
Abstract
The mechanisms underlying atrial fibrillation (AF), a type of heart arrhythmia, have not been fully identified. Long noncoding RNAs (lncRNAs) have been implicated in the progression of AF. The current study aimed to ascertain the means by which X-inactive specific transcript (XIST), a lncRNA, contributes to the pathogenesis of AF in an animal model or in atrial myocytes. Extracellular vesicles (EVs) derived from mouse adipose tissue-derived mesenchymal stem cells (AMSCs) were isolated, transfected with XIST, and either injected into AF mouse models or incubated with atrial myocytes. The in vitro and in vivo effects of EV-derived XIST on myocardial pyroptosis were determined by Western blot analysis of pyroptosis-related protein and an ELISA for inflammatory factors. Bioinformatics analysis revealed a relationship between XIST, microRNA (miR)−214-3p, and Arl2, which was subsequently verified by a dual luciferase assay and RNA immunoprecipitation. Functional experiments were performed to elucidate whether changes in miR-214-3p or Arl2 regulated the effect of XIST on myocardial pyroptosis. Overexpressed XIST from AMSC-EVs were found to decrease myocardial pyroptosis while alleviating inflammation, which was demonstrated by reduced expression of nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3), apoptosis-associated speck-like protein containing a CARD (ASC), cleared-caspase-1/caspase-1 and gasdermin D (GSDMD), as well as the amount of interleukin (IL)-1β and IL-18 in both the cardiomyocytes and AF mouse tissues. Mechanistically, XIST is a competing endogenous RNA (ceRNA) of miR-214-3p, triggering upregulation of its target gene Arl2. Silencing of Arl2 or overexpression miR-214-3p reversed the effects of XIST on inflammation and pyroptosis. Taken together, the key findings of our study suggest that XIST may blunt myocardial pyroptosis by absorbing miR-214-3p to promote Arl2 expression, providing encouraging insight into XIST-based targeted therapy for AF.
Similar content being viewed by others


Introduction
Atrial fibrillation (AF) is one of the most common clinical arrhythmias, occurring in fewer than 1% of people aged 60−65 years but in 8−10% of those older than 80 years [1] and has been reported to increase the risk of heart failure and hospitalization [2]. Current treatment modalities have brought about a distinct improvement in AF patient survival, however, the long-term prognosis remains poor with improved targeted therapy required [3]. Although significant studies into the pathogenesis of AF have provided a basis for present-day treatments, current anti-arrhythmia drugs are not effective enough, perhaps due to the fact that the mechanism of AF development is still not completely clear [4]. Further exploration into the pathogenesis of AF is required to identify novel biomarkers.
Mesenchymal stem cells (MSCs) are pluripotent stem cells for tissue repair and have received extensive attention in the field of regenerative medicine [5]. Paracrine extracellular vesicles (EVs) EVs contain numerous RNAs with various functions, including messenger RNAs (mRNAs), circular RNAs (circRNAs), and long noncoding RNAs (lncRNAs) [6]. EVs have been implicated in the development of several diseases including cancer [7] and cardiovascular diseases [8]. The most significant complication associated with AF is stroke, and several studies have suggested that EVs or EVs from MSCs may potentially alleviate or facilitate the progression of the disorders [9, 10]. Exosomal lncRNAs have been reported to contribute to tumor progression, immunomodulation, and cardiac disease [9, 11]. X-inactive specific transcript (XIST), a lncRNA encoded by XIST gene, inactivates mammal X chromosome [12]. The downregulation of XIST activates microRNA (miRNA)-mediated pyroptotic cell death [13]. At the time of writing, no reports exist regarding the function of XIST in AF.
Inflammation represents a crucial biological process, comprised of an acute response to infection and tissue damage harmful to the host [14]. Inflammasomes are a multi-protein complex, usually consisting of three proteins, such as NOD-like receptor (NLR), apoptosis-associated speck-like protein containing a CARD (ASC), and caspase-1 [15]. To date, the most thoroughly studied is the nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome [16]. When the innate immune system recognizes a series of inflammatory stimuli, such as pathogen-associated molecular patterns and damage-associated molecular patterns, the NLRP3 inflammasome in the cells activates caspase-1, promoting the production of the pro-inflammatory cytokine interleukin (IL)−1β and IL-18, ultimately leading to cell pyroptosis, inflammatory programmed cell death [17, 18]. Pyroptosis has been documented as a form of programmed lytic cell death initiated by inflammasomes, featuring with membrane rupture that is a consequence of a gasdermin pore and may be most effective in defense against bacteria evolving in hosts without inflammasomes [19]. Mechanistically, pyroptosis occurs due to two major signaling pathways: one mediated by caspase-1 and the other by caspase-4/5/11, and targeting the pyroptosis signaling pathways, highlighting their potential as therapeutic targets in cardiovascular diseases [20]. Although the role of pyroptosis in AF has been examined [21], the finer molecular mechanism underlying this process remains unclear.
In order to further clarify the role of XIST in AF, we established cell and mouse AF models, and found that adipose tissue-derived mesenchymal stem cell (AMSCs)-derived EVs containing XIST inhibited myocardial cell pyroptosis. The function of XIST was mediated by downstream miR-214-3p/Arl2. Based on these observations, targeting XIST/miR-214-3p/Arl2 may be a useful strategy for the protection of cardiomyocytes in AF.
Methods
Ethics statement
The current study was performed with the approval of the Ethics Committee of Affiliated Hospital of Nantong University and performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Extensive efforts were made to ensure minimal suffering as well as the number of animals used during the study.
AMSC isolation
Subcutaneous adipose tissues from the male C57BL/6 mice were collected, rinsed repeatedly with phosphate-buffered saline (PBS) to remove the blood and connective tissues, followed by digestion with an equal volume of 0.2% type I collagenase in a constant temperature water bath (37 °C). After filtering through a 200 μm metal screen and centrifugation, the pellet was resuspended in MesenPROR RSTM medium (1% antibiotic−antimycotic, Gibco BRL, Grand Island, NY, USA). The suspension was seeded into a culture flask in a 37 °C, 5% CO2 incubator, and passaged until 90% confluence was confirmed.
Flow cytometry
Cells were digested with trypsin and incubated with subsequent antibodies from BD Biosciences Pharmingen (San Diego, CA, USA) at 4 °C in the dark for 30 min, including phycoerythrin (PE)-labeled CD105 (Cat No.560839), PE-labeled CD106 (Cat No.555647), PE-labeled CD44 (Cat No.555479), fluorescein isothiocyanate (FITC)-labeled CD45 (Cat No.561874), FITC-labeled CD34 (Cat No.555821), and FITC-labeled CD90 (Cat No.555595). The cells were subsequently centrifuged at 700 r/min for 5 min, and resuspended in 300 µL PBS, followed by detection with a flow cytometer (Merck-Millipore, Germany).
Identification of the differentiation ability of AMSCs
Upon reaching 80% confluence, AMSCs at passage 3 were induced and cultured with adipogenic differentiation medium (Gibco). After 14 days, AMSCs were stained with Oil Red O staining and observed under an inverted phase-contrast microscope to assess adipogenic differentiation. In terms of osteogenic differentiation evaluation, the cells were cultured in an osteogenic medium (Gibco) for 21 days followed by Alizarin Red S staining to detect the calcification deposits. For chondrogenic differentiation, 1 × 106 cells were centrifuged in a 15 mL centrifuge tube and then transferred to a chondrogenic medium (Gibco). After 4 weeks, the cells were fixed with 4% paraformaldehyde and cut into 10 µm sections. The sections were stained with Toluidine blue and observed under a microscope.
Isolation and identification of AMSC-derived EVs
Fetal bovine serum (FBS) was centrifuged at 100 000 × g for 7 h at 4°C, after which the supernatant was removed in order to prepare an EV-free complete medium, where the AMSCs at passage 3 in the logarithmic growth phase were cultured for 48 h. The EVs in the supernatant were extracted via density gradient centrifugation, and stored at −80°C for subsequent use. Western blot analysis was performed to detect EV surface markers, including CD9 (1: 2000, ab92726, Abcam Inc., Cambridge, UK), CD63 (Cat No. 564221, BD Biosciences Pharmingen, San Diego, CA, USA), TSG101 (Cat No. 612697, BD), and Calnexin (Cat No. 610524, BD). AMSC-Dulbecco’s modified Eagle’s medium precipitation was regarded as a negative control (NC). The EVs were observed with Tecnai™ Spirit (T12) transmission electron microscope (HiLLsboro, Oregon, USA), with their size and concentration determined using a nanoparticle tracking analysis (NTA) with MaLvern’s NanoSight NS300. A Bicinchoninic acid (BCA) protein detection kit (Pierce Biotechnology Inc., Rockford, IL, USA) was used to determine the EV protein content. After the EVs had been incubated in Rnase and/or Triton X-100-treated medium, the presence of XIST inside the EVs was determined.
EV labeling
Quantified EV suspension (100 μL, 20 µg) was mixed with 1 mL of diluted PKH67 (4 µL/mL, Sigma-Aldrich Chemical Company, St Louis, MO, USA) and incubated at room temperature for 4 min. Next, 1 mL of 0.5% bovine serum albumin (BSA) was added to the suspension EV re-extraction, which was stained with PKH67 (green) under a fluorescence microscope. The PKH67-labeled EVs were incubated with HL-1 cells stained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) for 12 h. PKH67-labeled EVs were injected into the mouse atrium.
Culture and rapid pacing of atrial myocytes HL-1
HL-1 atrial myocytes from adult mice were obtained from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (Shanghai, China) and maintained in complete Claycomb medium (Sigma-Aldrich) with 10% FBS (PAA Laboratories GmbH, Austria), 100 U/mL penicillin (Gibco, Landsmeer, The Netherlands), 100 µg/mL streptomycin (Gibco), 4 mM L-glutamine (Gibco), 0.3 mM L-ascorbic acid (Sigma-Aldrich), and 100 µM norepinephrine (Sigma-Aldrich). HL-1 cells were cultured on coverslips coated with 0.02% gelatin (Sigma-Aldrich) in 5% CO2 at 37°C. C-Pace100TM-culture pacer (IonOptix Corporation, Amsterdam, The Netherlands) was employed to perform normal pacing (1 Hz, 40 V, and 20 ms pulse as a normal group) or rapid pacing (500 times/min rapid atrial stimulation, 6 Hz, 40 V, and 20 ms pulse as a model group) on HL-1 cells.
5-ethynyl-2′-deoxyuridine (EdU) incorporation assay
The EdU incorporation assay was performed in accordance with the manufacturer’s instructions (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA). Briefly, the HL-1 cells were fixed in 4% paraformaldehyde for 15 min, immersed in 1% Triton X-100 for 20 min, and incubated with EdU for 1.5 h. Following three PBS washes, the click-it reaction mixture was added to the cells and cultured at room temperature for 30 min. The cells were stained with Hoechst 33342 for 30 min, and observed under a fluorescence microscope (Olympus, Tokyo, Japan). The proportion of EdU-positive cells was calculated and expressed as proliferation rates.
AF induction
A total of 120 male C57BL/6 mice (24.2 ± 2.1 g and 8 weeks old) purchased from JOINN Laboratories (Suzhou, China), were randomly divided into 10 groups (n = 12 per group) and were separately housed in cages under conditions with 20−26 °C temperature and 40−70% humidity, exposed to a 12-h light/dark cycle and provided with free access to food and water. Wild-type (WT) mice were subjected to pentobarbital sodium (40 mg/kg) for anesthesia purposes with artificial respiration provided with body temperatures monitored (37°C) throughout the experiment. AF in mice was induced based on a previously reported method [22]. The sham-operated mice underwent jugular vein cannulation with no atrial pacing. AF induction was defined as a fast irregular atrial rhythm lasting at least 1 s.
Injection of EVs and transduction of lentivirus in mice
EVs (10 μg) or PBS were injected at different three sites in the right atrium using a 31-gauge Hamilton syringe. For the transduction of lentiviral vectors, mice were anesthetized, fixed, intubated, and ventilated. Then, thoracic surgery was performed in the 3−4 intercostal space in order to expose the right atrium, with 10 μL 108 TU of lentivirus short hairpin RNA (sh)-Arl2 or sh-NC, lentivirus (lenti)-miR-214-3p or lenti-NC injected at different injection points. At 48 h post skin suture, the transfection efficiency was detected by quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot analysis.
Enzyme-linked immunosorbent assay (ELISA)
The IL-1β and IL-18 levels in the mouse serum, as well as the IL-1β (#DY401) and IL-18 (#7625) levels in HL-1 in the cell supernatant, were determined using an ELISA kit 24 h post cell transfection (R&D Systems, Minneapolis, MN, USA), based on the manufacturer’s instructions.
Immunofluorescence staining
Paraffin-embedded heart sections were incubated with primary antibody Ki67 (1: 500, ab15580, Abcam) at 4 °C overnight followed by Alexa Fluor® 488-conjugated secondary antibody (1: 500, ab150077, Abcam) at room temperature for 1 h. After PBS washing, the sections were stained by DAPI, and imaged under a fluorescence microscope with three fields randomly selected to calculate the cell proliferation rate.
HL-1 cells were treated with 4% paraformaldehyde for 20 min, blocked with 1% BSA and 0.1% Triton-X for 2 h at room temperature, and incubated with primary antibody anti-NLRP3 (1: 100, ab9722, Abcam), caspase-1 (1: 100, ab138483, Abcam), and IL-1β (1: 200, # MAB7578, R&D Systems) at 4 °C overnight and subsequent secondary antibodies at room temperature for 1 h. The nuclei were stained with DAPI (Shanghai Beyotime Biotechnology Co. Ltd., Shanghai, China). The immunostaining results were analyzed by Image-Pro Plus 6.0.
Immunohistochemistry
The right atrium specimen was fixed with 4% paraformaldehyde, embedded in paraffin, sectioned, and incubated with primary antibodies: anti-NLRP3 (1: 100, ab9722, Abcam), caspase-1 (1: 100, ab138483, Abcam), and IL-1β (1: 200, # MAB7578, R&D Systems) overnight at 4 °C and secondary antibody. The sections were subsequently observed under a microscope.
Cell transduction
HL-1 cells were transduced with lentiviral vectors expressing the full-length mouse XIST, sh-XIST, sh-Arl2, or an empty lentiviral vector from Genechem Company (Shanghai, China) using the TransDux MAX Lentivirus Transduction Reagent (System Biosciences, Palo Alto, CA, USA) and polybrene (8 μg/mL; Sinopharm) at a multiplicity of infection between 20 and 30. The HL-1 cells were re-transduced with lentiviral vectors expressing lenti-anti-miR-214-3p or lenti-miR-214-3p obtained from the Genechem Company at a multiplicity of infection between 15 and 20. Forty-eight hours later, the cells were collected and subjected to a selection procedure in a culture medium containing blasticidin (10 μg/mL, Sinopharm) for 72 h.
Dual-luciferase reporter assay
The DNA sequence on XIST gene containing miR-214-3p binding site was subcloned into a psiCHECK-2 vector (Promega, Madison, WI, USA) to construct a luciferase plasmid Luc-XIST-WT. Alternatively, the DNA sequence on the XIST gene, containing a mutated miR-214-3p binding site was also subcloned into the luciferase vector in an attempt to create another luciferase plasmid Luc-XIST-mutant (MUT). The constructed luciferase plasmids were co-transfected with miR-214-3p mimic (GenePharma Co., Ltd., Shanghai, China) or mimic-NC into the HEK293T cells. Synthesized WT and MUT of 3′-untranslated region (3′-UTR) Arl2 genes were inserted into the luciferase reporter vector pmirGLO (Promega). The luciferase reporter plasmids WT and MUT were co-transfected into HEK293 cells with miR-214-3p mimic or mimic NC, respectively. After 48 h, relative luciferase activity was determined using a Dual-Luciferase Reporter Assay (Promega) and standardized to renin luciferase activity.
RNA binding protein immunoprecipitation (RIP) assay
The cells were collected and lysed in a complete RIP lysis buffer using an EZ-Magna RIP Kit (Millipore, Massachusetts, USA). The cell extract was subsequently incubated with RIP buffer containing magnetic beads bound to anti-Ago2 antibody (1:50, ab32381, Abcam), followed by incubation with proteinase K to isolate immunoprecipitated RNA. The concentration of the RNA was determined using a nanodrop spectrophotometer, with the purified RNA subsequently subjected to qPCR analysis.
qRT-PCR
Total RNA was extracted using TRIzol reagent (TAKARA, Dalian, China) to synthesize complementary DNA (cDNA) via reverse transcription using an RT-qPCR mixRNA PCR Core Kit (Applied Biosystems, Foster City, CA, USA). The synthesized cDNA was then subjected to qRT-PCR detection using the Fast SYBR Green PCR kit (Applied Biosystems) and ABI7900/illumina (Applied Biosystems), with three replicates set for each well. The miRcute plus miRNA first-strand cDNA synthesis kit (Tianjin Tianjian Biotechnology Co., Ltd., Beijing, China) and miRcute Plus miRNA PCR detection kit (TIANGEN) were employed for reverse transcription and quantification of miRNA, respectively. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was regarded as an internal reference for the standardization of both XIST and mRNA values, while U6 was employed as the internal reference for normalization of the miR-214-3p values. The relative RNA level was evaluated using the 2−ΔΔCt method. The primer design is illustrated in Supplementary Table 1.
Western blot analysis
The HL-1 cells and atrial tissues of each group were collected by trypsin digestion, and subsequently lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (Boster Biological Technology Co., Ltd., Wuhan, Hubei, China) containing protease inhibitors followed by concentration determination using a BCA protein quantification kit (Boster). Equal amounts of protein were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane and blocked with BSA. The membranes were subsequently blotted with diluted primary antibodies: NLRP3 (1: 500, ab214185, Abcam), ASC (1: 1000, ab47092, Abcam), cleaved-caspase-1 (1: 1000, # 893325, Cell Signaling Technology, Beverly, MA, USA), Caspase−1 (1: 1000, # 24232, Cell signaling technology), cleaved-gasdermin D (GSDMD) (1: 1000, ab255603, Abcam), GSDMD (1: 1000, ab209845, Abcam), Arl2 (1: 1000, ab183510, Abcam), and GAPDH (1: 1000, ab8245, Abcam, internal reference) overnight at 4 °C and horseradish peroxidase (HRP)-labeled goat anti-rabbit secondary antibody (ab205719; 1: 2000; Abcam) at room temperature. The signal was detected using enhanced chemiluminescence reagents (Amersham GE Healthcare, Chicago, Illinois, United States) and quantified by density meter (Syngene, GenetooLs). Each experiment was repeated three times.
Microarray-based gene expression profiling
The TargetScan (http://www.targetscan.org/mamm_31/) and starBase databases (http://starbase.info/) were explored to predict the potential target mRNAs of miR-214-3p. Briefly, the specific sequence of miR-214-3p was used as the input in the corresponding website. Based on the prediction score and the prediction intersection of the two databases, potential binding targets were selected for subsequent verification.
Statistical analysis
Data were analyzed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Measurement data were expressed as the mean ± standard deviation. Data obeying normal distribution and homogeneity of variance between two groups were compared using an unpaired t-test while data among multiple groups were compared using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests with corrections for multiple comparisons. p < 0.05 was considered to be indicative of a statistically significant difference.
Results
Identification of AMSCs and AMSC-derived EVs
In this study, mice AMSCs were isolated from subcutaneous adipose tissue. Inverted phase-contrast microscopy revealed that the primary cells grew in an adherent manner following 2−3 days of culture, and reached 80% confluence after 7 days. The third-generation AMSCs had a uniform long spindle shape, and were arranged in a spiral shape with dense growth. Cell surface markers were used to identify AMSCs. The results indicated that the expression of CD44, CD90, and CD105 was positive and that of CD106, CD34 and CD45 was negative in AMSCs (Fig. 1A), indicating that the AMSCs had indeed been successfully isolated from mouse adipose tissues. Adipogenic, osteogenic, and chondrogenic induction was performed for the AMSCs. The Oil Red O staining results indicated that a greater number of lipid droplets had formed and merged in the cytoplasm (Fig. 1B). Additionally, Alizarin Red S staining and Toluidine blue staining suggested that AMSCs had the potential of osteogenic and chondrogenic differentiation (Fig. 1C, D). Furthermore, our results revealed no evidence of the endoplasmic reticulum marker protein Calnexin, but CD9, CD63, and TSG101 were expressed in AMSC-derived EVs (Fig. 1E). Transmission electron microscopy observed that AMSC-derived EVs exhibited typical exosome morphology (Fig. 1F). These results indicated the successful isolation of EVs.

A Morphology of AMSCs was observed by an inverted phase-contrast microscope and surface markers were identified by flow cytometry. B Oil Red O staining observation after the third generation AMSCs had been induced for 14 days (×400). C Alizarin Red S staining observation after the third-generation AMSCs had been induced for 21 days (×200). D Toluidine blue staining observation after the third-generation AMSCs had been induced for 4 weeks (×200). E Western blot analysis of EV surface markers CD9, CD63, and TSG101 and endoplasmic reticulum marker protein Calnexin. F Morphology of EVs was observed under transmission electron microscopy (scale bar, 0.5 µm).
EV-derived XIST inhibited NLRP3 activation and myocardial pyroptosis in mice with AF
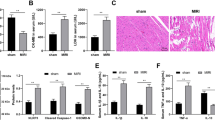
EVs are predominantly responsible for intercellular communication by transferring miRNAs, lncRNA, and proteins. It is reported that XIST can reduce H9c2 cardiomyocyte damage [23]. However, at present no evidence regarding the significance of AMSC-EV-derived XIST in AF. We evaluated the expression of XIST in the culture medium from AMSCs. qRT-PCR detection found that the expression of XIST in the culture medium decreased significantly after the addition of RNase plus Triton X-100 (Fig. 2A), indicating that XIST was in the membrane structure. Then, a plasmid expressing XIST was transfected into AMSCs cells, and its EVs were extracted. qRT-PCR demonstrated the expression of XIST was markedly higher post transfection (Fig. 2B). Immunofluorescence revealed PKH67 green fluorescence in the atrial tissue section of mice injected with PKH67-labeled EVs and no fluorescence in that with PBS (Fig. 2C), indicating the transfer of EVs. The mice were subsequently injected with EVs containing XIST (XIST EVs), NC EVs, EVs, or PBS. AF was induced in order to verify the effect of EV-derived XIST on AF. The qRT-PCR results demonstrated that the mice with AF had downregulated XIST, while upregulated XIST was detected in the AF mice administered with EV injection or XIST EV injection (Fig. 2D). Pyroptosis is a type of programmed cell death, characterized by the maturation of caspase-1, eventually leading to the cleavage of pore-forming protein GSDMD, and IL-1β and IL-18 release [24]. Inflammasome activation, particularly NLRP3 inflammasome, is related to pyroptosis [25]. The qRT-PCR and Western blot analysis illustrate that mice with AF exhibited upregulated NLRP3, ASC, caspase-1, and GSDMD, while AF mice with EV injection and with XIST EV injection exhibited downregulated NLRP3, ASC, caspase-1, and GSDMD (Fig. 2E, F). ELISA demonstrated upregulated IL-1β and IL-18, in mice with AF while AF mice with EV injection and with XIST EV injection exhibited downregulated levels of IL-1β and IL-18 (Fig. 2G). Immunohistochemistry assay results further confirmed that mice with AF exhibited upregulated IL-1β and NLRP3, while AF mice with EV injection and those with XIST EV injection exhibited diminished levels of IL-1β and NLRP3 (Supplementary Fig. 1A). Additionally, mice with AF exhibited reduced Ki67-positive cells, while AF mice with EV injection and with XIST EV injection displayed increased Ki67-positive cells (Supplementary Fig. 1B). The aforementioned results provided evidence verifying that EV-derived XIST attenuated NLRP3 inflammatory corpuscle activation and myocardial pyroptosis in mice with AF. Data related to the mouse model of AF are shown in the Supplementary Material.

Detection of XIST in ASMCs culture medium (A) and in EVs (B) through qRT-PCR. C Mouse atrial tissue sections were observed under immunofluorescence microscopy. D Detection of XIST expression in mouse atrial tissue through qRT-PCR. E Detection of expression of NLRP3, ASC, caspase-1, and GSDMD in mouse atrial tissue through qRT-PCR. F Detection of NLRP3, ASC, cleaved-caspase-1/caspase-1, and cleaved-GSDMD/GSDMD in mouse atrial tissue through Western blot analysis. G Detection of IL-1β and IL-18 by ELISA. *p < 0.05, ***p < 0.001 versus sham-operated mice; #p < 0.05 versus AF mice with PBS treatment; &p < 0.05 versus AF mice with NC EV injection. Data in multiple groups were compared by one-way ANOVA with Tukey’s post hoc test, n = 12 per group.
EV-derived XIST attenuated NLRP3 inflammasome activation and pyroptosis in HL-1 cells
Next, to elucidate the effect of EV-derived XIST on inflammatory response and pyroptosis of HL-1 cells, AMSCs were treated with conditioned medium (CM), and EV inhibitor GW4869 to inhibit the production and release of EVs (GW4869 + CM). The culture supernatant of the two groups of cells was collected and added to the HL-1 cell culture medium, after which pyroptosis-related factors were detected. The qRT-PCR, Western blot analysis, and ELISA revealed that HL-1 cells treated with GW4869 + CM exhibited higher expression levels of NLRP3, ASC, caspase-1, GSDMD, cleaved-caspase-1/caspase-1, cleaved-GSDMD/GSDMD, IL-1β, and IL-18 than those treated with CM alone (Fig. 3A−C). Immunochemistry provided further indication that HL-1 cells treated with GW4869 + CM had increased NLRP3, caspase-1, and IL-1β expression (Supplementary Fig. 2A). Additionally, HL-1 cells treated with GW4869 + CM exhibited a decreased number of EdU-positive cells (Supplementary Fig. 2B). Taken together, the effect of EVs on reducing the inflammatory response of HL-1 cells was reversed by GW4869, highlighting that the release of EVs can inhibit the activation of NLRP3 inflammasome and pyroptosis in HL-1 cells. Moreover, the flow cytometry results suggested that the uptake was increased as the culture time increased with saturated uptake occurring after 24 h of incubation (Fig. 3D).

A NLRP3, ASC, caspase-1, and GSDMD mRNA expression in HL-1 cells treated with CM and GW4869 + CM detected by qRT-PCR. B Protein expression of H, NLRP3, ASC, cleaved-caspase-1/caspase-1, and cleaved-GSDMD/GSDMD in HL-1 cells treated with CM and GW4869 + CM detected by Western blot analysis. C Detection of pro-inflammatory factors IL-1β and IL-18 in the supernatant of HL-1 cells treated with CM and GW4869 + CM by ELISA. D Flow cytometric analysis of EV uptake by HL-1 cells with the duration of the culture. E Detection of XIST expression in HL-1 cells treated with EVs, NC EVs, XIST EVs, or controls by qRT-PCR. F qRT-PCR analysis of NLRP3, ASC, caspase-1, and GSDMD mRNA expression in HL-1 cells treated with EVs, NC EVs, XIST EVs, or controls. G Western blot analysis of NLRP3, ASC, cleaved-caspase-1/caspase-1, and cleaved-GSDMD/GSDMD protein expression in HL-1 cells treated with EVs, NC EVs, XIST EVs, or controls. H Detection of pro-inflammatory factors IL-1β and IL-18 by ELISA in the supernatant of HL-1 cells treated with EVs, NC EVs, XIST EVs, or controls. *p < 0.05 versus normal HL-1 cells (normal or CM); #p < 0.05 versus HL-1 cells receiving AF induction and PBS; &p < 0.05 versus HL-1 cells receiving AF induction and NC EVs. Comparison between two groups was analyzed by unpaired t-test and among multiple groups was by one-way ANOVA with Tukey’s post hoc test. All experiments were repeated three times with similar results.
In order to explore the effect associated with XIST EVs on pyroptosis of HL-1 cells, XIST EVs, NC EVs, EVs, or PBS were used to incubate with HL-1 cells. HL-1 cells receiving AF induction exhibited downregulated XIST and upregulated NLRP3, ASC, caspase-1, GSDMD, cleaved-caspase-1/caspase-1, cleaved-GSDMD/GSDMD, IL-1β, and IL-18, while HL-1 cells receiving AF induction and EVs/XIST EVs incubation exhibited upregulated XIST while NLRP3, ASC, caspase-1, GSDMD, cleaved-caspase-1/caspase-1, cleaved-GSDMD/GSDMD, IL-1β, and IL-18 were all downregulated (Fig. 3E−H). Immunofluorescence staining provided further evidence confirming that the HL-1 cells that underwent AF induction had elevated NLRP3, caspase-1, and IL-1β, while in the HL-1 cells that underwent AF induction and EVs/XIST EVs incubation had decreased NLRP3, caspase-1, and IL-1β (Supplementary Fig. 2C). Additionally, HL-1 cells receiving AF induction exhibited reduced EdU-positive cells, while HL-1 cells receiving AF induction and EVs/XIST EVs incubation exhibited increased EdU-positive cells (Supplementary Fig. 2D). Moreover, decreased XIST in EVs reduced the attenuating effect of EVs on NLRP3 inflammasome activation and pyroptosis of HL-1 cells (Supplementary Fig. 2E−G). Altogether, the aforementioned results indicated that EV-derived XIST attenuated the activation of NLRP3 inflammasome and pyroptosis in HL-1 cells.
XIST increases Arl2 expression by acting as an endogenous RNA (ceRNA) for miR-214-3p in HL-1 cells
LncRNAs are said to function as miRNA sponges [26, 27]. Hence, we predicted the potential miRNAs, binding to XIST through bioinformatics analysis. Following the exploration of the starBase database, a potential binding site was found between XIST and miR-214-3p (Fig. 4A). We set out to ascertain whether XIST interacted with miR-214-3p in vitro. As depicted in Fig. 4B, the co-transfection of miR-214-3p mimic and XIST WT exhibited inhibited luciferase activity. The relationship between XIST and miR-214-3p with AGO2 was further verified by anti-AGO2 and RIP (Supplementary Fig. 3A). Furthermore, TargetScan indicated a potential binding site between miR-214-3p and 3′-UTR of Arl2 (Fig. 4C). As shown in Fig. 4D, co-transfection of miR-214-3p mimic and Arl2 WT exhibited reduced luciferase activity. We subsequently performed qRT-PCR and Western blot analysis methods to detect miR-214-3p and Arl2 expression in HL-1 cells. As illustrated in Fig. 4E, F, increased miR-214-3p and decreased Arl2 were detected in the HL-1 cells with AF induction, while EV and XIST EV treatment inhibited miR-214-3p and promoted Arl2, which was consistent with their expression levels in mouse myocardium (Supplementary Fig. 3B, C). To further elucidate the function of miR-214-3p in HL-1 cells, miR-214-3p was silenced, which led to a significant increase in the expression of XIST and Arl2 (Fig. 4G, H). Taken together, these results supported the idea that XIST increases the expression of Arl2 in HL-1 cells by absorbing miR-214-3p.

A StarBase database showed that XIST and miR-214-3p had potential binding sites. B Relative luciferase activity after co-transfection of XIST-WT/MUT and miR-214-3p/NC mimic into HEK293T cells. C TargetScan database predicted that Arl2 and miR-214-3p had potential binding sites. D Relative luciferase activity after co-transfection of Arl2-WT/MUT and miR-214-3p/NC mimic into HEK293T cells. E miR-214-3p expression in HL-1 cells detected by qRT-PCR. F Arl2 expression in HL-1 cells detected by qRT-PCR. G miR-214-3p, XIST, and Arl2 expression in HL-1 cells detected by qRT-PCR. H Arl2 expression in HL-1 cells detected by Western blot analysis. *p < 0.05 versus HEK293T transfected with NC mimic, HL-1 cells, or HL-1 cells receiving AF induction and NC inhibitor; #p < 0.05 versus HL-1 cells receiving AF induction and PBS; &p < 0.05 versus HL-1 cells receiving AF induction and NC EVs. Comparison between two groups was analyzed by unpaired t-test and among multiple groups was by one-way ANOVA with Tukey’s post hoc test. All experiments were repeated three times with similar results.
EV-derived XIST repressed pyroptosis of HL-1 cells by downregulating miR-214-3p
Next, to ascertain the impact of the interaction between XIST and miR-214-3p on AF, we transfected HL-1 cells with lenti-miR-214-3p or lenti-NC, and then the cells were incubated with XIST EVs or NC EVs, followed by rapid pacing induction. As illustrated in Fig. 5A, B, treatment with miR-214-3p led to a decrease in the expression of XIST and Arl2 in HL-1 cells while increasing miR-214-3p expression. Treatment with XIST EVs triggered a decrease in the expression of miR-214-3p while increasing XIST and Arl2 expression. Relative to XIST EVs + NC, XIST EVs + miR-214-3p exhibited a decrease XIST and Arl2 expression but increased miR-214-3p expression. Besides, activation of NLRP3 inflammasome was assessed by qRT-PCR and Western blot analysis. As a sequence, exogenously expressed XIST EVs decreased expression of NLRP3, ASC, caspase-1, GSDMD, while treatment of miR-214-3p exerted opposite activity, increasing expression of the molecules (Fig. 5C, D). Additionally, the addition of XIST EVs declined the expression of NLRP3, caspase-1, and IL-1β and IL-18 as well as the number of EdU-positive cells, while exogenously expressed miR-214-3p rescued the effects of XIST (Fig. 5E−G). The above results suggest that XIST downregulated miR-214-3p leading to an attenuation of pyroptosis and NLRP3 activation in HL-1 cells.

A qRT-PCR analysis of miR-214-3p, XIST, and Arl2 in HL-1 cells upon treatment with XIST EVs, miR-214-3p, or NC EVs. B Western blot analysis of XIST, and Arl2 in HL-1 cells upon treatment with XIST EVs, miR-214-3p, or NC EVs. C qRT-PCR analysis of NLRP3, ASC, caspase-1, and GSDMD in HL-1 cells upon treatment with XIST EVs, miR-214-3p, or NC EVs. D Western blot analysis of NLRP3, ASC, cleaved-caspase-1/caspase-1, and GSDMD in HL-1 cells upon treatment with XIST EVs, miR-214-3p, or NC EVs and corresponding quantification. E ELISA of IL-1β and IL-18 in HL-1 cells upon treatment with XIST EVs, miR-214-3p, or NC EVs. F Immunofluorescence of NLRP3, caspase-1, and IL-1β in HL-1 cells upon treatment with XIST EVs, miR-214-3p, or NC EVs. G EdU of the proliferation of HL-1 cells upon treatment with XIST EVs, miR-214-3p, or NC EVs. *p < 0.05 versus HL-1 cells receiving AF induction and NC EVs; #p < 0.05 versus HL-1 cells receiving AF induction and XIST EVs by one-way ANOVA with Tukey’s post hoc test. Error bars indicate mean ± standard deviation. All experiments were repeated three times with similar results.
EV-derived XIST suppressed pyroptosis of HL-1 cells by upregulating Arl2
We subsequently set out to evaluate whether the interaction between Arl2 and XIST attenuated pyroptosis and NLRP3 activation. HL-1 cells were transfected with sh-Arl2 or sh-NC, and incubated with XIST EVs or NC EVs, followed by rapid pacing induction. It was clear that treatment with sh-Arl2 led to a reduction in the expression of Arl2 and XIST EVs resulted in increased Arl2 expression, while both treatments of XIST EVs + sh-Arl2 reduced Arl2 expression (Fig. 6A, B). As depicted in Fig. 6C, D, the inhibitory effect of XIST EVs on NLRP3/ASC/caspase-1/GSDMD was reversed following Arl2 silencing. ELISA results indicated that XIST EVs treatment led to a decrease in the expression of NLRP3, caspase-1, and IL-1β and IL-18 as well as EdU-positive cells while additional treatment with sh-Arl2 restored the expression of NLRP3, caspase-1, and IL-1β and IL-18 as well as the number of EdU-positive cells (Fig. 6E−G). The above results indicated that XIST attenuates pyroptosis of HL-1 cells through upregulation of Arl2.

A qRT-PCR analysis of miR-214-3p, XIST, and Arl2 in HL-1 cells upon treatment with XIST EVs, sh-Arl2, or NC EVs. B Western blot analysis of XIST, and Arl2 in HL-1 cells upon treatment with XIST EVs, sh-Arl2, or NC EVs. C qRT-PCR analysis of NLRP3, ASC, caspase-1, and GSDMD in HL-1 cells upon treatment with XIST EVs, sh-Arl2, or NC EVs. D Western blot analysis of NLRP3, ASC, cleaved-caspase-1/caspase-1, and GSDMD in HL-1 cells upon treatment with XIST EVs, sh-Arl2, or NC EVs and corresponding quantification. E ELISA of IL-1β and IL-18 in HL-1 cells upon treatment with XIST EVs, sh-Arl2, or NC EVs. F Immunofluorescence of NLRP3, caspase-1, and IL-1β in HL-1 cells upon treatment with XIST EVs, miR-sh-Arl2, or NC EVs. G EdU of the proliferation of HL-1 cells upon treatment with XIST EVs, sh-Arl2, or NC EVs. *p < 0.05 versus HL-1 cells receiving AF induction and NC EVs; #p < 0.05 versus HL-1 cells receiving AF induction and XIST EVs by one-way ANOVA with Tukey’s post hoc test. Error bars indicate mean ± standard deviation. All experiments were repeated three times with similar results.
EV-derived XIST downregulated miR-214-3p and activated Arl2 to attenuate cardiac pyroptosis in mice with AF
In vivo experiments were performed to further elucidate the mechanism by which XIST functions in AF through mediating miR-214-3p and Arl2. EVs containing XIST were injected and lentiviral vectors (sh-Arl2/sh-NC, lenti-miR-214-3p/lenti-NC) were transduced into AF mice. qRT-PCR and Western blot analysis demonstrated that relative to the XIST EVs and NC, combined treatment of miR-214-3p and XIST EVs led to an increase in the expression of miR-214-3p and a decrease in the content of XIST and Arl2 (Fig. 7A, B). Based on XIST EVs, the treatment with miR-214-3p or sh-Arl2 increased the content of NLRP3, ASC, caspase-1, and GSDMD (Fig. 7C, D). Besides, in the presence of miR-214-3p or sh-Arl2, the expression of NLRP3, caspase-1, and IL-1β and IL-18 reduced by XIST EVs was elevated significantly (Fig. 7E, F). Consistently, immunofluorescence staining demonstrated that treatment with either miR-214-3p or sh-Arl2 led to a marked decline in the number of Ki67-positive cells (Fig. 7G). The aforementioned findings indicated that XIST attenuates cardiac pyroptosis via the miR-214-3p/Arl2 axis.

A qRT-PCR analysis of miR-214-3p, XIST, and Arl2 in HL-1 cells upon treatment with XIST EVs, sh-Arl2, miR-214-3p, or NC EVs. B Western blot analysis of XIST, and Arl2 in HL-1 cells upon treatment with XIST EVs, sh-Arl2, miR-214-3p, or NC EVs. C qRT-PCR analysis of NLRP3, ASC, caspase-1, and GSDMD in HL-1 cells upon treatment with XIST EVs, miR-214-3p, sh-Arl2, or NC EVs. D Western blot analysis of NLRP3, ASC, cleaved-caspase-1/caspase-1, and GSDMD in HL-1 cells upon treatment with XIST EVs, sh-Arl2, miR-214-3p, or NC EVs and corresponding quantification. E ELISA of IL-1β and IL-18 in HL-1 cells upon treatment with XIST EVs, sh-Arl2, miR-214-3p, or NC EVs. F Immunocytochemical staining of NLRP3, caspase-1, and IL-1β in HL-1 cells upon treatment with XIST EVs, miR-sh-Arl2, miR-214-3p, or NC EVs. G Representative images of Ki67 immunofluorescence staining of atrial tissues upon treatment with XIST EVs, sh-Arl2, miR-214-3p, or NC EVs. *p < 0.05 versus mice receiving AF, XIST EVs, and NC; #p < 0.05 versus mice receiving AF, XIST EVs, and sh-NC by one-way ANOVA with Tukey’s post hoc test, n = 12 per group. Error bars indicate mean ± standard deviation.
Discussion
Recent studies highlight the efficacy of MSC-exosome-based therapy in animal models of cardiovascular diseases [28, 29]. However, relatively little is known regarding the role of lncRNAs derived from MSC-exosome in the process of myocardial pyroptosis in patients with AF. The present study provides evidence, indicating low levels of XIST expression in cardiomyocytes subjected to rapid pacing, and EVs secreted by AMSCs transferred XIST to cardiomyocytes. The overexpression of XIST in the cardiomyocytes was found to promote the expression of Arl2 by inhibiting miR-214-3p, ultimately inhibiting myocardial pyroptosis (Fig. 8).

Schematic illustration of the proposed mechanism: the EVs from AMSCs containing XIST suppressed the activation of NLRP3 inflammasome and the myocardial cell pyroptosis in AF by impairing miR-214-3p-mediated Arl2 inhibition.
Various mechanisms have been proposed in relation to the development of AF. Studies have indicated that more cases of postoperative AF are being observed due to an increase in cardiac surgeries [30]. It has been reported that activated NLRP3 inflammasome signaling in cardiomyocytes can induce AF [31]. In addition, pyroptosis has been reported to contribute to the damage of cardiomyocytes and pyroptosis, highlighting its potential as a therapeutic target for cardiovascular diseases [24, 25]. Pyroptosis represents a type of programmed cell death associated with inflammation [32, 33], characterized by cell swelling, cell membrane damage, and leakage of proinflammatory cytokines IL-1β/IL-18 [34, 35]. AF progression is often associated with a strong inflammatory response [21, 36]. A strong correlation between pyroptosis and various human diseases, including atherosclerosis and diabetic nephropathy has been reported, with the proposed mechanism suggested to involve the regulation of certain non-coding RNAs and other different kinds of molecules, which also influences the proliferation, invasion, and metastasis of tumors [37]. However, the exact mechanism underlying the role of pyroptosis in AF has not been extensively investigated.
Therapeutic applications of MSC-derived extracellular vesicles have been proposed [38]. The key mechanism associated with the anti-inflammatory effect of MSCs is the secretion of EVs, delivering functional small RNAs [39]. Circulating EVs offer a noninvasive and virtually continuous entrance to circulating information on disease state, with the application of EVs suggested to be a promising novel therapeutic target for various cardiovascular diseases [40]. MSC-EV injections in rats have been reported to reduce apoptosis as well as myocardial infarct size, improving heart function following stroke [41]. Previous reports have suggested that exosomes derived from AMSCs transfer miR-320d to inhibit cardiomyocyte apoptosis in AF [42]. Existing literature has highlighted the key role played by lncRNAs in various biological processes, including cell proliferation, migration, inflammation, apoptosis, and autophagy with studies suggesting that lncRNAsa can regulate gene expression by competitively binding miRNAs [43,44,45]. Notably, lncRNAs have been widely implicated in the fundamental mechanisms that contribute to the development of AF, including electrical remodeling, structural remodeling, renin-angiotensin system effects, and calcium handling abnormalities, rendering them a therapeutic target for the treatment and control of AF [46]. The greater majority of research on XIST has focused on its functions in human cancers and diseases. The downregulation of XIST has been reported to inhibit the development of non-small-cell lung cancer by activating miR-335/SOD2/ROS axis-mediated pyroptosis [13]. XIST inhibits the activation of the NLRP3 inflammasome of bovine mammary epithelial cells [47]. Reports have indicated that XIST represses hypoxia-induced H9c2 cardiomyocyte injury by means of regulating the miR-122-5p/FOXP2 axis [23]. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analyses in a recent study highlighted XIST as well as five other factors that play notable roles in the pathophysiology of AF owing to their increased intramodular connectivity [48]. However, the specific mechanism of XIST in AF has not been reported. During the present study, HL-1 cells and mice were subjected to rapid pacing and they exhibited NLRP3 inflammasome activation, upregulated pyroptosis-related markers, and downregulated lncRNA XIST. By transfecting plasmids expressing XIST into AMSCs followed by extraction of EVs, we identified that XIST attenuates myocardial pyroptosis in AF, highlighting promising biomarkers and potential therapeutic targets for treating patients with AF.
Analysis using the starBase database revealed that miR-214-3p was a potential target of XIST while its level in HL-1 cells that had been subjected to rapid pacing was significantly increased. Luciferase reporter assay results indicated that XIST interacted with miR-214-3p in a direct manner while XIST was also found to downregulate miR-214-3p, thereby attenuating myocardial pyroptosis. Consistent with our results, a recent study has reported that miR-214-3p is a target of XIST and that XIST can bind to miR-214-3p to reduce the expression of miR-214-3p [49]. The implication of miRNAs in the control of AF development manifests in two distinct instances: firstly, the aberrant miRNA expression has been widely detected in animal models as well as in patients with AF, both in cardiac tissues as well as blood. Changes in the expression of miRNAs in cardiac tissues have been emphasized as a potential sign of remodeling, while the alteration in blood highlights the potential value of miRNAs as diagnostic and prognostic markers; secondly, a direct causal correlation between AF and individual miRNAs has been established, whereby alterations in the expression of miRNA can either enhance or reduce AF susceptibility in animal models, indicating the promise in targeting miRNAs as a potential approach for the management of AF [50]. miR-214-3p is significantly upregulated in the serum of AF patients, suggesting it may be a potential biomarker for AF [51]. Furthermore, the miR-214-3p/caspase-1 axis has been linked with pyroptosis [52, 53]. The current study is the first to report the XIST/miR-214-3p axis as a novel pathway involved in regulating pyroptosis following AF, providing novel insights into the pyroptosis of cardiomyocytes. Li-Min Long et al asserted that miR-214 negatively regulates the expression of Arl2 by means of directly targeting its 3′-UTR, while the overexpression of miR-214 has been reported to trigger the apoptosis of colon cancer cells [54]. Arl2 represents a member of the ADP-ribosylation factor family of GTP-binding proteins [55]. Previous studies have suggested that Arl2-knockdown leads to G1/S phase arrest and inhibition of cell proliferation [56]. Our results indicated that Arl2 was a potential target gene for miR-214-3p. The expression of Arl2 was elevated following the overexpression of XIST. Hence, we believe that XIST regulates the miR-214-3p/Arl2 axis to attenuate myocardial pyroptosis in vitro and in vivo.
In conclusion, the key findings of the present study highlight the role of XIST as a key regulator of myocardial pyroptosis. AMSC-derived EVs transfer XIST into cardiomyocytes, where XIST performs as a ceRNA to interact with miR-214-3p, thereby reducing the inhibitory effect of miR-214-3p on the expression of Arl2. Our results provide insight into the role of XIST/miR-214-3p/Arl2 in AF-induced myocardial pyroptosis and revealed XIST as a potential target for therapeutic intervention. However, additional studies are required to further establish a positive correlation between XIST and Arl2. Specimens from patients diagnosed with AF are also essential in validating these findings and to further elucidate their translational potential.
Data availability
The datasets generated analyzed during the current study are available from the corresponding author upon reasonable request.
References
Zimetbaum P. Atrial fibrillation. Ann Intern Med. 2017;166:ITC33–ITC48.
Seccia TM, Caroccia B, Maiolino G, Cesari M, Rossi GP. Arterial hypertension, aldosterone, and atrial fibrillation. Curr Hypertens Rep. 2019;21:94.
Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. 2014;114:1453–68.
Lau DH, Linz D, Sanders P. New findings in atrial fibrillation mechanisms. Card Electrophysiol Clin. 2019;11:563–71.
Keshtkar S, Azarpira N, Ghahremani MH. Mesenchymal stem cell-derived extracellular vesicles: novel frontiers in regenerative medicine. Stem Cell Res Ther. 2018;9:63.
Li Y, Zhao J, Yu S, Wang Z, He X, Su Y, et al. Extracellular vesicles long RNA sequencing reveals abundant mRNA, circRNA, and lncRNA in human blood as potential biomarkers for cancer diagnosis. Clin Chem. 2019;65:798–808.
Xie F, Zhou X, Fang M, Li H, Su P, Tu Y, et al. Extracellular vesicles in cancer immune microenvironment and cancer immunotherapy. Adv Sci. 2019;6:1901779.
Hafiane A, Daskalopoulou SS. Extracellular vesicles characteristics and emerging roles in atherosclerotic cardiovascular disease. Metabolism. 2018;85:213–22.
Bang OY, Kim EH. Mesenchymal stem cell-derived extracellular vesicle therapy for stroke: challenges and progress. Front Neurol. 2019;10:211.
Thulin A, Lindback J, Granger CB, Wallentin L, Lind L, Siegbahn A. Extracellular vesicles in atrial fibrillation and stroke. Thromb Res. 2020;193:180–9.
Kenneweg F, Bang C, Xiao K, Boulanger CM, Loyer X, Mazlan S, et al. Long noncoding RNA-enriched vesicles secreted by hypoxic cardiomyocytes drive cardiac fibrosis. Mol Ther Nucleic Acids. 2019;18:363–74.
Weakley SM, Wang H, Yao Q, Chen C. Expression and function of a large non-coding RNA gene XIST in human cancer. World J Surg. 2011;35:1751–6.
Liu J, Yao L, Zhang M, Jiang J, Yang M, Wang Y. Downregulation of LncRNA-XIST inhibited development of non-small cell lung cancer by activating miR-335/SOD2/ROS signal pathway mediated pyroptotic cell death. Aging. 2019;11:7830–46.
Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. 2013;13:59–66.
Kanneganti TD. The inflammasome: firing up innate immunity. Immunol Rev. 2015;265:1–5.
He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–21.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35.
Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–84.
Jia C, Chen H, Zhang J, Zhou K, Zhuge Y, Niu C, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. 2019;67:311–8.
Chen G, Chelu MG, Dobrev D, Li N. Cardiomyocyte inflammasome signaling in cardiomyopathies and atrial fibrillation: mechanisms and potential therapeutic implications. Front Physiol. 2018;9:1115.
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–87.
Peng H, Luo Y, Ying Y. lncRNA XIST attenuates hypoxia-induced H9c2 cardiomyocyte injury by targeting the miR-122-5p/FOXP2 axis. Mol Cell Probes. 2020;50:101500.
Mao Q, Liang XL, Zhang CL, Pang YH, Lu YX. LncRNA KLF3-AS1 in human mesenchymal stem cell-derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR-138-5p/Sirt1 axis. Stem Cell Res Ther. 2019;10:393.
Zhang L, Liu H, Jia L, Lyu J, Sun Y, Yu H, et al. Exosomes mediate hippocampal and cortical neuronal injury induced by hepatic ischemia-reperfusion injury through activating pyroptosis in rats. Oxid Med Cell Longev. 2019;2019:3753485.
Wei R, Zhang L, Hu W, Wu J, Zhang W. Long non-coding RNA AK038897 aggravates cerebral ischemia/reperfusion injury via acting as a ceRNA for miR-26a-5p to target DAPK1. Exp Neurol. 2019;314:100–10.
Cheng Y, Geng L, Wang K, Sun J, Xu W, Gong S, et al. Long noncoding RNA expression signatures of colon cancer based on the ceRNA network and their prognostic value. Dis Markers. 2019;2019:7636757.
Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013;10:301–12.
Witwer KW, Van Balkom BWM, Bruno S, Choo A, Dominici M, Gimona M, et al. Defining mesenchymal stromal cell (MSC)-derived small extracellular vesicles for therapeutic applications. J Extracell Vesicles. 2019;8:1609206.
Nair SG. Atrial fibrillation after cardiac surgery. Ann Card Anaesth. 2010;13:196–205.
Yao C, Veleva T, Scott L Jr., Cao S, Li L, Chen G, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. 2018;138:2227–42.
Broz P. Immunology: caspase target drives pyroptosis. Nature. 2015;526:642–3.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109.
de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6:a016287.
Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–39.
Harada M, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J. 2015;79:495–502.
Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121:109595.
Rani S, Ryan AE, Griffin MD, Ritter T. Mesenchymal stem cell-derived extracellular vesicles: toward cell-free therapeutic applications. Mol Ther. 2015;23:812–23.
Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci. 2014;15:4142–57.
Jansen F, Nickenig G, Werner N. Extracellular vesicles in cardiovascular disease: potential applications in diagnosis, prognosis, and epidemiology. Circ Res. 2017;120:1649–57.
Liu L, Jin X, Hu CF, Li R, Zhou Z, Shen CX. Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy Via AMPK and Akt pathways. Cell Physiol Biochem. 2017;43:52–68.
Liu L, Zhang H, Mao H, Li X, Hu Y. Exosomal miR-320d derived from adipose tissue-derived MSCs inhibits apoptosis in cardiomyocytes with atrial fibrillation (AF). Artif Cells Nanomed Biotechnol. 2019;47:3976–84.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14.
Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–504.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Babapoor-Farrokhran S, Gill D, Rasekhi RT. The role of long noncoding RNAs in atrial fibrillation. Heart Rhythm. 2020;17:1043–9.
Ma M, Pei Y, Wang X, Feng J, Zhang Y, Gao MQ. LncRNA XIST mediates bovine mammary epithelial cell inflammatory response via NF-kappaB/NLRP3 inflammasome pathway. Cell Prolif. 2019;52:e12525.
Li W, Wang L, Wu Y, Yuan Z, Zhou J. Weighted gene coexpression network analysis to identify key modules and hub genes associated with atrial fibrillation. Int J Mol Med. 2020;45:401–16.
Feng Y, Wan P, Yin L. Long noncoding RNA X-inactive specific transcript (XIST) promotes osteogenic differentiation of periodontal ligament stem cells by sponging MicroRNA-214-3p. Med Sci Monit. 2020;26:e918932.
Luo X, Yang B, Nattel S. MicroRNAs and atrial fibrillation: mechanisms and translational potential. Nat Rev Cardiol. 2015;12:80–90.
Natsume Y, Oaku K, Takahashi K, Nakamura W, Oono A, Hamada S, et al. Combined analysis of human and experimental murine samples identified novel circulating microRNAs as biomarkers for atrial fibrillation. Circ J. 2018;82:965–73.
Yang F, Li A, Qin Y, Che H, Wang Y, Lv J, et al. A novel circular RNA mediates pyroptosis of diabetic cardiomyopathy by functioning as a competing endogenous RNA. Mol Ther Nucleic Acids. 2019;17:636–43.
Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018;9:1000.
Long LM, He BF, Huang GQ, Guo YH, Liu YS, Huo JR. microRNA-214 functions as a tumor suppressor in human colon cancer via the suppression of ADP-ribosylation factor-like protein 2. Oncol Lett. 2015;9:645–50.
Kahn RA, Volpicelli-Daley L, Bowzard B, Shrivastava-Ranjan P, Li Y, Zhou C, et al. Arf family GTPases: roles in membrane traffic and microtubule dynamics. Biochem Soc Trans. 2005;33:1269–72.
Wang K, Li P, Dong Y, Cai X, Hou D, Guo J, et al. A microarray-based approach identifies ADP ribosylation factor-like protein 2 as a target of microRNA-16. J Biol Chem. 2011;286:9468–76.
Acknowledgements
We acknowledge and appreciate our colleagues for their valuable efforts and comments on this paper.
Funding
This study was supported by Nantong Municipal Science and Technology Plan (guidance) Project in 2019 (JCZ19058).
Author information
Authors and Affiliations
Contributions
BY, TL, and CY participated in the conception and design of the study. XL performed the analysis and interpretation of data. QD and BY contributed to drafting the article. CY revised it critically for important intellectual content. LP is the GUARANTOR for the article who accepts full responsibility for the work and/or the conduct of the study, had access to the data, and oversaw the decision to publish.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The current study was performed with the approval of the Ethics Committee of Affiliated Hospital of Nantong University and performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Extensive efforts were made to ensure minimal suffering as well as the number of animals used during the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yan, B., Liu, T., Yao, C. et al. LncRNA XIST shuttled by adipose tissue-derived mesenchymal stem cell-derived extracellular vesicles suppresses myocardial pyroptosis in atrial fibrillation by disrupting miR-214-3p-mediated Arl2 inhibition. Lab Invest 101, 1427–1438 (2021). https://doi.org/10.1038/s41374-021-00635-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-021-00635-0
This article is cited by
-
LncRNA XIST facilitates hypertrophy of ligamentum flavum by activating VEGFA-mediated autophagy through sponging miR-302b-3p
Biology Direct (2023)
-
Long non-coding RNA and circular RNA: new perspectives for molecular pathophysiology of atrial fibrillation
Molecular Biology Reports (2023)
-
Current Approaches in Cardiac Repair: Somatic and Stem Cell Exosomes
Current Treatment Options in Cardiovascular Medicine (2023)
-
Can Extracellular Vesicles as Drug Delivery Systems Be a Game Changer in Cardiac Disease?
Pharmaceutical Research (2023)
-
Long Non-coding RNA Involved in the Pathophysiology of Atrial Fibrillation
Cardiovascular Drugs and Therapy (2023)
