan City. Patients provided informed consent. The pathologic stage of all specimens was confirmed according to the 1997 NPC staging system of the WHO.
RNA isolation, reverse transcription and qRT-PCR
RNA isolation, reverse transcription and qRT-PCR were performed in NPC cell lines, tissues and normal nasopharynx tissues according to a previous description.13 Specific sense primers for miR-374a, CCND1 and PDCD4 are shown in Supplementary Tables S4.
Immunohistochemistry
Immunohistochemistry of CCND1 (Santa Cruz Biotechnology, Santa Cruz, USA) was performed in NPC tissues according to a previous description.6, 12, 13, 14, 15, 34 Stained tissue sections were evaluated separately by two pathologists blinded to the clinical parameters. For cytoplasmic staining, scoring was assessed based on the sum of cytoplasm staining intensity and the percentage of positive staining areas of cells. The staining intensity was scored as 0 (negative), 1 (weak), 2 (medium) and 3 (strong) and the percentage of positive staining areas of cells was defined as a scale of 0 to 3, where 0 represents <10%, 1 was 10–25%, 2 was 26–75% and 3 was ⩾76%. For nuclear staining, the score was defined according to the sum of nuclear staining intensity and numbers of cells. Nuclear staining intensity score was consistent with cytoplasm and positive nuclear staining scores were defined as follows: 0 represents <10%, 1 was 10–50%, 2 was 51–80% and 3 was ⩾80%. The sum of the cytoplasm and nuclear scores was used as the final CCND1 staining score (0−12). For statistical analysis, scores of 0−6 or 7−12 was respectively considered to be low or high expression.
Establishment of NPC cell lines with stable expression of miR-374a
Lentiviral (GV209, H1-MCS-CMV-EGFP) particles carrying miR-374a precursor and its flanking control sequence were constructed by GeneChem, Shanghai, China. SUNE1 and EBV-positive HONE1 NPC cells were infected with recombinant lentiviral particles according to the manufacturer’s protocol. The resulting cells were seeded onto 96-well plates and cultured for 3 weeks to produce a stable miR-374a-overexpressing SUNE1 and HONE1 cells. Overexpression of miR-374a was validated by quantitative RT-PCR.
Transient transfection with siRNA, miR-374a mimics/inhibitor, CCND1 or c-JUN plasmids and PI3K inhibitor Ly294002 treatment
Small-interfering RNA (siRNA) for c-JUN, CCND1 and c-MYC (Supplementary Table S5) (RiboBio Inc, Guangzhou, China), as well as miR-374a mimics and inhibitor were designed and synthesized (Supplementary Table S5) at GenePharma Co., Ltd (Shanghai, China). PI3K inhibitor Ly294002 was purchased from Sigma Inc, USA. CCND1 and c-JUN plasmid was purchased in Biosense Technologies (Guangzhou, China). Twenty-four hours prior to transfection, SUNE1 and HONE1 cells were plated onto a 6-well plate or a 96-well plate (Nest, Biotech,China) at 30–50% confluence. siRNA or miRNAs were then transfected at a working concentration of 100 nM using TurboFectTM siRNA Transfection Reagent (Fermentas, Vilnius, Lithuania) according to the manufacturer’s protocol. Cells were collected after 48–72 h for further experiments.
Cell proliferation analysis
Cell proliferation was analyzed using MTT assay according to a previous description.13 Experiments were performed three times.
Cell cycle analysis
Cell cycle analysis was performed according to a previous description.13 Each experiment was performed in triplicate.
In vitro cell migration and invasion assays
In vitro cell migration and invasion assays were performed according to a previous description.34 All assays were independently repeated at least three times.
Wound-healing assay
For the wound-healing assay, cells were grown to confluence in a six-well plate. Artificial wound tracks were created by scraping confluent cell monolayers with a pipette tip. The cells were fed with serum-free medium. The ability of the cells to migrate into the wound area was assessed at 0 and 24 h after scratching.
Apoptosis assay
Apoptosis assay was performed according to a previous description.13 Each experiment was performed in triplicate.
In vivo tumorigenesis and metastasis assays in nude mice
For in vivo tumorigenesis assays, a total of 1 × 106 logarithmically growing miR-374a-overexpressing SUNE1 and HONE1 cells or control parental cells in 0.1 ml RPMI 1640 medium were respectively subcutaneously injected into the left or right flank of 4- to 6-week-old 12-13 g male BALB/c nu/nu mice (N=5). After 18 days, mice were killed and tumor tissues were excised and weighed. For in vivo metastasis assays, 50 μl of SUNE1 and HONE1 cells (5 × 106) stably expressing GFP/miR-374a or an equal number of their respective control cells were injected under the liver capsule of each mouse (5 mice for each group). All mice were killed 3 weeks later. Liver and intestines were subjected to fluorescent image detection using LT-9MACIMSYSPLUS whole-body imaging system (Lighttools Research, Encintas, CA, USA). Mice were maintained in a barrier facility on HEPA-filtered racks and fed with an autoclaved laboratory rodent diet. All animal studies were conducted in accordance with the principles and procedures outlined in Southern Medical University Guide for the Care and Use of Animals.
Western blot analysis
Western blot analysis was performed according to a previous description.13 Antibodies included anti-PDCD4 (9535S), E-cadherin (3195P), N-cadherin (4061P), β-catenin (8480P), Snail (3879P), pRB (Ser780, 8180S), c-MYC (9402S), c-JUN (9165S), cleaved-Caspase 9 (7237S), PARP (5625S), PI3K (4249S), AKT (4691S), pAKT (Ser473, 4060S), pPI3K (Tyr458, 4228S) (1:1000; Cell Signaling Technology), β-actin (sc-8432), E2F1 (sc-193) and CCND1 (sc-753) antibody (1:400; Santa Cruz Biotechnology), β-Tublin antibody (CW0098A, 1:400; ComWin Biotechnology, Beijing, China). HRP-conjugated anti-rabbit and anti-mouse IgG antibody were used as secondary antibodies (Zhongshan, Beijing, China). Signals were detected using enhanced chemiluminescence reagents (Pierce, Rockford, IL, USA).
miRNA array for sh-PDCD4
miRNA array was used to examine the differential expression of miRNAs in knocking down PDCD4 NPC cells.13 Experiments were repeated four times and data were analyzed using and BRB ArrayToolsV3.7.2 software (National Cancer Institute, Bethesda, MD, USA).
Luciferase reporter assay
CCND1 and c-MYC were predicted to be directly regulated targets of miR-374a by RNAhybrid and miRwalk software. A 452-bp or 460-bp fragment of CCND1 and c-MYC 3′UTR amplified by PCR primers (Supplementary Table S4) was cloned into psiCHECK-2 vectors (named wild-type (wt)). Site-directed mutagenesis of the miR-374a binding site in CCND1 3’UTR was performed using GeneTailor Site-Directed Mutagenesis System (Invitrogen) (named mutant (mt)). For reporter assays, SUNE1 cells were cotransfected with miR-374a mimics, inhibitor or their corresponding negative control, and wt vector or mt vector, or only transfected with wt or mt. Luciferase activity was measured 48 h after transfection using the Dual-Luciferase Reporter Assay System (Promega).
To generate a miR-374a promoter vector, a 1888-bp fragment containing the 3 binding sites of c-JUN (labeled as P1, P2 and P3) was PCR-amplified and inserted into a psiCHECK-2 luciferase reporter vector. In addition, vectors with mutated c-JUN-binding sites were constructed. These psiCHECK-2-derived and c-JUN-expressing vectors were cotransfected into SUNE1 cells using Lipofectamine 2000 Reagent (Invitrogen). Luciferase activities were assayed as the aforementioned methods. The primer sequences used for PCR amplification of plasmid construction are listed in Supplementary Table S4.
Chromatin immunoprecipitation assay
DNA–protein complexes were immunoprecipitated from SUNE1 and PDCD4-overexpressing 5−8 F cells by using the Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA, USA) according to the manufacturer’s protocol with the following polyclonal antibodies: anti-c-JUN and normal mouse IgG (Millipore), with the latter serving as a control for nonspecific DNA binding. Precipitated DNA was subjected to qPCR analysis using specific primers. Primers (Supplementary Table S4) were utilized to amplify across the miR-374a promoter region.
MTT cytotoxicity assay
Cis-diamminedichloroplatinum (cisplatin, DDP) (Qilu Pharmo Co. Ltd, Jinan,China) was resuspended in PBS (0.5 mg/ml) and stored at −20 °C. Drug sensitivity was determined by MTT assay. Cells were seeded in 96-well plates in 100 μl RPMI-1640 medium supplemented with 10% FBS at 5 × 103 cells/well. Once attached, cells were treated with 2.5, 5, 10, 20 or 40 μM Cisplatin (0.5 mg/ml) and incubated at 37 °C in 5% CO2 for 48 h. Assays were then performed according to a previous description13 three times.
Treatment experiments in nude mice
In vivo experiments were approved by the Animal Care and Use Committee of Southern Medical University. Forty 4-week old male mice weighing 12−13 g were provided by the Central Animal Facility of Southern Medical University. To establish an NPC mouse model, 6 × 105 miR-374a-overexpressing SUNE1 cells (N=20) or control cells (N=20) were intraperitoneal injected in 0.2 ml buffered saline into nude mice (BALB/C, nu/nu, 4weeks). Tumors were allowed to grow for 3 days and then the animals were randomly divided into four groups: control cell group (NC)+Normal saline (NS), miR-374+NS, NC+DDP or miR-374+DDP (N=10 for each group). Mice were intraperitoneal injected with NS or DDP every 3 days. Bodyweight and survival times were measured regularly.
Statistical analysis
SPSS 13.0 was used for statistical analysis. No statistical methods were used to predetermine sample size, but our sample sizes are similar to those reported in previous publications.6, 12, 13, 14, 15, 34 The Pearson test was applied to examine the relationship among miR-374a, PDCD4 and CCND1 mRNA expression. Spearman test was utilized to evaluate the relationship between PDCD4 and CCND1 protein expression. Survival analysis was performed using the Kaplan-Meier method. Two-tailed Student’s t-test was used for comparisons of two independent groups. One-way analysis of variance was used to determine the differences between groups for all in vitro analyses. A P value of less than 0.05 was considered statistically significant. Data met the assumptions of the statistical tests used, and equal variances were formally tested. The criteria for excluding data points were established before data collection. An outlier was defined as a value outside the mean±3 s.d.
A Supplementary Methods checklist is available.
References
Lin KT, Yeh YM, Chuang CM, Yang SY, Chang JW, Sun SP et al. Glucocorticoids mediate induction of microRNA-708 to suppress ovarian cancer metastasis through targeting Rap1B. Nat Commun 2015; 6: 5917.
Bu P, Wang L, Chen KY, Rakhilin N, Sun J, Closa A et al. miR-1269 promotes metastasis and forms a positive feedback loop with TGF-β. Nat Commun 2015; 6: 6879.
Zhang P, Wang L, Rodriguez-Aguayo C, Yuan Y, Debeb BG, Chen D et al. miR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat Commun 2014; 5: 5671.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 2014; 5: 5241.
Keklikoglou I, Hosaka K, Bender C, Bott A, Koerner C, Mitra D et al. MicroRNA-206 functions as a pleiotropic modulator of cell proliferation, invasion and lymphangiogenesis in pancreatic adenocarcinoma by targeting ANXA2 and KRAS genes. Oncogene 2015; 34: 4867–4878.
Que T, Song Y, Liu Z, Zheng S, Long H, Li Z et al. Decreased miRNA-637 is an unfavorable prognosis marker and promotes glioma cell growth, migration and invasion via direct targeting AKT1. Oncogene 2015; 34: 4952–4963.
Miko E, Czimmerer Z, Csanky E, Boros G, Buslig J, Dezso B et al. Differentially expressed microRNAs in small cell lung cancer. Exp Lung Res 2009; 35: 646–664.
Cai J, Guan H, Fang L, Yang Y, Zhu X, Yuan J et al. MicroRNA-374a activates Wnt/beta-catenin signaling to promote breast cancer metastasis. J Clin Invest 2013; 123: 566–579.
Wang Y, Xia H, Zhuang Z, Miao L, Chen X, Cai H . Axl-altered microRNAs regulate tumorigenicity and gefitinib resistance in lung cancer. Cell Death Dis 2014; 5: e1227.
Võsa U, Vooder T, Kolde R, Fischer K, Välk K, Tõnisson N et al. Identification of miR-374a as a prognostic marker for survival in patients with early-stage nonsmall cell lung cancer. Genes Chromosomes Cancer 2011; 50: 812–822.
Li JY, Zhang Y, Zhang WH, Jia S, Kang Y, Tian R . Effects of differential distribution of microvessel density, possibly regulated by miR-374a on breast cancer prognosis. Asian Pac J Cancer Prev 2013; 14: 1715–1720.
Yu X, Zhen Y, Yang H, Wang H, Zhou Y, Wang E et al. Loss of connective tissue growth factor as an unfavorable prognosis factor activates miR-18b by PI3K/AKT/C-JUN and C-Myc and promotes cell growth in nasopharyngeal carcinoma. Cell Death Dis 2013; 4: e634.
Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S et al. Tumor suppressor PDCD4 modulates miR-184-mediated direct suppression of C-MYC and BCL2 blocking cell growth and survival in nasopharyngeal carcinoma. Cell Death Dis 2013; 4: e872.
Wu A, Wu B, Guo J, Luo W, Wu D, Yang H et al. Elevated expression of CDK4 in lung cancer. J Transl Med 2011; 9: 38.
Cai LM, Lyu XM, Luo WR, Cui XF, Ye YF, Yuan CC et al. EBV-miR-BART7-3p promotes the EMT and metastasis of nasopharyngeal carcinoma cells by suppressing the tumor suppressor PTEN. Oncogene 2015; 34: 2156–2166.
Lu J, He ML, Wang L, Chen Y, Liu X, Dong Q et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res 2011; 71: 225–233.
Zhao M, Luo R, Liu Y, Gao L, Fu Z, Fu Q et al. miR-3188 regulates nasopharyngeal carcinoma proliferation and chemosensitivity through a FOXO1-modulated positive feedback loop with mTOR-p-PI3K/AKT-c-JUN. Nat Commun 2016; 7: 11309.
Afonja O, Juste D, Das S, Matsuhashi S, Samuels HH . Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER-2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene 2004; 23: 8135–8145.
Bitomsky N, Wethkamp N, Marikkannu R, Klempnauer KH . siRNA-mediated knockdown of Pdcd4 expression causes upregulation of p21(Waf1/Cip1) expression. Oncogene 2008; 27: 4820–4829.
Dweep H, Gretz N . miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods 2015; 12: 697.
Dweep H, Sticht C, Pandey P, Gretz N . miRWalk–database: prediction of possible miRNA binding sites by "walking" the genes of three genomes. J Biomed Inform 2011; 44: 839–847.
Ye Y, Jin L, Wilmott JS, Hu WL, Yosufi B, Thorne RF et al. PI(4,5)P2 5-phosphatase A regulates PI3K/AKT signalling and has a tumour suppressive role in human melanoma. Nat Commun 2013; 4: 1508.
Sorokin AV, MEMO1 Chen J. a new IRS1-interacting protein, induces epithelial-mesenchymal transition in mammary epithelial cells. Oncogene 2013; 32: 3130–3138.
Walsh AM, Kapoor GS, Buonato JM, Mathew LK, Bi Y, Davuluri RV et al. Sprouty2 Drives Drug Resistance and Proliferation in Glioblastoma. Mol Cancer Res 2015; 13: 1227–1237.
Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discov 2011; 1: 524–538.
Zhou Y, Liang C, Xue F, Chen W, Zhi X, Feng X et al. Salinomycin decreases doxorubicin resistance in hepatocellular carcinoma cells by inhibiting the beta-catenin/TCF complex association via FOXO3a activation. Oncotarget 2015; 6: 10350–10365.
Liao H, Bai Y, Qiu S, Zheng L, Huang L, Liu T et al. MiR-203 downregulation is responsible for chemoresistance in human glioblastoma by promoting epithelial-mesenchymal transition via SNAI2. Oncotarget 2015; 6: 8914–8928.
Miow QH, Tan TZ, Ye J, Lau JA, Yokomizo T, Thiery JP et al. Epithelial-mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015; 34: 1899–1907.
Lei K, Townsend DM, Tew KD . Protein cysteine sulfinic acid reductase (sulfiredoxin) as a regulator of cell proliferation and drug response. Oncogene 2008; 27: 4877–4887.
Jiang W, Kahn SM, Zhou P, Zhang YJ, Cacace AM, Infante AS et al. Overexpression of cyclin D1 in rat fibroblasts causes abnormalities in growth control, cell cycle progression and gene expression. Oncogene 1993; 8: 3447–3457.
Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G . Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 1993; 7: 812–821.
Zhong Z, Yeow WS, Zou C, Wassell R, Wang C, Pestell RG et al. Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Res 2010; 70: 2105–2114.
Ju X, Casimiro MC, Gormley M, Meng H, Jiao X, Katiyar S et al. Identification of a cyclin D1 network in prostate cancer that antagonizes epithelial-mesenchymal restraint. Cancer Res 2014; 74: 508–519.
Wang H, Wu Q, Liu Z, Luo X, Fan Y, Liu Y et al. Downregulation of FAP suppresses cell proliferation and metastasis through PTEN/PI3K/AKT and Ras-ERK signaling in oral squamous cell carcinoma. Cell Death Dis 2014; 5: e1155.
Larue L, Bellacosa A . Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene 2005; 24: 7443–7454.
Mihailovich M, Bremang M, Spadotto V, Musiani D, Vitale E, Varano G et al. miR-17-92 fine-tunes MYC expression and function to ensure optimal B cell lymphoma growth. Nat Commun 2015; 6: 8725.
Ebert MS, Sharp PA . Roles for microRNAs in conferring robustness to biological processes. Cell 2012; 149: 515–524.
Acknowledgements
This study was supported by National Nature Science Fund of China (No. 81372184 and No. 81401906) (http://www.nsfc.gov.cn), the Outstanding Young Teacher Training Project of Colleges and Universities in Guangdong Province (No. Yq2013136), Guangdong Province Education Foundation (No. 2014KTSCX107), and Yangcheng Scholar Research Projects from Universities of Guangzhou (No. 12A011D). We are also grateful to Dr Douglas E Linn (Brigham & Women’s Hospital, Boston, MA, USA) for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhen, Y., Fang, W., Zhao, M. et al. miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop modulated by PDCD4 suppresses cell growth, metastasis, and sensitizes nasopharyngeal carcinoma to cisplatin. Oncogene 36, 275–285 (2017). https://doi.org/10.1038/onc.2016.201
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.201
This article is cited by
-
MicroRNA-181b-5p Facilitates Thyroid Cancer Growth via Targeting Programmed Cell Death 4
Molecular Biotechnology (2024)
-
MicroRNAs as the pivotal regulators of cisplatin resistance in head and neck cancers
Cancer Cell International (2023)
-
Precision medicine in nasopharyngeal carcinoma: comprehensive review of past, present, and future prospect
Journal of Translational Medicine (2023)
-
The role of long non-coding RNA HCG18 in cancer
Clinical and Translational Oncology (2022)
-
NAP1L1 interacts with hepatoma-derived growth factor to recruit c-Jun inducing breast cancer growth
Cancer Cell International (2021)
