
Abstract
The gut is connected to the CNS by immunological mediators, lymphocytes, neurotransmitters, microbes and microbial metabolites. A mounting body of evidence indicates that the microbiome exerts significant effects on immune cells and CNS cells. These effects frequently result in the suppression or exacerbation of inflammatory responses, the latter of which can lead to severe tissue damage, altered synapse formation and disrupted maintenance of the CNS. Herein, we review recent progress in research on the microbial regulation of CNS diseases with a focus on major gut microbial metabolites, such as short-chain fatty acids, tryptophan metabolites, and secondary bile acids. Pathological changes in the CNS are associated with dysbiosis and altered levels of microbial metabolites, which can further exacerbate various neurological disorders. The cellular and molecular mechanisms by which these gut microbial metabolites regulate inflammatory diseases in the CNS are discussed. We highlight the similarities and differences in the impact on four major CNS diseases, i.e., multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, and autism spectrum disorder, to identify common cellular and molecular networks governing the regulation of cellular constituents and pathogenesis in the CNS by microbial metabolites.
Similar content being viewed by others
Introduction
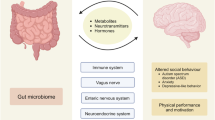
The central nervous system (CNS) is known as an immune-privileged tissue system but remains susceptible to inflammatory responses. While well protected by the blood–brain barrier (BBB), nevertheless the CNS hosts many types of inflammatory responses. A mounting body of evidence indicates the presence of close interactions between the neural system and gut microbes. These interactions involve endocrine, neurological, metabolic and immunological communication. The CNS and enteric nervous system (ENS) communicate via neurotransmitters, hormones, and metabolites1,2. Gut microbial metabolites are transported to the CNS, wherein they regulate CNS cells and immune cells. This review will examine our current understanding of how gut microbial metabolites impact four major CNS diseases and highlight the common disease-modifying functions of major microbial metabolites.
Multiple sclerosis (MS) is the most common form of inflammatory disease in the CNS and is associated with autoimmune-mediated demyelination3,4. Other less frequent inflammatory diseases include Rasmussen’s encephalitis5 and neuromyelitis optica (NMO)6. These diseases involve immune cell infiltrates in the brain and spinal cord, resulting in neuritis and myelitis7,8. Antigen-specific T and B lymphocytes are frequent immune cell infiltrates in CNS lesions. Inflammatory responses in the CNS are induced or suppressed by different types of T lymphocytes, such as Th1 and Th17 cells and Tregs9,10. Antibodies and B cells can augment or alter the pathogenesis of CNS inflammation11. Other cell types, such as microglia, macrophages and astrocytes, actively participate in immune cell regulation and neuroinflammation12. An increasing body of evidence indicates that other neurological diseases, such as Parkinson’s disease (PD), Alzheimer’s disease, and autism spectrum disorder (ASD), also involve significant inflammatory responses13,14,15. Therefore, we focus this review on these four CNS diseases, which have distinct tissue involvement, inflammatory responses and pathogenesis.
In recent years, we have witnessed a plethora of research on the functions of gut microbiota and their metabolites in regulating inflammatory responses in the CNS16,17,18,19. The intestine is rich in dietary materials and host secretions such as bile acids and mucins, which are subsequently metabolized by the cooperative activities of host enzymes and then by the gut microbiome. Various dietary fibers and complex carbohydrates can reach the terminal ileum and colon, which host the majority of the gut microbiome in the body. Microbial fermentation of sugars derived from carbohydrates in the colon produces short-chain fatty acids (SCFAs) such as acetate (C2), propionate (C3) and butyrate (C4)20. Another major group of metabolites includes tryptophan (Trp) metabolites. Dietary tryptophan is converted into indole derivatives, including indole-3-acetic acid (IAA), indole-3-propionic acid (IPA), indole-3-aldehyde (IAld), indole-3-acetaldehyde (IAAld), and indoleacrylic acid. Serotonin (5-HT), a neurotransmitter, is also produced in the gut lumen from Trp21,22,23,24. Primary bile acids are produced from cholesterol in the liver and are dehydroxylated by gut microbes to generate secondary bile acids25. These gut microbial metabolites function as agonists for various host receptors and intracellular molecules, such as G-protein-coupled receptors (GPCRs) and transcription factors, including GPR43, GPR41, TGR5, aryl hydrocarbon receptor (AhR), pregnane X receptor (PXR), farnesoid X receptor (FXR), vitamin D receptor (VDR), and others26. Moreover, many of these metabolites, particularly SCFAs, are metabolized in host cells to generate energy. Microbial metabolites regulate numerous host enzymes, such as histone deacetylases (HDACs). In general, microbial metabolites regulate host gene expression and metabolic activity in both immune cells and tissue cells. Importantly, these metabolites directly and indirectly affect the CNS to exert their regulatory functions19,27,28.
Basic functions of gut microbial metabolites
The most abundant metabolites in the colon are SCFAs, which have a combined luminal concentration of greater than 0.1 M in the human colon29. The three major SCFAs, C2, C3 and C4, function through four G-protein-coupled receptors (GPCRs), GPR41, GPR43, GPR109A and Olfr7830,31,32. In addition, SCFAs are natural HDAC inhibitors, and therefore, they can increase the acetylation and phosphorylation of host proteins such as histones, leading to increased gene expression and cell signaling33,34. SCFAs directly inhibit Type I and II HDACs and can also indirectly affect certain Type III HDACs, such as Sirtuin 135. Moreover, SCFAs are metabolized in host cells for energy production and thus have significant regulatory effects on host cell metabolism, leading to increased ATP levels, mTOR activity and fatty acid synthesis36. SCFAs are known to boost the differentiation of Th1 and Th17 cells and Tregs from naïve CD4 T cells, depending on the immunological milieu36. SCFAs increase IL-10 expression by lymphocytes (T and B cells) and macrophages37,38 in a manner largely mediated by their HDAC inhibitory activity. SCFAs regulate the activity of innate lymphocytes; they increase the number and activity of Group 3 innate lymphoid cells (ILC3s) but suppress Group 2 innate lymphoid cells (ILC2s) through GPCR activation and HDAC inhibition, respectively39,40,41. SCFAs also induce tolerogenic macrophages and dendritic cells by both GPCR activation and HDAC inhibition42.
Certain amino acid metabolites produced by microbes are also important regulators of host cells. Phenylalanine and tyrosine are precursors of dopamine, a key neurotransmitter, in the host, and some commensal bacteria produce amines related to these amino acids that activate host GPR5643. Trp is metabolized by both host cells and certain gut bacteria to serotonin (5-hydroxytryptamine), kynurenine (Kyn) and indole derivatives, which function as neurotransmitters and metabolic regulators24. Certain indole derivatives, such as IAA, IPA, and IAld, function as ligands for AhR, a member of the basic helix-loop-helix transcription factor family. These metabolites can also regulate host cells through EIF5a, IR76B, and PXR44. Certain microbial species, such as Peptostreptococcus russellii45 and Lactobacillus species46,47, can effectively produce indole derivatives45,46,47. Dietary indole derivatives, such as indole-3-carbinol (I3C) and diindolylmethane (DIM), can increase the activity of Tregs but suppress the generation of myelin oligodendrocyte glycoprotein (MOG)-specific Th17 cells48. Polyamines such as putrescine and spermidine are produced by intestinal bacteria and are present at 0.5 to 1 mM in the human colon49. Polyamines support the hypusine modification of eukaryotic translation initiation factor 5A (eIF5A) and promote polypeptide translation and cell proliferation50. Polyamines also have suppressive effects on Th17 cells51. Similarly, phytochemical metabolites, such as ginsenosides, resveratrol, and I3C, activate AhR to exert some of their regulatory functions52,53. Ginsenosides suppress Th1 and Th17 cells but increase Treg activity54. Resveratrol suppresses Th17 activity55.
Primary bile acid metabolites, such as cholic acid and chenodeoxycholic acid (CDCA), are made from cholesterol in the liver. Secondary bile acid metabolites, such as deoxycholate (DCA) and lithocholate (LCA), are formed by bacterial 7α-dehydroxylation of primary bile acids. Secondary bile acids activate multiple host receptors, such as PXR, VDR, LXR, FXR, TGR5, retinoid-related orphan receptor-γt (RORγt), and FoxP325,56. 3-OxoLCA inhibits Th17 polarization by binding to RORγt, whereas isoallo-LCA enhances Treg generation, in part by producing mitochondrial reactive oxygen species that induce FoxP3 expression57. Bile acid metabolites have both beneficial and pathogenic functions58,59,60,61. Primary and secondary bile acids activate the Nod-like receptor protein 3 (NLRP3) inflammasome and IL-1β production in macrophages61, which can increase innate immunity but cause chronic inflammation. However, activation of the bile acid receptors FXR and TGR5 is often associated with decreased inflammatory responses61,62.
Some microbial metabolites are pathogenic, causing inflammatory diseases and cancer. Pathogenic metabolites include trimethylamine N-oxide (TMAO)63, hydrogen sulfide64, phenol, p-cresol, N-nitrosamine, ammonia, 4-ethylphenylsulfate65, and uric acid66. These metabolites are risk factors for MS, AD, and other neurological disorders67,68,69. Certain environmental contaminants, such as the AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), are also pathogenic70.
Regulation of experimental neuroinflammation by microbial metabolites
The regulatory effects of microbiota on experimental neuroinflammation have been reported71,72,73,74. Rodent experimental autoimmune encephalitis (EAE) models are most frequently used75 because they largely mimic human MS. Antigen-induced EAE can be induced by immunization with several neuronal cell antigens, such as MOG, myelin basic protein (MBP), and proteolipid protein (PLP), along with potent adjuvants, such as complete Freund’s adjuvant and pertussis toxin75. Demyelination similar to that in MS is also induced by chemicals such as cuprizone (a copper-chelating agent) and lysolecithin76,77,78. Infection with Theiler’s murine encephalomyelitis virus induces demyelinating encephalitis79,80. Moreover, the engineered expression of T and B cell receptors specific for CNS antigens causes spontaneous EAE responses in mice81,82.
While dysbiosis exacerbates EAE responses83,84, studies with germ-free (GF) animals or broad-spectrum antibiotics revealed that the microbiota is required for optimal EAE pathogenesis85,86, perhaps because of the general adjuvant effect of the microbiota in inducing immune responses. When specific microbial species or their products are suppressed, EAE activity may either increase or decrease, depending on the function of the affected microbial species87,88. For example, segmented filamentous bacteria (SFB), S. aureus and P. heparinolytica promote the generation of Th17 cells and exacerbate EAE responses89,90,91. Elimination of these Th17-inducing microbial species ameliorated EAE inflammation85,86. In contrast, treatment with SCFA-producing bacteria such as Clostridium tyrobutyricum ameliorated EAE immune responses92. Thus, the impact of the microbiota on CNS inflammation depends on the function of the altered microbial species in the gut84.
It is thought that the microbiota affects neuroinflammation by producing immune regulatory products and metabolites. For example, polysaccharide A (PSA), a capsular polysaccharide produced by Bacteroidetes fragilis, has significant immune regulatory function93. Oral administration of PSA suppressed demyelination and delayed the onset of EAE activity94. In this previous study, PSA increased the number of CD103+ dendritic cells (DCs) in draining lymph nodes and induced IL-10-producing T cells. PSA promoted Toll-like receptor 2 (TLR2) activation, which induced CD39+ regulatory T cells, in part by modulating DCs. In addition, poly-γ-glutamic acid (γ-PGA) has suppressive effects on EAE activity95. This finding is in line with decreased Th17 cell but increased Treg activity in the CNS following the administration of γ-PGA, which activates TLR4-MyD88-dependent and TLR4-MyD88-independent pathways to regulate T cell activity95. In contrast, some bacterial products can exacerbate CNS inflammation. Lipopolysaccharide (LPS) treatment accelerated inflammation-mediated demyelination in rats96. It is well established that peptidoglycans, the major cell wall component of gram-positive bacteria, promote DC maturation through TLR2 activation, which increases the secretion of proinflammatory cytokines and promotes effector T cell generation during EAE development89. Thus, microbial TLR ligands can have both inflammatory and suppressive roles in neuroinflammation.
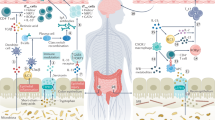
BBB disruption is observed in EAE and MS patients, and this can cause harmful exposure of CNS tissues to microbial products and microbial invasion following gut dysbiosis, leading to amplified inflammatory responses97,98. Under normal conditions, the microbiota and SCFAs support the integrity of the BBB99; it is important to protect the CNS from the harmful effects of inflammatory responses and toxicants. The BBB integrity is weakened in GF mice, and this defect is normalized by SCFA treatment in drinking water99 (Fig. 1). SCFAs, such as C2, C3, or C4, administered in drinking water ameliorated EAE activity. Interestingly, SCFAs appear to support CNS development beyond their immunoregulatory roles100,101. Microglial cells in GF mice had defective microglia with altered cell proportions and an immature phenotype, which were corrected by SCFA feeding in a GPR43-dependent manner100. In addition, C4 promoted oligodendrocyte maturation in the brain101. Thus, SCFAs support the functional maturation of CNS cells (Fig. 1). Increased levels of secondary bile acids, such as DCA, can weaken BBB function, perhaps because these compounds are amphiphilic steroids that loosen the cell membrane102,103. Thus, BBB function and associated CNS pathogenesis can be altered by changes in microbial metabolites.

Microbial metabolites positively and negatively influence CNS development and inflammatory responses. In the best case, beneficial metabolites are produced in symbiosis with a balanced population of diverse microbes in the gut. Together, these microbes produce myriad metabolites that are beneficial for the host. In dysbiosis, the production of harmful metabolites is increased while that of beneficial metabolites is decreased. In general, beneficial metabolites, such as SCFAs and Trp metabolites, reinforce the integrity of the gut barrier and BBB and support the functional maturation of CNS cells such as microglia, oligodendrocytes and astrocytes. Thus, these metabolites support CNS formation, neurological function, and the development of regulatory immune cells for immune tolerance. Moreover, these metabolites suppress harmful immune responses, such as the generation of pathogenic Th17 cells. These functions are mediated in part by various host receptors, such as GPCRs, transcription factors, nuclear ligand receptors (PXR and FXR), and TLRs. Conversely, harmful metabolites weaken the gut barrier and BBB and cause systemic inflammatory responses, neuronal cell death and tissue injury (e.g., demyelination), leading to inflammatory conditions that exacerbate CNS diseases. Not only harmful microbial metabolites but also pathogenic bacterial cells and T cells travel from the gut to the CNS to increase inflammation under pathological conditions.
Multiple groups have reported that SCFAs have EAE-suppressing activities (Table 1). Mice with elevated levels of SCFAs after feeding with SCFA water or dietary fiber had increased numbers of regulatory T cells in the intestine and CNS upon MOG immunization104,105. In addition, when mice were fed C4, cuprizone-mediated demyelination was ameliorated.18,74,101,105. The protective effect of C4 was further validated in a lysolecithin-induced demyelination model101. In addition to the major SCFAs (i.e., C2, C3, and C4), valerate (C5) can also regulate EAE activity27. C5 increased both regulatory B cells (Bregs) and T cells (Tregs). C5-treated B cells showed increased expression of IL-10, and adoptive transfer of these B cells ameliorated EAE activity. The protective effect was accompanied by decreased Th17 cell activity in the small intestine. C5, well known for its potent HDAC inhibitory activity, increased mTOR activation and glycolysis in lymphocytes27. Thus, both epigenetic (HDAC inhibition) and metabolic (mTOR) regulation appears to be involved in the protective effects of SCFAs in EAE pathogenesis.
The effects of SCFA-producing fiber-rich diets on EAE are mixed, and this appears to be due to considerable variation in the dietary fiber composition of high- and low-fiber diets used by different groups. Some groups used a diet containing no fiber (soluble or insoluble) as the control diet106,107,108. In general, soluble fibers such as pectin and inulin produce high levels of SCFAs, whereas insoluble fibers such as cellulose are not efficient sources of SCFAs. Other groups, including ours, compared the effects of high- and low-fiber diets containing the same amount of cellulose but different levels (0, 5, and 20%) of soluble fiber. When a high-fiber diet was compared with a zero-fiber diet, the protective effect was significant105. However, when the animal group fed a diet high in soluble fiber (pectin and inulin) was compared with that fed a diet containing zero soluble dietary fiber but the same level of cellulose, the difference was not significant18. This finding suggests the involvement of mechanisms other than SCFA production in the beneficial effects of dietary fiber. Despite its low bioavailability and SCFA-producing ability, cellulose seems to have a suppressive effect on EAE activity84. Cellulose decreased the number of neutrophils and Th2 cells in a spontaneous opticospinal encephalomyelitis mouse model expressing MOG-specific T and B cell antigen receptors84. While cellulose did not significantly change SCFA levels, it changed the microbiome composition in the gut, indicating that the protective effect of certain dietary fibers, such as cellulose, appears to be more dependent on microbial changes than on SCFA production108.
SCFAs appear to regulate EAE responses via several cellular and molecular mechanisms. SCFAs may function through astrocytes and microglial cells, which produce both suppressive and inflammatory cytokines and influence EAE responses. SCFAs can also act through immune cells such as Th17 cells and regulatory T cells, which differentially regulate inflammatory responses. Several cell types can produce IL-10 in response to SCFAs. Major cell types that produce IL-10 are T cells and macrophages, and this production can be further increased by SCFAs38,109,110,111 (Fig. 1). In our own study, SCFAs increased IL-10 production by microglial cells, and IL-10 production was required for the protective effect of SCFAs on EAE activity18.
While SCFAs have suppressive effects on EAE activity, their G-protein-coupled receptors, such as GPR43, can function unexpectedly to exacerbate EAE activity18. The mechanism is not entirely clear at this point, but the data imply that the protective function of SCFAs is perhaps mediated mainly through non-GPCR-dependent mechanisms. In this regard, the HDAC inhibitory activity of SCFAs may be important. GPCR activation by SCFAs can increase immune activity, including invigorating epithelial responses and activating inflammasomes in the gut112,113. We speculate that GPCR activation by SCFAs in the CNS could promote similar inflammatory responses. In this regard, the function of SCFAs in regulating EAE activity appears to be complex. When transferred into host mice, SCFA-treated MOG-specific effector Th17 cells have greater EAE activity than similarly prepared control Th17 cells18, indicating that SCFAs can affect both the inflammatory and regulatory arms of the CNS immune system. More work is required to understand the effects of SCFAs on key immune and CNS cell types, such as T and B cells, ILCs, glial cells, neurons, macrophages, and dendritic cells.
Indole derivatives and certain environmental contaminants, such as the AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), have been studied for their regulatory effects on EAE activity. AhR activation by FICZ and 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) (Trp-derived indole metabolites) suppressed EAE responses114,115, whereas AhR activation by TCDD exacerbated EAE pathogenesis70. Similarly, FICZ and TCDD differentially affected Th17 cells, Tregs, DCs, and astrocytes. The administration of dietary indoles was effective at suppressing EAE development48. Moreover, dietary indoles significantly ameliorated existing EAE48,116; this protection is thought to be mediated by the AhR-mediated expansion of regulatory T cells. These Trp-derived metabolites appear to suppress CNS inflammation by decreasing proinflammatory cytokine expression and inflammatory monocyte infiltration47,48,116, which may involve activation of the type-I IFN pathway in astrocytes to limit CNS inflammation in an AhR-dependent manner. Similarly, cruciferous plant-derived AhR ligands such as glucosinolates suppressed EAE pathogenesis117.
Significant associations between microbial metabolites and MS in humans
In humans, MS manifests in several ways with different patterns of disease progression and relapse; the common categorizations of MS include clinically isolated syndrome (CIS), relapsing-remitting MS (RRMS), primary progressive MS (PPMS), and secondary progressive MS (SPMS)118,119. RRMS, with repeated relapses and remissions, is most common (~85%). Most people with RRMS progress to SPMS with steadily worsening symptoms with or without remissions and relapses. PPMS is rarer (~10%), with slowly worsening symptoms from disease onset without remissions and relapses.
Changes in gut luminal microbial composition have been observed along with the abnormal invasion of microbes in the brains of RRMS patients120. RRMS patients have increased levels of Actinobacteria but decreased levels of Firmicutes and Bacteroidetes in feces. Within the Firmicutes family, fourteen species within Clostridium clusters XIVa and IV were found to be decreased in MS patients121. A cohort study found decreased Prevotella_9 but increased Streptococcus species, along with reduced levels of fecal SCFAs, in MS patients. In general, Tregs and Th17 cells in the blood indicate tolerogenic and inflammatory activity, respectively122. A positive correlation between Streptococcus species and Tregs was reported, and an inverse correlation between Prevotella_9 and Th17 cells was observed. Increased numbers of microbial peptidoglycan-containing macrophages and dendritic cells were detected in the brains of MS patients. Thus, MS patients have both dysbiosis and microbial invasion of the brain120,123. CCR9 is the small intestine-homing receptor for lymphocytes124,125 and is important for thymocyte localization in the thymic medulla126,127. Interestingly, CCR9+CD4+ T cells were found in the cerebrospinal fluid (CSF) of MS patients. T cell migration into inflamed CNS tissues involves another chemokine receptor, CCR6, that is highly expressed by Th17 cells128. CCR9+CD4+ T cells express RORγ and secrete IL-17 and IFN-γ in MS patients. Antibiotic treatment decreased these Th17 cells, suggesting that inflammatory CCR9+CD4+ T cells are induced by microbes129. While further study is required to determine the origin of these T cells, they appear to be generated in the intestine following dysbiosis and neural inflammation. These data imply the active movement of microbes and immune cells from the gut to CNS tissues under inflammatory conditions.
Normally, antigen-induced EAE induction in animals requires the use of potent adjuvants to break immune tolerance to self-antigens75. Fecal microbial transfer (FMT) from MS patients to animals induced EAE development following immunization without any adjuvant. FMT decreased certain microbial species, such as those in the Sutterella genus, and decreased IL-10 expression by T cells16,130. The effect of SCFA-generating dietary fibers on MS activity has been studied. A diet rich in plant fibers increased the fecal presence of the Lachnospiraceae phylum in MS patients and increased the numbers of Tregs and IL-10-producing PD-L1+ monocytes in the blood. While a fiber-rich diet failed to change MS clinical activity, patients on a Western diet had increased MS clinical activity131.
Negative correlations have been found between SCFA levels and T cell activity in MS patients. Higher fecal and blood SCFA levels were associated with increased Treg and suppressed Th1 cell activity122,131,132. Compared with healthy individuals, MS patients have decreased levels of blood C4 and SCFA-producing microbial species118. In line with the dysbiosis and reduced levels of SCFAs in MS patients118,133, decreased levels of all major SCFAs, such as C2, C3, and C4, were found in the blood of SPMS patients compared with that of healthy controls18. In addition, gut SCFA levels were decreased in RRMS patients122. In this regard, C3 increased Treg activity and decreased episodes of MS relapse134. In another study, fecal C3 levels were decreased in both RRMS and SPMS patients132. The therapeutic effect of C3 in MS patients was recently reported134. Regardless of MS subtype, C3 supplementation alleviated the clinical symptoms of MS, and MS patients with increased levels of SCFA-producing gut bacteria showed limited inflammatory T cell activity. C3 also increased regulatory T cells that produce IL-10, altered the expression of many genes in T cells, and increased oxygen consumption in T cell mitochondria. However, increased levels of plasma C2 and increased numbers of circulating Th17 cells were observed in MS patients with severe disability135,136. Because C2 is also produced by host cells under inflammatory and infection conditions137, the increased levels of C2 could originate from the host rather than microbial cells. These data, while contradictory to other results, seem to be in line with the effect of C2 on Th17 cell generation109. Therefore, more studies are required to establish the possible associations between SCFA levels and MS subtypes. More data may enable the utilization of SCFAs as biomarkers for MS activity and gut microbial status in MS patients.
Oral Trp administration boosted serotonin but decreased plasma cortisol levels in patients with neurological diseases. Acute MS and other neurologic symptoms were alleviated138,139. Dietary Trp can be metabolized by gut microbes or host cells after enteric absorption. While gut microbes produce various indole metabolites, host cells produce Kyn, kynurenic acid (KA) and quinolinic acid (QA) via the kynurenine pathway. The levels of urinary Kyn were decreased in RRMS patients140. KA levels were increased in RRMS patients but decreased in PPMS patients, while the level of QA was increased in most MS patients141. Trp administration improved the cognition and memory function of MS patients140,142,143,144. Pediatric MS is less frequent than adult MS, but relapses are more frequent and possibly more inflammatory in affected pediatric patients145. Increased Actinobacteria but decreased Clostridiales were observed in pediatric MS patients146. Decreased levels of Trp and indole lactate were also observed in pediatric MS patients. Interestingly, higher Kyn levels were associated with an increased relapse rate142. Many Trp metabolites, such as I3S, tryptamine (TA), IAA, Kyn and KA, function as AhR ligands147,148. KA also activates another host receptor, GPR35149. Trp metabolites increase AhR expression on microglia and astrocytes and control the inflammatory activities of these cells116,150. Secondary bile acids also regulate astrocytes by upregulating GPBAR1. Tauroursodeoxycholic acid (TUDCA), a taurine conjugate of ursodeoxycholic acid (UDCA), suppressed microglial and astrocytic polarization (Table 2)143. Overall, multiple gut metabolites can modulate microglial cells and astrocytes to suppress MS pathogenesis (Fig. 1).
Microbial metabolites and Parkinson’s disease
Parkinson’s disease (PD) is caused by the loss of dopaminergic neurons in the substantia nigra (SN), leading to dopamine deficiency in the midbrain; in this region, dopamine is important for the regulation of body movement151. A number of genes and their polymorphisms have been implicated in PD pathogenesis152. The presence of neurotoxic protein inclusions called Lewy bodies, which are composed of α-synuclein oligomers, in the midbrain is a pathological feature of PD153,154. Th1 and Th17 cell-driven inflammation, potentially induced by Lewy bodies, plays significant roles in PD pathogenesis155,156. Because of the inflammatory nature of PD and the potential roles of microbiota and gut metabolites in regulating PD, we also include a discussion of PD in this review (Tables 1, 2). Both genetics and environmental factors, such as diet and exposure to neurotoxic chemicals, play significant roles in PD pathogenesis. Genetic risk factors include autosomal dominant mutations in the alpha-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2) and vacuolar protein sorting ortholog 35 (VPS35) genes as well as autosomal recessive mutations in the PTEN-induced kinase 1 (PINK1), DJ-1 (also known as PARK7) and PARKIN genes157. Interestingly, polymorphisms in the LRRK2 gene are commonly found in PD and inflammatory bowel disease patients158,159.
Gut dysbiosis has been observed in PD patients71,160,161. Gut microbiota can regulate PD pathogenesis in animal models71,162,163,164. Mice colonized with fecal microbiota from PD patients developed PD-like pathological features, including α-synuclein aggregation in the brain71. Metabolite profiling of PD patients revealed decreased metabolism of unsaturated fatty acids and increased levels of secondary bile acid metabolites such as deoxycholic acid (DCA) and lithocholic acid (LCA). Importantly, significant decreases in SCFA-producing bacteria and decreased microbial carbohydrate processing activity were detected in a group of PD patients with reduced mobility165,166,167.
Among the LRRK2 gene mutations associated with PD, the G2019S mutation that affects the kinase domain is the most frequent168. The immunological characteristics of transgenic rats expressing human LRRK2 G2019S were examined169. Defective myelopoiesis and a decreased Th17 cell frequency were observed in these rats when they were challenged with colitis-inducing agents, such as 2,4,6-trinitrobenzene sulfonic acid (TNBS) and dextran sulfate sodium (DSS). Interestingly, we observed increased levels of Bacteroidetes in this animal model169. The MitoPark (MP) mouse model was created by deleting the mitochondrial transcription factor A (TFAM) gene in dopaminergic neurons170. Decreased gastrointestinal tract motility and gut dysbiosis were observed in this model. Moreover, increased levels of Prevotella were observed in this model and in PD patients. Young rats fed permethrin, a pesticide, developed PD-like symptoms and increased gut permeability. These animals also developed dysbiosis and showed decreased SCFA production.
The protective effects of valproic acid (a SCFA-related HDAC inhibitor) and C4 were observed in a model of PD induced with the chemicals manganese and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine160,163,171,172,173,174. Interestingly, a synthetic FFAR3 (GPR41) agonist (AR420626) corrected the movement disorder and neuronal loss in a 6-hydroxydopamine (OHDA, a neurotoxin)-induced PD mouse model163. In contrast, the microbiota and SCFAs were reported to promote PD pathogenesis in α-synuclein-transgenic mice71. Other researchers have also reported that SCFA administration exacerbates PD pathogenesis in another animal model175. The pathogenic role of SCFAs is unexpected but corresponds with the role of these metabolites in promoting the generation of inflammatory Th1 and Th17 cells, as reported previously by our group109,176. While these animal models provide insight into PD pathogenesis, the experimental conditions and pathogenesis in these models differ greatly from each other and from PD patients. Thus, the effects of SCFAs on PD pathogenesis are unclear, and more studies are required.
A previous study reported potential changes in bile acid metabolism with increased levels of secondary bile acids, such as LCA and DCA165. In a latent PD model in which α-syn fibrils were injected into the olfactory bulb to induce PD-like pathogenesis, the levels of bile acids such as ω-muricholic acid, TUDCA and UDCA were decreased in the blood177. Beneficial roles of TUDCA in acute, progressive and nigral transplant animal models of PD have been reported178,179,180. TUDCA ameliorated the decrease in dopaminergic fibers, mitochondrial dysfunction and neuroinflammation in an MPTP-induced animal model179,181,182. Moreover, TUDCA suppressed α-synuclein-induced oxidative stress via the activation of Nrf2, JNK and AKT179,182. In addition, UDCA decreased mitochondrial dysfunction in skin fibroblasts from patients harboring the LRRK2 G2019S mutation183. Thus, secondary bile acids have protective effects against PD pathogenesis.
Microbial metabolites and Alzheimer’s disease
AD is a progressive neural disease induced by the accumulation of toxic misfolded amyloid β-protein (Aβ) plaques in the brain. In this disease, extracellular β-amyloid plaques form in the basal, temporal, and orbitofrontal neocortex. In severe AD cases, the plaques spread to the hippocampus184. Dysbiosis is a risk factor for AD development. Mice humanized with stool from AD patients showed defects in cognitive function185. In addition to the gut bacterial composition, the diversity of gut fungal species (mycobiome) in patients with mild cognitive disorder was lower than that in control subjects. Dietary intervention with a Mediterranean ketogenic diet enhanced fungal microbiome diversity and decreased the levels of AD biomarkers in cerebral spinal fluid186.
Similar to MS and PD patients, AD patients have decreased levels of SCFAs186,187,188,189. In particular, AD patients with brain amyloid accumulation and endothelial dysfunction had decreased blood levels of C4187. AD pathology is associated with increased blood LPS and inflammatory cytokines. Interestingly, blood C4 and IL-10, but not acetate and valerate, were negatively associated with AD pathology, suggesting a role for SCFAs in AD pathogenesis. Aging and oxidative stress have been suggested to alter the gut microbial community and thus reduce the levels of SCFAs. Caloric restriction and dietary antioxidant supplementation can diminish AD development, in part by modulating microbial composition190,191,192. Dietary fiber and SCFAs have been reported to have beneficial effects on AD pathogenesis193,194,195. SCFA treatment decreased the polymerization of Aβ1-40 or Aβ1-42 peptides into neurotoxic multimeric Aβ forms188. Probiotic supplementation increased the hippocampal concentration of SCFAs and reduced anxiety-like behavior in a humanized AD mouse model73. The microbiota has been shown to regulate AD pathogenesis in several animal models. For example, amyloidosis with defective long-term synaptic potentiation develops in the APP/PS1 mouse model, created by overexpressing amyloid precursor protein (APP) and a mutant form of presellin 1 (PS1)196,197,198. Fecal microbiota transplantation into APP/PS1 mice increased C4 levels in the colon and increased the expression of synaptic plasticity-related proteins (PSD-95 and synapsin I) in the brain, indicating a decrease in synaptic disorder. The administration of C4-producing bacteria suppressed β-amyloid deposition and effector cytokine release from microglial cells73,195,199,200. In AD patients, the blood Kyn/Trp ratio was found to be increased due to decreased Trp levels, indicating an abnormality in Trp metabolism201. The levels of secondary bile acids such as DCA, TCA, 3-DCA, and UDCA were increased in AD patients202,203. Thus, AD patients have an imbalance in the levels of metabolites that regulate AD pathogenesis. SCFAs have both protective and pathogenic effects, Trp metabolites have protective effects, and bile acid metabolites have overall pathogenic effects on AD (Table 2).
Impacts on autism spectrum disorder
Autism spectrum disorder (ASD) is a group of developmental disabilities that involve social, behavioral and communication challenges. Both genetic and environmental factors have been identified as risk factors for the development of ASD, which is heritable. For example, chromosome 16p11.2 deletion and 15q11-q13 duplication are risk factors for ASD development. In addition, environmental factors such as maternal age, health status, infection/inflammation, perinatal hypoxia, medication, nutrition, and toxic exposure during fetal or early development are associated with ASD204,205. Pathogenic maternal immune activation of the IL-17A pathway was shown to promote ASD development in offspring206. The injection of synthetic dsRNA into pregnant dams, mimicking viral infection during pregnancy, induced placental IL-17A expression and fetal brain IL-17 receptor expression in animal models206. Th17 inflammation is associated with abnormal cortical development in animal models.
While ASD is different from MS, PD and AD in that it primarily involves CNS developmental issues rather than an ongoing inflammatory disease, a significant body of literature indicates an active role of dysbiosis in ASD. Some ASD patients report GI problems, such as abdominal pain, diarrhea, and food sensitivities207,208. Decreased levels of Firmicutes, Fusobacteria and Verrucomicrobia but increased levels of Bacteroidetes have been reported in ASD patients209. Elevated levels of Th17-inducing gut microbial species, such as segmented filamentous bacteria (SFB), Bifidobacterium adolescentis, and certain E. coli isolates, during pregnancy were shown to increase ASD development in offspring210. Intestinal CD11c+ DCs, which recognize microbial TLR3 ligands and produce IL-1β, IL-23, and IL-6, can induce pathogenic Th17 responses206,210. The significance of the microbial community in the onset of ASD has been validated in an FMT model72. When germ-free mice were humanized with microbes from ASD patient stool, their offspring showed behavioral disorders and decreased locomotion and interactions with other animals. The abundance of Bacteroides spp. and P. merdae was correlated with increased social behavior. Increased E. tayi, however, was associated with decreased social interaction in offspring72. Thus, dysbiosis can affect ASD at both the developmental and effector phases.
An altered microbial composition can impact metabolite production. It appears that microbial metabolites can bidirectionally regulate ASD activity. Decreased levels of fecal SCFAs (C2, C3, and C5) were observed in ASD patients211,212. Moreover, C4-producing bacteria and fecal SCFA levels were reported to be increased in ASD patients compared with unaffected individuals (Table 2). In a rat ASD model, C3 administration altered the patterning of human neural stem cells, leading to gliosis, disturbed neurocircuitry, inflammatory responses in the hippocampus and increased ASD-like behavior213,214. On the other hand, C4 administration improved social behavior and regulated autism-related genes related to the excitatory/inhibitory balance in the prefrontal cortex of mice215 (Table 1). Thus, the effect of SCFAs on ASD is currently equivocal. It is interesting that different SCFAs exert different effects on ASD development. Offspring from female animals that received an FMT from human ASD patients had lower levels of 5-aminovaleric acid (5AV) and taurine in the colon; these metabolites are gamma-aminobutyric acid (GABA) receptor agonists. When 5AV and taurine were administered to a mouse model of ASD (i.e., BTBR T+ tf/J mice), ASD-related behavior was suppressed72. Thus, microbial metabolites have significant regulatory effects on ASD activity.
Implications and concluding remarks
The gut is functionally linked to the CNS by immunological mediators, lymphocytes, neurotransmitters, microbes and microbial metabolites. Indeed, it is apparent that most organs and tissues in the body are regulated by gut microbiota and their metabolites, and the CNS is no exception. Importantly, because of the unique (i.e., neurological) functions of the CNS, the impact of gut microbes and their metabolites is manifested quite uniquely in this system. However, the effects of the microbiota and metabolites on different CNS diseases appear to be heterogeneous. Overall, the microbiota and their metabolites influence and strengthen CNS function in the healthy state; for example, SCFAs strengthen the BBB to maintain CNS integrity and protect the CNS from toxic and immunological damage. Various microbial metabolites regulate both inflammatory responses and neuronal activity in the CNS. Gut microbial metabolites promote the maturation of key CNS cells, such as oligodendrocytes, microglial cells and astrocytes, and regulate synapse formation. In addition, the gut microbial metabolites serotonin, dopamine, 5AV and taurine regulate neurotransmission. The production of these metabolites by microbes partly explains the changes in neurological activity following FMT from patients to experimental animals. Certain microbial metabolites, such as SCFAs, secondary bile acids and Trp metabolites, promote immune tolerance by increasing the expression of IL-10 and IL-22 to prevent inflammatory diseases. Under the highly variable pathological conditions of different diseases and hosts, SCFAs and other metabolites induce the generation of effector cells that produce neuroinflammatory cytokines. In this regard, microbial metabolites have important functions in regulating T cells, antigen-presenting cells, microglial cells and astrocytes.
We discussed the effects of microbiota and their metabolites on the pathogenesis of four different CNS diseases or disorders. A common pattern of regulation emerging from the discussed information is that disease pathogenesis is accelerated under conditions of dysbiosis (Fig. 2). While dysbiosis alone would not cause overt inflammatory responses in the CNS in normal hosts, it appears to increase pathological processes in hosts with underlying disease activity or risk factors. Changes in microbial metabolites are significant modifiers of pathogenesis in the CNS; these changes either positively or negatively affect disease onset and pathogenesis. Moreover, the altered production of microbe-derived neurotransmitters in dysbiosis appears to significantly affect neurological activity.

The common initiating factors for the four neurological diseases are genetic predisposition and environmental factors, which include diet and lifestyle. Under pathogenic conditions, the intestinal barrier can be breached, and systemic inflammatory responses can occur. These changes can be followed by dysbiosis (i.e., decreased gut microbial diversity leading to decreased levels of beneficial microbes). For example, consumption of a diet high in calories and fat but low in dietary fiber can accelerate pathogenic dysbiosis. As a result, decreased levels of beneficial gut microbial metabolites such as SCFAs, Trp metabolites and phytochemicals are produced, and simultaneously, the production of harmful metabolites such as long-chain fatty acids (LCFAs), certain bile acid metabolites, and toxic microbial metabolites increases, thereby affecting immune and tissue cells in both the intestine and CNS. These changes can decrease immune tolerance, which is important for preventing autoimmune diseases, and exacerbate pathogenic immune responses mediated by inflammatory cells such as Th17 and Th1 cells. These pathogenic inflammatory responses can contribute to tissue damage (demyelination in MS), neuronal cell death (PD and AD), and neuronal synapse development (ASD). Moreover, certain microbial metabolites regulate neurotransmission and, therefore, can directly affect neurological activity.
Despite research progress in this field, significant controversies remain regarding the functions of microbiota and their metabolites in regulating various CNS diseases. We still lack an understanding of the detailed functions of microbial metabolites. Importantly, microbes and microbial metabolites have both pro- and anti-inflammatory roles. What underlies the apparent bidirectional activities of gut microbial metabolites? SCFAs, which are largely anti-inflammatory and disease-suppressing, are sometimes associated with increased CNS disease activity. While more information is needed to elucidate the mechanisms underlying the apparent bidirectional regulation, it is clear that gut microbiota and their metabolites are significant modifiers rather than primary inducers of CNS diseases. Their effects on CNS pathogenesis can be altered depending on host factors, such as the type of inflammatory responses and stage of pathogenesis. Thus, an improved understanding of CNS regulation by microbial metabolites is required. The precise mechanisms by which these metabolites regulate immune responses individually or in combination to suppress or increase pathogenesis in the CNS have yet to be elucidated, particularly for relatively understudied diseases such as PD, AD, and ASD.
References
Sampson, T. R. & Mazmanian, S. K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 17, 565–576 (2015).
Silva, Y. P., Bernardi, A. & Frozza, R. L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 11, 25 (2020).
Lassmann, H., Brück, W. & Lucchinetti, C. F. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 17, 210–218 (2007).
Lucchinetti, C. et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707–717 (2000).
Bien, C. G. et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann. Neurol. 51, 311–318 (2002).
Misu, T. et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 125, 815–827 (2013).
Kuhlmann, T., Lassmann, H. & Brück, W. Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol. 115, 275–287 (2008).
Höftberger, R. & Lassmann, H. Inflammatory demyelinating diseases of the central nervous system. Handb. Clin. Neurol. 145, 263–283 (2017).
Bradl, M. et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 66, 630–643 (2009).
Linington, C., Bradl, M., Lassmann, H., Brunner, C. & Vass, K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am. J. Pathol. 130, 443 (1988).
Cross, A. H., Trotter, J. L. & Lyons, J.-A. B cells and antibodies in CNS demyelinating disease. J. Neuroimmunol. 112, 1–14 (2001).
Carson, M. J., Thrash, J. C. & Walter, B. The cellular response in neuroinflammation: the role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 6, 237–245 (2006).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015).
Hirsch, E. C. & Hunot, S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397 (2009).
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W. & Pardo, C. A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81 (2005).
Berer, K. et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl Acad. Sci. U. S. A. 114, 10719–10724 (2017).
Mestre, L. et al. Manipulation of gut microbiota influences immune responses, axon preservation, and motor disability in a model of progressive multiple sclerosis. Front. Immunol. 10, 1374 (2019).
Park, J., Wang, Q., Wu, Q., Mao-Draayer, Y. & Kim, C. H. Bidirectional regulatory potentials of short-chain fatty acids and their G-protein-coupled receptors in autoimmune neuroinflammation. Sci. Rep. 9, 8837 (2019).
Haase, S. et al. The role of the gut microbiota and microbial metabolites in neuroinflammation. Eur. J. Immunol. 50, 1863–1870 (2020).
Wong, J. M., De Souza, R., Kendall, C. W., Emam, A. & Jenkins, D. J. Colonic health: fermentation and short chain fatty acids. J. Clin. Gastroenterol. 40, 235–243 (2006).
Yano, J. M. et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276 (2015).
Alexeev, E. E. et al. Microbiota-derived indole metabolites promote human and murine intestinal homeostasis through regulation of interleukin-10 receptor. Am. J. Pathol. 188, 1183–1194 (2018).
Taleb, S. Tryptophan dietary impacts gut barrier and metabolic diseases. Front. Immunol. 10, 2113 (2019).
Agus, A., Planchais, J. & Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 23, 716–724 (2018).
Sayin, SamaI. et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR Antagonist. Cell Metab. 17, 225–235 (2013).
Kim, C. H. Immune regulation by microbiome metabolites. Immunology 154, 220–229 (2018).
Luu, M. et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat. Commun. 10, 760 (2019).
Parker, A., Fonseca, S. & Carding, S. R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 11, 135–157 (2020).
Macfarlane, S. & Macfarlane, G. T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 62, 67–72 (2003).
Nøhr, M. K. et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 154, 3552–3564 (2013).
Pluznick, J. L. Microbial short-chain fatty acids and blood pressure regulation. Curr. Hypertens. Rep. 19, 25 (2017).
Thangaraju, M. et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 69, 2826–2832 (2009).
Sealy, L. & Chalkley, R. The effect of sodium butyrate on histone modification. Cell 14, 115–121 (1978).
Kobayashi, M. et al. Short-chain fatty acids, GPR41 and GPR43 ligands, inhibit TNF-α-induced MCP-1 expression by modulating p38 and JNK signaling pathways in human renal cortical epithelial cells. Biochem. Biophys. Res. Commun. 486, 499–505 (2017).
Yu, X. et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J. Virol. 88, 4466–4479 (2014).
Kim, C. H., Park, J. & Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 14, 277–288 (2014).
Säemann, M. D. et al. Anti‐inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL‐12 and up‐regulation of IL‐10 production. FASEB J. 14, 2380–2382 (2000).
Sun, M. et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 9, 3555 (2018).
Kim, C. H. Control of lymphocyte functions by gut microbiota-derived short-chain fatty acids. Cell. Mol. Immunol. 18, 1161–1171 (2021).
Sepahi, A., Liu, Q., Friesen, L. & Kim, C. H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 14, 317–330 (2021).
Chun, E. et al. Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut. Immun. Immun. 51, 871–884 e876 (2019).
Goverse, G. et al. Diet-derived short chain fatty acids stimulate intestinal epithelial cells to induce mucosal tolerogenic dendritic cells. J. Immunol. 198, 2172–2181 (2017).
Chen, H. et al. A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 177, 1217–1231 e1218 (2019).
Proietti, E., Rossini, S., Grohmann, U. & Mondanelli, G. Polyamines and kynurenines at the intersection of immune modulation. Trends Immunol. 41, 1037–1050 (2020).
Wlodarska, M. et al. Indoleacrylic acid produced by commensal peptostreptococcus species suppresses inflammation. Cell Host Microbe 22, 25–37 e26 (2017).
Lamas, B. et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 22, 598–605 (2016).
Zelante, T. et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 (2013).
Rouse, M., Singh, N. P., Nagarkatti, P. S. & Nagarkatti, M. Indoles mitigate the development of experimental autoimmune encephalomyelitis by induction of reciprocal differentiation of regulatory T cells and Th17 cells. Br. J. Pharmacol. 169, 1305–1321 (2013).
Matsumoto, M. & Benno, Y. The relationship between microbiota and polyamine concentration in the human intestine: a pilot study. Microbiol. Immunol. 51, 25–35 (2007).
Landau, G., Bercovich, Z., Park, M. H. & Kahana, C. The role of polyamines in supporting growth of mammalian cells is mediated through their requirement for translation initiation and elongation. J. Biol. Chem. 285, 12474–12481 (2010).
Tersey, S. A., Colvin, S. C., Maier, B. & Mirmira, R. G. Protective effects of polyamine depletion in mouse models of type 1 diabetes: implications for therapy. Amino Acids 46, 633–642 (2014).
Chitrala, K. N., Yang, X., Nagarkatti, P. & Nagarkatti, M. Comparative analysis of interactions between aryl hydrocarbon receptor ligand binding domain with its ligands: a computational study. BMC Struct. Biol. 18, 15 (2018).
Hu, Q. et al. Ginsenosides are novel naturally-occurring aryl hydrocarbon receptor ligands. PLoS One 8, e66258 (2013).
Lee, M. J. et al. Korean red ginseng and ginsenoside-Rb1/-Rg1 alleviate experimental autoimmune encephalomyelitis by suppressing Th1 and Th17 cells and upregulating regulatory T Cells. Mol. Neurobiol. 53, 1977–2002 (2016).
Xuzhu, G. et al. Resveratrol modulates murine collagen-induced arthritis by inhibiting Th17 and B-cell function. Ann. Rheum. Dis. 71, 129–135 (2012).
Campbell, C. et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479 (2020).
Hang, S. et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148 (2019).
Reddy, B. S., Watanabe, K., Weisburger, J. H. & Wynder, E. L. Promoting effect of bile acids in colon carcinogenesis in germ-free and conventional F344 rats. Cancer Res 37, 3238–3242 (1977).
Wang, Y. D., Chen, W. D., Yu, D., Forman, B. M. & Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology 54, 1421–1432 (2011).
Sakanaka, T. et al. The effects of a TGR5 agonist and a dipeptidyl peptidase IV inhibitor on dextran sulfate sodium-induced colitis in mice. J. Gastroenterol. Hepatol. 30(Suppl 1), 60–65 (2015).
Hao, H. et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 25, 856–867 e855 (2017).
Guo, C. et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45, 944 (2016).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 (2011).
Yazici, C. et al. Race-dependent association of sulfidogenic bacteria with colorectal cancer. Gut 66, 1983–1994 (2017).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013).
Guo, Z. et al. Intestinal microbiota distinguish gout patients from healthy humans. Sci. Rep. 6, 20602 (2016).
Vogt, N. M. et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res. Ther. 10, 124 (2018).
Shao, X. et al. Uric acid induces cognitive dysfunction through hippocampal inflammation in rodents and humans. J. Neurosci. 36, 10990–11005 (2016).
Liu, Y., Hou, Y., Wang, G., Zheng, X. & Hao, H. Gut microbial metabolites of aromatic amino acids as signals in host–microbe interplay. Trends Endocrinol. Metab. 31, 818–834 (2020).
Veldhoen, M. et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 (2008).
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s Disease. Cell 167, 1469–1480 e1412 (2016).
Sharon, G. et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 177, 1600–1618 e1617 (2019).
Kaur, H. et al. Effects of probiotic supplementation on short chain fatty acids in the AppNL-G-F mouse model of Alzheimer’s Disease. J. Alzheimers Dis. 76, 1083–1102 (2020).
Haghikia, A. et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 43, 817–829 (2015).
Constantinescu, C. S., Farooqi, N., O’Brien, K. & Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 164, 1079–1106 (2011).
Sun, X., Liu, Y., Liu, B., Xiao, Z. & Zhang, L. Rolipram promotes remyelination possibly via MEK-ERK signal pathway in cuprizone-induced demyelination mouse. Exp. Neurol. 237, 304–311 (2012).
Blakemore, W. F. Demyelination of the superior cerebellar peduncle in the mouse induced by cuprizone. J. Neurol. Sci. 20, 63–72 (1973).
Pavelko, K. D., Van Engelen, B. G. & Rodriguez, M. Acceleration in the rate of CNS remyelination in lysolecithin-induced demyelination. J. Neurosci. 18, 2498–2505 (1998).
Lipton, H. L. Theiler’s virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect. Immun. 11, 1147–1155 (1975).
Liu, C., Collins, J. & Sharp, E. The pathogenesis of Theiler’s GD VII encephalomyelitis virus infection in mice as studied by immunofluorescent technique and infectivity titrations. J. Immunol. 98, 46–55 (1967).
Krishnamoorthy, G., Lassmann, H., Wekerle, H. & Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Invest. 116, 2385–2392 (2006).
Pöllinger, B. et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 206, 1303–1316 (2009).
Johanson, D. M. et al. Experimental autoimmune encephalomyelitis is associated with changes of the microbiota composition in the gastrointestinal tract. Sci. Rep. 10, 1–14 (2020).
Berer, K. et al. Dietary non-fermentable fiber prevents autoimmune neurological disease by changing gut metabolic and immune status. Sci. Rep. 8, 10431 (2018).
Lee, Y. K., Menezes, J. S., Umesaki, Y. & Mazmanian, S. K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. U. S. A. 108(Suppl 1), 4615–4622 (2011).
Ochoa-Reparaz, J. et al. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol. 183, 6041–6050 (2009).
Benedek, G. et al. Estrogen protection against EAE modulates the microbiota and mucosal-associated regulatory cells. J. Neuroimmunol. 310, 51–59 (2017).
Cekanaviciute, E. et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl Acad. Sci. U. S. A. 114, 10713–10718 (2017).
Visser, L. et al. Proinflammatory bacterial peptidoglycan as a cofactor for the development of central nervous system autoimmune disease. J. Immunol. 174, 808–816 (2005).
Ivanov, I. I. et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349 (2008).
Calcinotto, A. et al. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nat. Commun. 9, 4832 (2018).
Chen, H. et al. Gut microbiota interventions with clostridium butyricum and norfloxacin modulate immune response in experimental autoimmune encephalomyelitis mice. Front. Immunol. 10, 1662 (2019).
Ramakrishna, C. et al. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat. Commun. 10, 2153 (2019).
Ochoa-Reparaz, J. et al. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 3, 487–495 (2010).
Lee, K. et al. Bacillus-derived poly-gamma-glutamic acid reciprocally regulates the differentiation of T helper 17 and regulatory T cells and attenuates experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 170, 66–76 (2012).
Felts, P. A. et al. Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain 128, 1649–1666 (2005).
Minagar, A. & Alexander, J. S. Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. 9, 540–549 (2003).
Dopkins, N., Nagarkatti, P. S. & Nagarkatti, M. The role of gut microbiome and associated metabolome in the regulation of neuroinflammation in multiple sclerosis and its implications in attenuating chronic inflammation in other inflammatory and autoimmune disorders. Immunology 154, 178–185 (2018).
Braniste, V. et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 6, 263ra158–263ra158 (2014).
Erny, D. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977 (2015).
Chen, T., Noto, D., Hoshino, Y., Mizuno, M. & Miyake, S. Butyrate suppresses demyelination and enhances remyelination. J. Neuroinflamm. 16, 165 (2019).
Mikov, M., Kevresan, S., Kuhajda, K., Jakovljevic, V. & Vasovic, V. 3Alpha,7alpha-dihydroxy-12-oxo-5beta-cholanate as blood-brain barrier permeator. Pol. J. Pharmacol. 56, 367–371 (2004).
Greenwood, J., Adu, J., Davey, A. J., Abbott, N. J. & Bradbury, M. W. The effect of bile salts on the permeability and ultrastructure of the perfused, energy-depleted, rat blood-brain barrier. J. Cereb. Blood Flow. Metab. 11, 644–654 (1991).
Haghikia, A. et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 44, 951–953 (2016).
Mizuno, M., Noto, D., Kaga, N., Chiba, A. & Miyake, S. The dual role of short fatty acid chains in the pathogenesis of autoimmune disease models. PLoS One 12, e0173032 (2017).
Schneeman, B. O. & Gallaher, D. Changes in small intestinal digestive enzyme activity and bile acids with dietary cellulose in rats. J. Nutr. 110, 584–590 (1980).
Levrat, M.-A., Behr, S. R., Rémésy, C. & Demigné, C. Effects of soybean fiber on cecal digestion in rats previously adapted to a fiber-free diet. J. Nutr. 121, 672–678 (1991).
Fischer, F. et al. Dietary cellulose induces anti-inflammatory immunity and transcriptional programs via maturation of the intestinal microbiota. Gut Microbes 12, 1–17 (2020).
Park, J. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol. 8, 80–93 (2015).
Cox, M. A. et al. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J. Gastrol. 15, 5549–5557 (2009).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Kim, M. H., Kang, S. G., Park, J. H., Yanagisawa, M. & Kim, C. H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 145, 396–406 (2013). e391–310.
Macia, L. et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 6, 6734 (2015).
Quintana, F. J. et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71 (2008).
Quintana, F. J. et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. U. S. A. 107, 20768–20773 (2010).
Rothhammer, V. et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 (2016).
Giacoppo, S. et al. Protective role of (RS)-glucoraphanin bioactivated with myrosinase in an experimental model of multiple sclerosis. CNS Neurosci. Ther. 19, 577–584 (2013).
Saresella, M. et al. Alterations in circulating fatty acid are associated with gut microbiota dysbiosis and inflammation in multiple sclerosis. Front. Immunol. 11, 1390 (2020).
Correale, J., Gaitan, M. I., Ysrraelit, M. C. & Fiol, M. P. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 140, 527–546 (2017).
Branton, W. G. et al. Brain microbiota disruption within inflammatory demyelinating lesions in multiple sclerosis. Sci. Rep. 6, 37344 (2016).
Miyake, S. et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to Clostridia XIVa and IV Clusters. PLoS One 10, e0137429 (2015).
Zeng, Q. et al. Gut dysbiosis and lack of short chain fatty acids in a Chinese cohort of patients with multiple sclerosis. Neurochem. Int. 129, 104468 (2019).
Schrijver, I. A. et al. Bacterial peptidoglycan and immune reactivity in the central nervous system in multiple sclerosis. Brain 124, 1544–1554 (2001).
Campbell, D. J. & Butcher, E. C. Intestinal attraction: CCL25 functions in effector lymphocyte recruitment to the small intestine. J. Clin. Invest. 110, 1079–1081 (2002).
Kang, S. G., Wang, C., Matsumoto, S. & Kim, C. H. High and low vitamin A therapies induce distinct FoxP3+ T-cell subsets and effectively control intestinal inflammation. Gastroenterology 137, 1391–1402 (2009).
Liu, C. et al. Coordination between CCR7- and CCR9-mediated chemokine signals in prevascular fetal thymus colonization. Blood 108, 2531–2539 (2006).
Uehara, S., Song, K., Farber, J. M. & Love, P. E. Characterization of CCR9 expression and CCL25/thymus-expressed chemokine responsiveness during T cell development: CD3(high)CD69+ thymocytes and gammadeltaTCR+ thymocytes preferentially respond to CCL25. J. Immunol. 168, 134–142 (2002).
Wang, C., Kang, S. G., Lee, J., Sun, Z. & Kim, C. H. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol. 2, 173–183 (2009).
Kadowaki, A., Saga, R., Lin, Y., Sato, W. & Yamamura, T. Gut microbiota-dependent CCR9+CD4+ T cells are altered in secondary progressive multiple sclerosis. Brain 142, 916–931 (2019).
Miller, P. G., Bonn, M. B., Franklin, C. L., Ericsson, A. C. & McKarns, S. C. TNFR2 deficiency acts in concert with gut microbiota to precipitate spontaneous sex-biased central nervous system demyelinating autoimmune disease. J. Immunol. 195, 4668–4684 (2015).
Saresella, M. et al. Immunological and clinical effect of diet modulation of the gut microbiome in multiple sclerosis patients: a pilot study. Front. Immunol. 8, 1391 (2017).
Takewaki, D. et al. Alterations of the gut ecological and functional microenvironment in different stages of multiple sclerosis. Proc. Natl Acad. Sci. U. S. A. 117, 22402–22412 (2020).
Chen, J. et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 6, 28484 (2016).
Duscha, A. et al. Propionic acid shapes the multiple sclerosis disease course by an immunomodulatory mechanism. Cell 180, 1067–1080 (2020).
Perez-Perez, S. et al. Acetate correlates with disability and immune response in multiple sclerosis. PeerJ 8, e10220 (2020).
Moussallieh, F. M. et al. Serum analysis by 1H nuclear magnetic resonance spectroscopy: a new tool for distinguishing neuromyelitis optica from multiple sclerosis. Mult. Scler. 20, 558–565 (2014).
Crabtree, B., Souter, M. J. & Anderson, S. E. Evidence that the production of acetate in rat hepatocytes is a predominantly cytoplasmic process. Biochem. J. 257, 673–678 (1989).
Hyyppä, M. T., Jolma, T., Riekkinen, P. & Rinne, U. K. Effects of L-tryptophan treatment on central indoleamine metabolism and short-lasting neurologic disturbances in multiple sclerosis. J. Neural Transm. 37, 297–304 (1975).
Hyyppä, M. T., Jolma, T., Långvik, V.-A., Kytömäki, O. & Syvälahti, E. l-Tryptophan and neuroendocrine regulation in neurologic patients: Hormone responses to l-tryptophan loading in patients with hypothalamic lesions. Psychoneuroendocrinology 2, 349–357 (1977).
Gaetani, L. et al. Host and microbial tryptophan metabolic profiling in multiple sclerosis. Front . Immunol. 11, 157 (2020).
Lim, C. K. et al. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 7, 41473 (2017).
Nourbakhsh, B. et al. Altered tryptophan metabolism is associated with pediatric multiple sclerosis risk and course. Ann. Clin. Transl. Neurol. 5, 1211–1221 (2018).
Bhargava, P. et al. Bile acid metabolism is altered in multiple sclerosis and supplementation ameliorates neuroinflammation. J. Clin. Invest. 130, 3467–3482 (2020).
Lieben, C. K. et al. Intake of tryptophan-enriched whey protein acutely enhances recall of positive loaded words in patients with multiple sclerosis. Clin. Nutr. 37, 321–328 (2018).
Gorman, M. P., Healy, B. C., Polgar-Turcsanyi, M. & Chitnis, T. Increased relapse rate in pediatric-onset compared with adult-onset multiple sclerosis. Arch. Neurol. 66, 54–59 (2009).
Tremlett, H. & Waubant, E. The gut microbiota and pediatric multiple sclerosis: recent findings. Neurotherapeutics 15, 102–108 (2018).
Heath-Pagliuso, S. et al. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry 37, 11508–11515 (1998).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 (2011).
Wang, J. et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 281, 22021–22028 (2006).
Rothhammer, V. et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018).
Maetzler, W. & Berg, D. Parkinson disease in 2017: changing views after 200 years of Parkinson disease. Nat. Rev. Neurol. 14, 70–72 (2018).
Singh, M. et al. Polymorphism in environment responsive genes and association with Parkinson disease. Mol. Cell. Biochem. 312, 131–138 (2008).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Winner, B. et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl Acad. Sci. U. S. A. 108, 4194–4199 (2011).
Kustrimovic, N. et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflamm. 15, 205 (2018).
Chen, S. et al. Increased abundance of myeloid-derived suppressor cells and Th17 cells in peripheral blood of newly-diagnosed Parkinson’s disease patients. Neurosci. Lett. 648, 21–25 (2017).
Hernandez, D. G., Reed, X. & Singleton, A. B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 139(Suppl 1), 59–74 (2016).
Fujioka, S. et al. Occurrence of Crohn’s disease with Parkinson’s disease. Parkinsonism Relat. Disord. 37, 116–117 (2017).
Franke, A. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 42, 1118–1125 (2010).
Unger, M. M. et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 32, 66–72 (2016).
Keshavarzian, A. et al. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 30, 1351–1360 (2015).
Perez-Pardo, P. et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 68, 829–843 (2019).
Hou, Y. F. et al. Gut microbiota-derived propionate mediates the neuroprotective effect of osteocalcin in a mouse model of Parkinson’s disease. Microbiome 9, 34 (2021).
Bordoni, L. et al. Positive effect of an electrolyzed reduced water on gut permeability, fecal microbiota and liver in an animal model of Parkinson’s disease. PLoS One 14, e0223238 (2019).
Li, P. et al. Gut microbiota dysbiosis is associated with elevated bile acids in Parkinson’s Disease. Metabolites 11, 29 (2021).
Shao, Y. et al. Comprehensive metabolic profiling of Parkinson’s disease by liquid chromatography-mass spectrometry. Mol. Neurodegener. 16, 4 (2021).
Cirstea, M. S. et al. Microbiota composition and metabolism are associated with gut function in Parkinson’s Disease. Mov. Disord. 35, 1208–1217 (2020).
Funayama, M. et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann. Neurol. 57, 918–921 (2005).
Park, J. et al. Parkinson disease-associated LRRK2 G2019S transgene disrupts marrow myelopoiesis and peripheral Th17 response. J. Leukoc. Biol. 102, 1093–1102 (2017).
Ekstrand, M. I. et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl Acad. Sci. U. S. A. 104, 1325–1330 (2007).
Liu, J. et al. Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J. Neurol. Sci. 381, 176–181 (2017).
Johnson, J. Jr et al. Valproate and sodium butyrate attenuate manganese-decreased locomotor activity and astrocytic glutamate transporters expression in mice. Neurotoxicology 64, 230–239 (2018).
Kidd, S. K. & Schneider, J. S. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 194, 189–194 (2011).
Aho, V. T. E. et al. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol. Neurodegener. 16, 6 (2021).
Qiao, C. M. et al. Sodium butyrate exacerbates Parkinson’s disease by aggravating neuroinflammation and colonic inflammation in MPTP-Induced Mice Model. Neurochem. Res. 45, 2128–2142 (2020).
Park, J., Goergen, C. J., HogenEsch, H. & Kim, C. H. Chronically elevated levels of short-chain fatty acids induce T cell-mediated ureteritis and hydronephrosis. J. Immunol. 196, 2388–2400 (2016).
Graham, S. F. et al. Metabolomic profiling of bile acids in an experimental model of prodromal Parkinson’s Disease. Metabolites 8, 71 (2018).
Cuevas, E. et al. Tauroursodeoxycholic acid (TUDCA) is neuroprotective in a chronic mouse model of Parkinson’s disease. Nutr. Neurosci. 1–18 (2020).
Castro-Caldas, M. et al. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol. Neurobiol. 46, 475–486 (2012).
Duan, W. M., Rodrigues, C. M., Zhao, L. R., Steer, C. J. & Low, W. C. Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson’s disease. Cell Transpl. 11, 195–205 (2002).
Rosa, A. I. et al. Tauroursodeoxycholic acid improves motor symptoms in a mouse model of Parkinson’s Disease. Mol. Neurobiol. 55, 9139–9155 (2018).
Moreira, S. et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 295, 77–87 (2017).
Mortiboys, H. et al. UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology 85, 846–852 (2015).
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A. & Nair, M. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 14, 5541–5554 (2019).
Fujii, Y. et al. Fecal metabolite of a gnotobiotic mouse transplanted with gut microbiota from a patient with Alzheimer’s disease. Biosci. Biotechnol. Biochem. 83, 2144–2152 (2019).
Nagpal, R. et al. Gut mycobiome and its interaction with diet, gut bacteria and alzheimer’s disease markers in subjects with mild cognitive impairment: a pilot study. EBioMedicine 59, 102950 (2020).
Marizzoni, M. et al. Short-chain fatty acids and lipopolysaccharide as mediators between gut dysbiosis and amyloid pathology in Alzheimer’s Disease. J. Alzheimers Dis. 78, 683–697 (2020).
Ho, L. et al. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert. Rev. Neurother. 18, 83–90 (2018).
Liu, S., Gao, J., Zhu, M., Liu, K. & Zhang, H. L. Gut microbiota and dysbiosis in Alzheimer’s Disease: implications for pathogenesis and treatment. Mol. Neurobiol. 57, 5026–5043 (2020).
Luchsinger, J. A., Tang, M.-X., Shea, S. & Mayeux, R. Caloric intake and the risk of Alzheimer Disease. Arch. Neurol. 59, 1258–1263 (2002).
Engelhart, M. J. et al. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 287, 3223–3229 (2002).
Spychala, M. S. et al. Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann. Neurol. 84, 23–36 (2018).
Govindarajan, N., Agis-Balboa, R. C., Walter, J., Sananbenesi, F. & Fischer, A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimers Dis. 26, 187–197 (2011).
Martins, I. J. & Fernando, W. M. A. D. B. High fibre diets and Alzheimer’s Disease. Food Nutr. Sci. 05, 410–424 (2014).
Sun, J. et al. Effect of Clostridium butyricum against microglia-mediated neuroinflammation in Alzheimer’s Disease via regulating gut microbiota and metabolites Butyrate. Mol. Nutr. Food Res. 64, e1900636 (2020).
Da Silva, S. V. et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A 2A receptors. Nat. Commun. 7, 1–11 (2016).
Spires, T. L. & Hyman, B. T. Transgenic models of Alzheimer’s disease: learning from animals. NeuroRx 2, 423–437 (2005).
Trinchese, F. et al. Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 55, 801–814 (2004).
Colombo, A. V. et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 10, e59826 (2021).
Sun, J. et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatr. 9, 189 (2019).
Widner, B. et al. Tryptophan degradation and immune activation in Alzheimer’s disease. J. Neural Transm. (Vienna) 107, 343–353 (2000).
MahmoudianDehkordi, S. et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimers Dement 15, 76–92 (2019).
Baloni, P. et al. Metabolic network analysis reveals altered bile acid synthesis and metabolism in Alzheimer’s Disease. Cell Rep. Med 1, 100138 (2020).
Lord, C., Elsabbagh, M., Baird, G. & Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 392, 508–520 (2018).
Bölte, S., Girdler, S. & Marschik, P. B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell. Mol. Life Sci. 76, 1275–1297 (2019).
Choi, G. B. et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939 (2016).
Chaidez, V., Hansen, R. L. & Hertz-Picciotto, I. Gastrointestinal problems in children with autism, developmental delays or typical development. J. Autism Dev. Disord. 44, 1117–1127 (2014).
Madra, M., Ringel, R. & Margolis, K. G. Gastrointestinal issues and autism spectrum disorder. Child Adolesc. Psychiatr. N. Am. 29, 501–513 (2020).
De Angelis, M. et al. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One 8, e76993 (2013).
Kim, S. et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532 (2017).
Adams, J. B., Johansen, L. J., Powell, L. D., Quig, D. & Rubin, R. A. Gastrointestinal flora and gastrointestinal status in children with autism – comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 11, 22 (2011).
Liu, S. et al. Altered gut microbiota and short chain fatty acids in Chinese children with autism spectrum disorder. Sci. Rep. 9, 1–9 (2019).
Choi, J. et al. Pathophysiological and neurobehavioral characteristics of a propionic acid-mediated autism-like rat model. PLoS One 13, e0192925 (2018).
Abdelli, L. S., Samsam, A. & Naser, S. A. Propionic acid induces gliosis and neuro-inflammation through modulation of PTEN/AKT pathway in Autism Spectrum Disorder. Sci. Rep. 9, 8824 (2019).
Kratsman, N., Getselter, D. & Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 102, 136–145 (2016).
Hammer, A. et al. Impact of combined sodium chloride and saturated long-chain fatty acid challenge on the differentiation of T helper cells in neuroinflammation. J. Neuroinflamm. 14, 184 (2017).
Platten, M. et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 310, 850–855 (2005).
Sun, M. F. et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-alpha signaling pathway. Brain Behav. Immun. 70, 48–60 (2018).
Shin, C., Lim, Y., Lim, H. & Ahn, T. B. Plasma short-chain fatty acids in patients With Parkinson’s Disease. Mov. Disord. 35, 1021–1027 (2020).
Chen, S. J. et al. The gut metabolite trimethylamine N-oxide is associated with Parkinson’s Disease severity and progression. Mov. Disord. 35, 2115–2116 (2020).
Coretti, L. et al. Gut microbiota features in young children with Autism Spectrum Disorders. Front. Microbiol. 9, 3146 (2018).
Wang, L. et al. Elevated fecal short chain fatty acid and ammonia concentrations in children with Autism Spectrum Disorder. Dig. Dis. Sci. 57, 2096–2102 (2012).
Acknowledgements
CHK is the Judy and Kenneth Betz Endowed Professor in the Mary H Weiser Food Allergy Center.
Funding
This study was supported, in part, by the NIH (R01AI121302, R21AI14889801, R01AI074745, and R01AI080769 to CHK) and the National Research Foundation of Korea (2020R1G1A1099715 to JP).
Author information
Authors and Affiliations
Contributions
CHK and JP collected information and wrote the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, J., Kim, C.H. Regulation of common neurological disorders by gut microbial metabolites. Exp Mol Med 53, 1821–1833 (2021). https://doi.org/10.1038/s12276-021-00703-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00703-x
This article is cited by
-
The Role of Gut-derived Short-Chain Fatty Acids in Multiple Sclerosis
NeuroMolecular Medicine (2024)
-
Gut Microbiota Modulation and Its Implications on Neuropathic Pain: A Comprehensive Literature Review
Pain and Therapy (2024)
-
Roles of bile acids signaling in neuromodulation under physiological and pathological conditions
Cell & Bioscience (2023)
-
Role of neuroinflammation in neurodegeneration development
Signal Transduction and Targeted Therapy (2023)
-
Small molecule metabolites: discovery of biomarkers and therapeutic targets
Signal Transduction and Targeted Therapy (2023)
