
Abstract
Dual-hereditary jaundice (Dubin–Johnson syndrome (DJS) and Gilbert’s syndrome (GS)) is a rare clinical entity resulting from defects of the ATP binding cassette subfamily C member 2 (ABCC2) and UDP glucuronosyltransferase family 1 member A1 (UGT1A1) genes with autosomal recessive inheritance. In this study, we aimed to investigate the mutation profiles and characterize the phenotypes in a Han Chinese family with DJS and GS. Genetic screening for variants in the ABCC2 and UGT1A1, immunohistochemistry for expression of ABCC2, and histopathological examination were carried out. The proband and his brother had unconjugated and conjugated hyperbilirubinemia after birth. The proband’s sister had only conjugated hyperbilirubinemia after birth. The proband developed into pleural effusions and ascites, pericardial thickening, intrahepatic and extrahepatic biliary duct dilatation, and enlarged gallbladder at age 50. Hepatocellular carcinoma occurred in the proband’s brother at age 46. Seven compound defects of the ABCC2 gene [c.2414delG, p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln)] and the UGT1A1 gene (c.-3279T>G, p.(Gly71Arg), and p.(Pro451Leu)) were identified in family members. Accumulation of pigment in hepatocytes characteristic of that in DJS was present in the proband and his brother. Expression of ABCC2 protein was markedly diminished in the patient’s liver. Our results show a different genetic profile of DJS and GS in a Han Chinese family, indicating a more complex pattern of dual-hereditary jaundice among different populations. The present study illuminates the underpinnings of DJS and GS and extends the mutation profiles and phenotypes of these two syndromes in dual-hereditary jaundice.
Similar content being viewed by others


Introduction
Hereditary hyperbilirubinemia syndromes are caused by defects in bilirubin transport or conjugation in the liver [1]. Two genes for these syndromes are involved: UDP-glucuronosyl transferase1A1 (UGT1A1) and ATP binding cassette subfamily C member 2 (ABCC2). Defects in UGT1A1 may impair the glucuronidation of bilirubin and may be responsible for Gilbert’s syndrome (GS; On-line Mendelian Inheritance in Man database [OMIM] No. 143500) [1, 2]. Defects in ABCC2 are responsible for Dubin–Johnson syndrome (DJS; OMIM No. 237500) [3]. Jaundice is a common clinical presentation of both syndromes.
The prevalence of GS is high, varying from 2 to 20% among persons of various ethnicities [4]. The clinical manifestation of GS is mildly increased unconjugated bilirubin values (total serum bilirubin >20 μmol/L but usually <80 μmol/L) without hepatocellular disease or hemolysis [5]. More than 100 variants in UGT1A1 have been reported; among them, the linked polymorphisms A (TA)7TAA (rs8175347) and c.-3279T>G (rs4124874) in the promoter region are most strongly associated with GS [6]. DJS is a rare disorder, characterized by conjugated hyperbilirubinemia (>7 μmol/L), melanin-like pigment deposition in hepatocytes, and normal liver function [7]. Thirty-four pathogenic variants in ABCC2 have been identified for DJS [8].
Dual-hereditary jaundice (GS and DJS) is an atypical phenotype, described in a few studies on Europeans, that may be confused with common liver diseases. Moreover, to our knowledge, no long-term follow-up of patients with dual-hereditary jaundice has been reported, so understanding of the outcome of patients with the disease is limited. Here, we present the first reported Chinese family with dual-hereditary hyperbilirubinemia, with characterization of the molecular mechanism and clinical features of the disorder.
Patients and methods
Patients
All participants were members of a Han Chinese family and signed the informed consent.
Mutational analysis
Sequences of ABCC2 and UGT1A1 genes were analyzed by polymerase chain reaction and direct sequencing from genomic DNA extracted from 200 µL whole blood. All exons of ABCC2 and UGT1A1 were amplified by polymerase chain reaction with corresponding primers [9, 10]. All sequence variants were annotated according to reference sequences (NG_011798.1 for ABCC2 and NG_002601.2 for UGT1A1). Novel mutations of ABCC2 were verified in 211 unrelated control subjects without a family history of hyperbilirubinemia.
To explore the transcriptional status of these mutations found in genomic DNA of the ABCC2 gene, total RNA was extracted from peripheral blood mononuclear cell by using the phenol-chloroform method after TRIzol treatment (Invitrogen; 15596018). The A260/A280 ratio and concentration of total RNA was measured on a micro-spectrophotometer (NanoDrop 2000, Thermo Scientific). The integrity of RNA samples based on 28S and 18S rRNA was evaluated on a 1.5% agarose gel. cDNA was synthesized from about 500 ng total RNA by using PrimeScriptTM RT reagent Kit with gDNA Eraser (TaKaRa, RR047A). Exon 18 and exon 31 of the ABCC2 gene of all family members and the full-length ABCC2 coding sequence (CDS) of the proband were amplified from cDNA. The primers used for coding region amplification and sequencing were listed in Supplementary Table S1. All variants were recorded according to the reference sequence (NM_000392.4). TA cloning was used to verify the three consecutive heterozygous mutations in exon 31.
Histology and immunohistochemistry
Formalin-fixed and paraffin-embedded sections of the liver were stained with hematoxylin-eosin, periodic acid Schiff (PAS), and silver ammonium complex (Masson’s method). Liver biopsy of the proband’s brother (II:9) was immunohistochemically stained for CD34, glypican, and KI-67.
Immunohistochemistry and confocal laser scanning microscopy (CLSM)
After being deparaffinized and pretreated, microsections were double immunohistochemical stained for ABCC2 (R260, dilution1:800; Cell Signaling Technology, Farmingdale, New York, USA) and CEA-related adhesion molecule 1 (CEACAM1; clone #283324, dilution1:200; R&D systems, Minnesota, USA). Slides were next incubated with appropriate secondary antibodies conjugated to Alexa Fluor® 488 or 594 (Jackson, USA), both diluted to 1:200 with phosphate-buffered saline. Slides were mounted with Mowiol, and nuclei were stained with 4’, 6-diamidino-2-phenylindole (Boster, Wuhan, China). Slides incubated without primary/secondary antibodies were used as negative controls. Slices were processed and imaged by microscopy (Carl Zeiss AxioScope. A1) with laser at the wavelength of 350 nm for blue fluorescence (nuclei), 488 nm for green fluorescence (ABCC2), and 594 nm for red fluorescence (CEACAM1). All biopsy specimens were evaluated by two pathologists. Only strong staining in a canalicular pattern was considered positive in the immunohistochemical examination of ABCC2 and CEACAM1.
Results
Clinical data
All family members had normal serum values of total protein, albumin, globulins, aspartate aminotransferase (AST), and alanine aminotransferase (ALT) (Table 1). Three of the 11 members had been previously diagnosed with suspected hereditary hyperbilirubinemia (Fig. 1 and Table 1); all three had hyperbilirubinemia after birth, with no history of drinking, smoking, or exposure to special drugs/poisons or abnormal BMI, but the proband’s brother (II:9) had a long-term medication. The proband (II:3) and his brother (II:9) had unconjugated and conjugated hyperbilirubinemia, while the proband’s sister (II:1) had only conjugated hyperbilirubinemia.

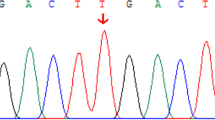
Pedigree of GS and DJS families with ABCC2 and UGT1A1 mutations, sequencing chromatogram, evolutionary conservation analysis, and liver histology. A Family with ABCC2 (c.2414delG, p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln)) and UGT1A1 mutations (c.-3279T>G, p.(Gly71Arg), and p.(Pro451Leu)). The proband is marked by arrow; black symbols denote affected members; white symbols denote unaffected members; squares denote men; and circles denote women. The sequencing chromatogram is shown. B Sequence alignment of ABCC2 and UGT1A1 is conserved among vertebrates (box). C Liver histology of the proband and his brother. C1 The proband, II:3. Hematoxylin-eosin (H-E) staining. Brown granular deposits of lipofuscin are evident. C2 The proband’s brother, II:9. Scale bar, 20 μm
The male proband (II:3) had cutaneous and scleral icterus at birth without a known cause. At age 50, he developed into pleural effusions, ascites, pericardial thickening, intrahepatic and extrahepatic biliary duct dilatation, and an enlarged gallbladder seen on computed tomography scan. Liver ultrasonography demonstrated normal liver size and morphology and uniform density. A mediastinal lymph node biopsy revealed reactive hyperplasia. Serologic tests were negative for Epstein–Barr virus, cytomegalovirus, and hepatitis A, B, C, D, or E viruses. He had no hematologic disorder, skin rash, or hepatosplenomegaly. Serum values of AST, ALT, α1-antitrypsin, copper, ceruloplasmin, thyroid hormones, sweat chloride, and screening biochemistries for metabolic diseases were within the normal range. However, severe hyperbilirubinemia was detected: total bilirubin 164 µmol/L (normal: 3.0–21.0 µmol/L) and direct bilirubin 133 µmol/L (normal: 0–7.0 µmol/L). In all, 2+ urinary bilirubin and 1+ urobilinogen were also recorded. Gamma-glutamyl transpeptidase activity was 136 U/L (normal: 11.0–50.0 U/L). After treatment, the total bilirubin stabilized at 80 µmol/L.
The proband’s brother (II:9) also had cutaneous and scleral icterus at birth without a known cause. At age 43, he had chronic cholecystitis and cholelithiasis. He took Xiaoyan Lidan tablets (Chinese patent medicine) regularly and amlodipine and metoprolol to control blood pressure. He developed cirrhosis and primary hepatocellular carcinoma at age 48 and died within 5 months of the emergence of the liver cancer. Hepatitis B infection was diagnosed at age 18, but HBeAb and HBcAb tests were positive without HBV DNA, suggesting spontaneous resolution of the hepatitis B infection. Serological tests for other hepatotropic viruses (hepatitis A, C, and D; Epstein–Barr virus; and cytomegalovirus) were negative. No hematologic disorders, autoimmune liver diseases, abnormal ceruloplasmin or serum ferritin, skin rash, or hepatosplenomegaly were detected. Mild hyperbilirubinemia (total bilirubin 63.5 µmol/L, direct bilirubin 45.8 µmol/L) at age 46 increased to severe hyperbilirubinemia (total bilirubin 371.4 µmol/L, direct bilirubin 286.1 µmol/L) with development of the liver cancer. Hepatocellular carcinoma was diagnosed by liver biopsy (Supplementary Fig. S2).
The proband’s sister (II:1) had conjugated hyperbilirubinemia (total bilirubin 30.8 µmol/L, direct bilirubin 24.5 µmol/L) without known disease or other causes of persisting hyperbilirubinemia (Table 1).
Detection of UGT1A1 and ABCC2 mutations
The pedigree analysis of the family was conducted (Fig. 1). The genotypes of all family members were consistent. Genetic analysis of the proband revealed two mutations in ABCC2: a heterozygous deletion of c.2414delG in exon 18, inherited from his mother, and three heterozygous mutations [p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln)] in exon 31, inherited from his father (Fig. 1 and Table 2). An evolutionary conservation analysis revealed that mutations p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln) led to a highly conserved amino acid change (Fig. 1). Two disease-causing mutations were found by MutationTaster (https://www.mutationtaster.org/), but they were not observed in the SNP database (https://www.ncbi.nlm.nih.gov/snp/). The alterations were not identified in 211 unrelated control subjects from the same southwest Chinese population (data not shown) or in the 1000 Genome Project dataset (http://browser.1000genomes.org/index.html).
For the UGT1A1 gene, the proband had a polymorphic mutation c.-3279T>G in the promoter, a heterozygous mutation p.(Gly71Arg) in exon 1, and another heterozygous mutation, p.(Pro451Leu), in exon 5 (Fig. 1 and Table 2). Mutation p.(Pro451Leu) is an evolutionary conserved amino acid change (Fig. 1). Other family members were also investigated for defects in the ABCC2 and UGT1A1 genes. The proband and his brother (II:9) had the same genetic defect in ABCC2 and UGT1A1. The proband and his sister (II:1) had similar mutations in the ABCC2 gene. Four members of the family were found to have another mutation in UGT1A1 [p.(Pro364Leu)], which was absent in the proband (Table 2).
Verification of ABCC2 mutations in CDS
To investigate the transcriptional mutation of the ABCC2 mutations in our samples, exon 18 and exon 31 were amplified from cDNA of all the family-based samples. The heterozygous deletion mutation c.2414delG in exon 18 of II:1, II:3, II:7, and II:9 were the same as that in the genomic DNA but were not available in I:2 and III:2 (Table 2). We got two PCR bands of exon 31 in cDNA from samples who carry those mutations in genomic DNA, but only one band in wild-type controls (Supplementary Fig. S1A). One of the 2 bands covered the full length of exon 31 with the three consecutive heterozygous mutations [p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln)], and the other band has a 195-bp deletion (c.4314-4508del) (Supplementary Fig. S1B). The three consecutive heterozygous mutations were verified by TA cloning (Supplementary Fig. S1C). Sequencing of the full-length CDS of the proband showed that the heterozygous deletion mutation c.2414delG in exon 18 do not occur in the full-length 4638-bp CDS of the ABCC2 gene. While, the three consecutive mutations were homozygous in the full-length 4638-bp CDS of the proband (Supplementary Fig. S1D), which were different from that of genomic DNA.
Histology and ABCC2 protein expression
For the proband, liver histology revealed normal structure and intense, brown parenchymal deposits of lipofuscin with a centrilobular and midzonal accentuation in liver cells (Fig. 1C). The pigment was strongly PAS positive (Supplementary Fig. S3) but was negative in Masson’s reaction. The accumulation of pigment was suggestive of that seen in DJS [8], but with distinctive brown granules. Perl Prussian blue staining for hemosiderin was negative, indicating normal hepatic iron load. The proband’s brother (II:9) had liver histopathological characteristics like those of the proband (Fig. 1C2 and Supplementary Fig. S3). Supplementary Fig. S4 illustrates that the lipofuscin pigment in the hepatocytes of the proband’s brother (II:9) was present in hepatocytes adjacent to cancer cells (Supplementary Fig. S4A) but not in the cancer cells (Supplementary Fig. S4B).
ABCC2 protein expression was analyzed with CLSM double immunofluorescence staining. In the normal liver, ABCC2 and CEACAM1 had similarly high expression (Fig. 2A–C), but in the patient’s liver, the expression of ABCC2 was lower than that in the control group (Fig. 2D–F).

Localization of ABCC2 (A and D, green) and CEACAM1 (B and E, red) by immunofluorescence staining in liver tissues of control and proband. Cell nuclei are stained with 4’, 6-diamidino-2-phenylindole (DAPI) (C and F, blue). Hepatocytes positive for CEACAM1 as well as ABCC2 are more numerous in control tissue than in the proband’s tissues. Cells positive for the target proteins and the cell nuclei are stained in the same sections (C and F). Scale bar, 25 μm
Discussion
The concurrence of mutation characteristics of DJS and GS has been designated dual-hereditary jaundice [9, 10]. Herein, we report a compound defect of ABCC2 [c.2414delG, p.(Ile1489Gly), p.(Thr1490Pro) and p.(Ile1491Gln)] and UGT1A1 genes [c.-3279T>G, p.(Gly71Arg) and p.(Pro451Leu)] causing DJS and GS in a Han Chinese family, which are not exactly the same mutations as identified in reported dual-hereditary jaundice patients [7, 9, 10]. Although many genetic alterations of the ABCC2 gene have been identified in DJS patients, hotspot mutations are not among them [11]. The new variant, c.2414delG, is in the exon 18 of the ABCC2 gene. The deletion leads to frameshift and truncation of the encoded ABCC2 protein from the original 1545 amino acids to 809 residues. The truncated protein damages three ATP binding cassettes and three transmembrane domains and probably degrades newly formed mRNA rapidly [12]. In this family, we also found three continuous coding variants [p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln)] in exon 31 of ABCC2, which is a rare mutational profile. p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln) have been predicted to be “probably damaging” to protein function and structure, with scores of 0.996, 1, and 1 base on PolyPhen-2 (Harvard Medical School, Boston, Massachusetts, USA), which had been reported [9]. We also found diminished expression of ABCC2 in the brother of the proband (II:9) with indel and mutations, a finding that may indicate that defective ABCC2 caused impaired protein function. Furthermore, indel and mutations were co-segregated with the phenotype of congenital conjugated hyperbilirubinemia for family members II:3, II:9, and II:1, which suggests that this mutation severely disrupts its transporter activity. However, the transcriptional status may be different between the family members who harbor the same mutation(s) in the genomic DNA according to the CDS sequences. The different transcriptional results may have a role in the different disease phenotype among the family members who share the same genomic DNA mutation(s). Unfortunately, we failed to acquire the full-length CDS of the ABCC2 gene of the other family members. Defective ABCC2 is responsible for DJS with melanin-like pigment in the hepatocytes. Unlike this black pigment, the liver of DJS has deposits of brown lipopigment, which are consistent with the results of Cebecauerova et al. [10] in a dual-hereditary jaundice case. The pigment in the hepatocytes of our proband’s brother was present in hepatocytes adjacent to cancer cells but not in the cancer cells, suggesting that liver cancer cells have a different metabolic mechanism for handling pigment deposits or lipofuscin. More evidence is needed to explain these findings.
Three coding variants [p.(Gly71Arg), p.(Pro364Leu), and p.(Pro451Leu)] and one promoter variant c.-3279T>G were identified in the UGT1A1 gene. Sense polymorphic mutations, A(TA)7TAA and c.-3279T>G in UGT1A1, were most strongly associated with GS, whereas mutations p.(Gly71Arg) and p.(Tyr486Asp) were more strongly associated with CNS-II [6]. Thus, we identified a different mutation profile with GS. Three variants [c.-3279T>G, p.(Gly71Arg), and p.(Pro451Leu)] were co-segregated with the phenotype of congenital unconjugated hyperbilirubinemia, which may account for GS. c.-3279T>G, p.(Gly71Arg), and p.(Pro364Leu) have been reported to contribute to GS, with bilirubin UGT activity reduced to 62%, 40%, and 36.6% of normal, respectively [13]. Though the role of p.(Pro451Leu) in UGT1A1 isoenzyme activity has not been reported, sorting intolerant from tolerant and polymorphism phenotyping analyses [14] and the correlation of phenotype-genotype observed in our pedigrees make it persuasive that this mutation was the cause of unconjugated hyperbilirubinemia. Although some studies suggest that compound variants [c.-3279T>G, p.(Gly71Arg), and p.(Pro364Leu)] make UGT1A1 protein failure, some family members (I:2, II:1, II:9, and III:4) who had these mutations had normal unconjugated bilirubin values. This finding suggests that the mutations have variable phenotypes among populations. We speculate that because of some compensatory mechanism in the family described herein, down-regulated UGT1A1 isoenzyme activity was not consistent with unconjugated hyperbilirubinemia.
Dual-hereditary jaundice has been described infrequently [7, 10], and we are aware of the lack of study which has followed such patients from infancy. Here, we found that the proband and his brother, who have the same defects in two genes and similar unconjugated and conjugated hyperbilirubinemia, developed different diseases in a span of 50 years. Bilirubin has two clinical behaviors: mild hyperbilirubinemia has health-promoting effects, which can protect from type 2 diabetes mellitus, cardiovascular diseases, and some cancers [15, 16]. However, severe hyperbilirubinemia can have adverse outcomes, such as jaundice, neurotoxicity, and cholestasis [17]. Few studies have explored the clinical outcomes of unconjugated and conjugated hyperbilirubinemia in GS and DJS. The proband in our study had 50 years of dual jaundice yet did not develop into severe liver-related disease. The proband’s brother, who had a long medication history, hypertension, and cholelithiasis, developed into hepatocellular carcinoma at age 48. So far, at least three studies had suggested that DJS coexisted with HCC [18,19,20]. The medication history of the two patients was compared; the proband’s brother had long-term medication for liver diseases, while the proband had no long medication history. Because UGT1A1 and ABCC2 proteins are responsible for drug biotransformation, defects in them may impair their ability to metabolize certain drugs and increase their toxicity. More similar cases or function verification may reinforce the conclusion.
In conclusion, we describe a Chinese family with two brothers who had combined GS and DJS. Compound defects of the ABCC2 gene [c.2414delG, p.(Ile1489Gly), p.(Thr1490Pro), and p.(Ile1491Gln)] and the UGT1A gene [c.-3279T>G, p.(Gly71Arg), and p.(Pro451Leu)] were identified. One brother developed hepatocellular carcinoma after long-term treatment with multiple medications. The present study illuminates the underpinnings of GS and DJS, and extends mutation profiles and phenotypes of the syndromes.
References
Erlinger S, Arias IM, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014;146:1625–38.
Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med. 1995;333:1171–5.
Corpechot C, Barbu V, Chazouilleres O, Broue P, Girard M, Roquelaure B, et al. Genetic contribution of ABCC2 to Dubin-Johnson syndrome and inherited cholestatic disorders. Liver Int. 2020;40:163–74.
Wagner KH, Shiels RG, Lang CA, Seyed Khoei N, Bulmer AC. Diagnostic criteria and contributors to Gilbert’s syndrome. Crit Rev Clin Lab Sci. 2018;55:129–39.
Sugatani J, Yamakawa K, Yoshinari K, Machida T, Takagi H, Mori M, et al. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem Biophys Res Commun. 2002;292:492–7.
Sun L, Li M, Zhang L, Teng X, Chen X, Zhou X, et al. Differences in UGT1A1 gene mutations and pathological liver changes between Chinese patients with Gilbert syndrome and Crigler-Najjar syndrome type II. Medicine (Baltimore). 2017;96:e8620.
Slachtova L, Seda O, Behunova J, Mistrik M, Martasek P. Genetic and biochemical study of dual hereditary jaundice: Dubin-Johnson and Gilbert’s syndromes. Haplotyping and founder effect of deletion in ABCC2. Eur J Hum Genet. 2016;24:704–9.
Togawa T, Mizuochi T, Sugiura T, Kusano H, Tanikawa K, Sasaki T, et al. Clinical, pathologic, and genetic features of neonatal Dubin-Johnson syndrome: a multicenter study in Japan. J Pediatr. 2018;196:161–7 e1.
Jiang J, Wang HG, Wu WL, Peng XX. Mixed Dubin-Gilbert syndrome: a compound heterozygous phenotype of two novel variants in ABCC2 gene. Chin Med J (Engl). 2017;130:1003–5.
Cebecauerova D, Jirasek T, Budisova L, Mandys V, Volf V, Novotna Z, et al. Dual hereditary jaundice: simultaneous occurrence of mutations causing Gilbert’s and Dubin-Johnson syndrome. Gastroenterology. 2005;129:315–20.
Wu L, Zhang W, Jia S, Zhao X, Zhou D, Xu A, et al. Mutation analysis of the ABCC2 gene in Chinese patients with Dubin-Johnson syndrome. Exp Ther Med. 2018;16:4201–6.
Keitel V, Nies AT, Brom M, Hummel-Eisenbeiss J, Spring H, Keppler D. A common Dubin-Johnson syndrome mutation impairs protein maturation and transport activity of MRP2 (ABCC2). Am J Physiol Gastrointest Liver Physiol. 2003;284:G165–74.
Li L, Deng G, Tang Y, Mao Q. Spectrum of UGT1A1 variations in Chinese patients with Crigler-Najjar Syndrome Type II. PLoS One. 2015;10:e0126263.
Di YM, Chan E, Wei MQ, Liu JP, Zhou SF. Prediction of deleterious non-synonymous single-nucleotide polymorphisms of human uridine diphosphate glucuronosyltransferase genes. AAPS J. 2009;11:469–80.
Kwon YJ, Lee YJ, Park BJ, Hong KW, Jung DH. Total serum bilirubin and 8-year incident type 2 diabetes mellitus: The Korean Genome and Epidemiology Study. Diabetes Metab. 2018;44:346–53.
Wagner KH, Wallner M, Molzer C, Gazzin S, Bulmer AC, Tiribelli C, et al. Looking to the horizon: the role of bilirubin in the development and prevention of age-related chronic diseases. Clin Sci (Lond). 2015;129:1–25.
Watchko JF, Tiribelli C. Bilirubin-induced neurologic damage-mechanisms and management approaches. N Engl J Med. 2013;369:2021–30.
Aoki H, Morihiro T, Arata T, Kanaya N, Takeda S, Ninomiya T, et al. Hepatectomy in a hepatocellular carcinoma case with Dubin-Johnson syndrome and indocyanine green excretory defect. Clin J Gastroenterol. 2013;6:69–74.
Sakamoto A, Mori I, Kawai K, Tsuchiyama H. Dubin-Johnson syndrome associated with hepatocellular carcinoma--report of an autopsy case. Gan No Rinsho. 1987;3:1361–7.
Shikada Y, Matsumata T, Suehiro T, Sugimachi K. Hepatocellular carcinoma in a case of Dubin-Johnson syndrome treated successfully with a central bilateral segmentectomy. Hepatogastroenterology. 2004;51:833–6.
Acknowledgements
This work was supported by grants from the Training Plans for Medical Reserve Talents of Yunnan Provincial Health Commission (H-2018102) and Young and middle-aged academic and technical leaders reserve talents in Yunnan Province (202105AC160048), Natural Science Foundation of Yunnan Province (202001AT070085), and National Natural Science Foundation of China (81960116). We thank Prof. Li-Jun Shen from Kunming Medical University, Kunming, China for pathologic analysis.
Author information
Authors and Affiliations
Contributions
JW, LZ, and TCZ designed the study. HL and JHF collected the samples and clinical information. XL and FWL performed the experimental procedures. TCZ and YLY performed the data analysis. TCZ, SHZ and LZ drafted the manuscript. All authors contributed to and have approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, TC., Li, X., Li, H. et al. Concurrence of novel mutations causing Gilbert’s and Dubin–Johnson syndrome with poor clinical outcomes in a Han Chinese family. J Hum Genet 68, 17–23 (2023). https://doi.org/10.1038/s10038-022-01086-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-022-01086-1
