
Abstract
Pleomorphic (PLCIS) and florid (FLCIS) lobular carcinoma in situ are rare histologic variants of LCIS that are considered more aggressive than classic LCIS (CLCIS), but optimal treatment is controversial. The genetic drivers of these lesions and their clonal relationships to paired CLCIS and ILC have not been characterized. We used capture-based next-generation sequencing to profile 16 LCIS variants (ten PLCIS, six FLCIS), including paired synchronous ILC and CLCIS in 11 and nine cases, respectively. Recurrent pathogenic alterations included CDH1 (9/10 PLCIS, 6/6 FLCIS), PIK3CA (7/10 PLCIS, 2/6 FLCIS), ERBB2 (6/10 PLCIS, 2/6 FLCIS; six mutations, two amplifications), ERBB3 (1/10 PLCIS, 2/6 FLCIS), FOXA1 (4/10 PLCIS, 1/6 FLCIS), TP53 (3/10 PLCIS), and CCND1 (2/10 PLCIS, 1/6 FLCIS). Mutational profiles and mean copy number alterations (CNA) were similar between LCIS variants with and without ILC. Compared with ILC in The Cancer Genome Atlas (TCGA), PLCIS, FLCIS, and associated ILC were enriched for ERBB2 mutations, and PLCIS was enriched for TP53 and FOXA1 mutations. Shared pathogenic mutations and CNA were identified between the LCIS variant and ILC in all cases, and between CLCIS and the LCIS variant/ILC in 89%. CLCIS to PLCIS progression was associated with increased mean nonsynonymous mutations and additional pathogenic alterations and/or CNA in 80%. Mean nonsynonymous mutations and CNA were similar between PLCIS and ILC, although additional pathogenic mutations were associated with invasion in a subset (43%). FLCIS harbored additional clonal pathogenic mutations in only 1/3 cases, and these were not shared with ILC, which was genetically divergent. In another case, ILC was genetically more similar to CLCIS than FLCIS. The results highlight clonal relationships between PLCIS/FLCIS and CLCIS, and implicate PLCIS as a genetically advanced ILC precursor. Frequent ERBB2/ERBB3 alterations in PLCIS and FLCIS are consistent with more aggressive behavior and may have prognostic and therapeutic implications.
Similar content being viewed by others
Introduction
Classic lobular carcinoma in situ (CLCIS) is often an incidental and multifocal finding that has long been considered a risk factor for development of invasive carcinoma and is managed accordingly. Pleomorphic (PLCIS) and florid (FLCIS) lobular carcinoma in situ are rare LCIS variants that can be distinguished from CLCIS histologically based on the presence of increased nuclear pleomorphism in PLCIS [1, 2] and marked ductal and lobular distention in FLCIS [3,4,5] with or without comedo-type necrosis. In contrast to CLCIS, PLCIS and FLCIS are often the targeted lesion on core biopsy and show high upgrade rates to invasive carcinoma or DCIS on surgical excision [6,7,8,9,10,11,12,13,14]. Indeed, PLCIS and FLCIS have been considered higher risk lesions than CLCIS, based on a more aggressive biomarker profile (more often ER− and/or HER2+), increased numbers of chromosomal copy number alterations (CNA), and frequent association with invasive carcinoma, which is most often of lobular phenotype [1, 14]. These features, together with unifocal co-localization of PLCIS and FLCIS with invasive lobular carcinoma (ILC), indirectly suggest that these variants are more advanced precursor lesions to ILC than CLCIS [14]. Recent genetic studies support the notion that some CLCIS are also nonobligate precursors clonally related to paired ILC [15]. However, the genetics and molecular drivers of PLCIS and FLCIS remain largely unknown, with studies limited to CNA analysis by array comparative genomic hybridization (aCGH). Furthermore, because these studies primarily analyzed nonpaired PLCIS/FLCIS and CLCIS, clonal relationships of these lesions with CLCIS have only been indirectly inferred. The nature of the genetic relationships between CLCIS, LCIS variants, and ILC with respect to neoplastic progression remains unknown.
In this study, we utilized a targeted capture-based next-generation DNA sequencing panel of 479 cancer-related genes to characterize the genomics of PLCIS and FLCIS and to analyze the clonal relationships of these LCIS variants with synchronous CLCIS and synchronous ILC. We also analyzed the genetics of pure LCIS variants not associated with invasive carcinoma. The findings advance our understanding of the origins and biology of PLCIS and FLCIS and provide a genetic basis for their more aggressive behavior and increased risk of progression to invasive carcinoma.
Materials and methods
Study population and histologic classification
This study was approved by the Institutional Review Board of the University of California San Francisco (UCSF). Sixteen cases of LCIS variants (seven PLCIS, five FLCIS, and four mixed PLCIS and FLCIS) were selected from the UCSF Pathology Department archives for genomic analysis. Preferential selection of cases was based on the presence of synchronous CLCIS and/or ILC components, technical feasibility of separating components to be analyzed without cross-contamination, and availability of sufficient tissue for analysis. A larger clinicopathologic analysis including a subset of these cases has been previously published [14]. Five PLCIS, all FLCIS, and all mixed PLCIS/FLCIS were associated with CLCIS. Five PLCIS, three FLCIS, and three mixed PLCIS/FLCIS were associated with ILC (designated P1–P7, F1–F4), whereas five LCIS variants (two apocrine PLCIS, two FLCIS, and one mixed PLCIS/FLCIS) were ultimately pure, without associated invasive cancer or DCIS (designated pP1–pP3, pF1, pF2). All specimens were fixed in 10% neutral buffered formalin and embedded in paraffin.
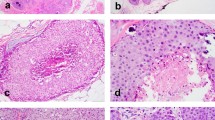
All LCIS lacked complete membranous E-cadherin immunostaining. CLCIS showed expansion (≥8 cells in diameter) of at least 50% of the acini in a terminal duct lobular unit (TDLU) by one or a mixture of the following two cell types: type A cells (uniform, round, and small nuclei up to 1.5× the size of a lymphocyte nucleus with inconspicuous nucleoli), and type B cells (more abundant cytoplasm and larger nuclei up to 2× the size of a lymphocyte nucleus, with more variability in size and shape and more prominent nucleoli) [14]. PLCIS was defined by marked nuclear pleomorphism (>2–3× variation in nuclear size) with nuclei equivalent to high-grade DCIS and at least some nuclei >4× the size of a lymphocyte (Fig. 1a) [1, 2]. Apocrine differentiation was defined by abundant eosinophilic, granular cytoplasm, and enlarged rounded nuclei with prominent nucleoli (Fig. 1c) [1]. FLCIS had cytologic features of CLCIS (often type B cells, less commonly mixed type A and type B cells) but were distinguished by marked expansion of TDLUs or larger ducts with at least one of two architectural features: (1) little to no intervening stroma between expanded ductules of a TDLU, or (2) expansion of an acinus/duct filling approximately one high-power field (Fig. 1e) [14]. Each case was evaluated for the following histologic features: the presence of CLCIS, comedonecrosis associated with the LCIS variant, the size of the LCIS variant, and, when applicable, the size, histologic type, and grade of associated invasive carcinoma. Comedonecrosis was defined as confluent necrosis comprising at least 30% of the duct lumen [16].

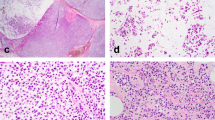
a, b Pleomorphic LCIS (a) (case P6) showed marked nuclear pleomorphism, equivalent to that of high-grade DCIS, and associated invasive lobular carcinoma (b) typically showed pleomorphic cytology and growth pattern. c, d This pure pleomorphic LCIS (pP2) had apocrine cytology, characterized by large cells with abundant eosinophilic granular cytoplasm (c). The neoplastic cells lack estrogen receptor expression (d). e, f Florid LCIS (case F1) had similar cytology to classic LCIS but showed marked expansion of terminal duct lobular units or ducts, with little to no intervening stroma between expanded lobules (e). The associated invasive lobular carcinoma in this case showed classic cytology and growth pattern (f).
Clinical information was obtained from the UCSF electronic medical record. Overall survival was calculated from the date of first pathologic diagnosis until the date of death or the date of last known alive for patients still alive or lost to follow-up.
Immunohistochemistry
For all cases, ER, PR, and HER2 biomarker expression was examined in the LCIS variant, in synchronous CLCIS, and in associated invasive carcinoma. In cases with mixed PLCIS/FLCIS, biomarker expression was only evaluated in the sequenced components. Immunohistochemistry was performed on whole tissue sections using the following antibodies and dilutions: ER (SP1, prediluted, Roche, Ventana Medical Systems, Tucson, AZ, USA); PR (IE2, prediluted, Roche, Ventana Medical Systems); HER2 (4B5, prediluted, Roche, Ventana Medical Systems); and E-cadherin (HECD-1, 1:100, Invitrogen, Carlsbad, CA, USA). Antigen retrieval was as follows: for ER, PR, and HER2—CC1 (Roche, Ventana Medical Systems); for E-cadherin—bond epitope retrieval solution 1 (Leica Biosystems, Buffalo Grove, IL, USA). For ER, PR, and HER2, positive staining was defined according to ASCO/CAP guidelines [17, 18].
Capture-based next-generation DNA sequencing
For capture-based next-generation DNA sequencing, LCIS variants and matched normal tissue were selected from all 11 cases that were associated with invasive carcinoma. Only LCIS variant tissue was sequenced for the five cases of pure LCIS variants not associated with invasive carcinoma. Synchronous ILC was sequenced in all 11 cases associated with invasive carcinoma, and synchronous CLCIS was sequenced in nine cases (four PLCIS, two FLCIS, and three mixed PLCIS/FLCIS). In one case of mixed PLCIS/FLCIS (case F3), two geographically distinct foci of CLCIS were separately sequenced, and the FLCIS but not PLCIS component was sequenced due to inability to accurately dissect PLCIS from ILC. For the other mixed PLCIS/FLCIS cases (cases P3, P5, and pP1), PLCIS was sequenced.
DNA sequencing was performed at the UCSF Clinical Cancer Genomics Laboratory using an assay that targets the coding regions of 479 cancer-related genes, select introns from 42 genes, and the TERT promoter, with a total sequencing footprint of 2.8 Mb (UCSF500 panel; Supplementary Table S1). Sequencing libraries were prepared from genomic DNA extracted from tumor and normal formalin-fixed paraffin-embedded tissue. Target enrichment was performed by hybrid capture using a custom oligonucleotide library. Sequencing was performed on an Illumina HiSeq 2500. Duplicate sequencing reads were removed computationally to allow for accurate allele frequency determination and copy number calling. The analysis was based on the human reference sequence UCSC build hg19 (NCBI build 37), using the following software packages: BWA: 0.7.13, Samtools: 1.1 (using htslib 1.1), Picard tools: 1.97 (1504), GATK: Appistry v2015.1.1–3.4.46-0-ga8e1d99, CNVkit: 0.7.2, Pindel: 0.2.5b8, SATK:Appistry v2015.1.1-1-gea45d62, Annovar: v2016 Feb01, Freebayes: 0.9.20 and Delly: 0.7.2 [19,20,21,22,23,24,25,26,27,28,29]. Only insertions/deletions up to 100 base pairs in length were included in the mutational analysis. Somatic single-nucleotide variants and insertions/deletions were visualized and verified using Integrated Genome Viewer. Genome-wide copy number analysis based on on-target and off-target reads was performed by CNVkit and Nexus Copy Number (Biodiscovery, Hawthorne, CA, USA) [24].
Statistical analysis was performed using paired and unpaired Student’s t tests and Fisher exact test as appropriate, with p < 0.05 considered significant.
Results
Clinicopathologic features of pleomorphic and florid lobular carcinoma in situ variants and synchronous invasive carcinomas
The clinicopathologic features of cases in the study population are listed in Table 1. All patients were women, with an average age of 63 years (range 53–81) for PLCIS, 53 years (range 40–68) for FLCIS, and 67 years for mixed PLCIS/FLCIS (range 60-74). Fourteen LCIS variants (five PLCIS, five FLCIS, and all four mixed PLCIS/FLCIS) arose in a background of CLCIS, and 11 LCIS variants (five PLCIS, three FLCIS, and three mixed PLCIS/FLCIS) were associated with synchronous ILC. Three PLCIS and one mixed PLCIS/FLCIS also had synchronous DCIS, and one mixed PLCIS/FLCIS had synchronous DCIS and a small focus of invasive ductal carcinoma.
All patients with associated ILC underwent surgical excision and axillary lymph node sampling. The average size of the LCIS variant was 4.3 cm (range 1.8–10 cm), and the average size of synchronous ILC was 3.4 cm (range 0.3–9.1 cm). Invasive carcinomas associated with PLCIS or mixed PLCIS/FLCIS were pleomorphic ILC in all but one case (88%, n = 8) (Fig. 1b). All invasive carcinomas associated with FLCIS were classic ILC (Fig. 1f). Nine (82%) ILC were modified Scarff-Bloom-Richardson (mSBR) grade 2, and two (18%) were mSBR grade 3. Two patients with ILC associated with mixed PLCIS/FLCIS had lymph node metastasis; the remaining patients were all node negative. One patient with lymph node metastasis died of disease (survival 20 months); all other patients with invasive carcinoma are alive with no evidence of disease (mean follow-up 47 months).
Five LCIS variants (two PLCIS, two FLCIS, and one mixed PLCIS/FLCIS) were not associated with either invasive carcinoma or DCIS (designated pure PLCIS or pure FLCIS). Both pure PLCIS had apocrine differentiation. All patients are alive with no evidence of disease (mean follow-up 55 months).
Biomarker expression
Biomarker expression was evaluated in the sequenced LCIS variants (ten PLCIS, six FLCIS) and sequenced synchronous CLCIS and ILC. Seven PLCIS were ER-positive and three were ER-negative, the latter including both cases of apocrine PLCIS (Fig. 1c, d); only 3/10 were PR-positive. All FLCIS were ER-positive and 5/6 were PR-positive. Two PLCIS had ERBB2 amplification with HER2 protein overexpression (Supplementary Fig. S1). Hormone receptor and HER2 status of paired ILC correlated with the respective LCIS variant in 9/11 cases (Table 1). Of the exceptions, one PLCIS (case P6) was weakly ER-positive, whereas the synchronous ILC was triple negative, and one FLCIS (case F4) was PR-positive, whereas the synchronous ILC was PR-negative. Hormone receptor and HER2 status of paired CLCIS correlated with the respective LCIS variant in 3/5 PLCIS and 3/3 FLCIS; two cases of PR-negative PLCIS had synchronous PR-positive CLCIS (cases P5 and P6). In one case (P3), paired CLCIS, PLCIS, and ILC were all triple (ER, PR, and HER2) positive (Supplementary Fig. S1C–F).
Genetics of pleomorphic and florid lobular carcinoma in situ variants
The mean target coverage of next-generation DNA sequencing was 533 (±246) unique reads per target interval for PLCIS, 718 (±310) unique reads per target interval for FLCIS, 519 (±318) unique reads per target interval for ILC, and 621 (±356) unique reads per target interval for CLCIS (Supplementary Table S2).
Predicted pathogenic and/or recurrent alterations identified in PLCIS and FLCIS are shown in Fig. 2, and a list of all identified nonsynonymous coding mutations is provided in Supplementary Table S3. The mean number of nonsynonymous coding mutations in UCSF500 panel genes per case was similar between PLCIS (7.3 ± 3.1) and FLCIS (8.0 ± 1.8) (p = 0.693, n = 7 and 4, respectively), and the mean number of predicted pathogenic alterations per case was also similar (4.3 ± 0.9 in PLCIS, n = 10, vs. 5 ± 2.1 in FLCIS, n = 6) (p = 0.371).

aPLCIS indicates apocrine PLCIS, FLCIS florid LCIS, LCIS lobular carcinoma in situ, PLCIS pleomorphic LCIS. Asterisk indicates sequenced LCIS variant component of mixed PLCIS/FLCIS case.
Ninety-four percent (15/16) of cases had frameshift, nonsense, or missense mutations in CDH1, with loss of heterozygosity in 93% (14/15), consistent with lobular differentiation. The single case (PLCIS, P2) without CDH1 mutation had monoallelic 16q loss, and E-cadherin protein loss was confirmed by immunohistochemistry (not shown). All PLCIS and FLCIS harbored pathogenic alterations in ERBB2, ERBB3, and/or downstream phosphatidyl inositol-3 kinase (PI-3K) pathway genes. Pathogenic alterations in PIK3CA and/or PIK3R1 were present in 70% PLCIS and 67% FLCIS and most frequently included constitutively activating hotspot mutations in the helical or kinase domain of PIK3CA (seven PLCIS and two FLCIS). Sixty percent of PLCIS (6/10) harbored pathogenic ERBB2 alterations, including known hotspot mutations in the ERBB2 kinase domain (p.L755S and p.T862A, two cases each) [30,31,32,33,34] in four cases and ERBB2 amplification in two additional cases (Fig. 2 and Supplementary Fig. S2A). Two of the three sequenced pure PLCIS had ERBB2 kinase domain mutations. Hotspot ERBB2 mutations (p.L755S and p.G778_P780dup) were also identified in 33% (2/6) FLCIS, none of which had ERBB2 amplification [34]. One PLCIS and two FLCIS harbored ERBB3 mutations, which included known pathogenic kinase domain (p.E928G, PLCIS case P6) and extracellular domain (p.T355I, FLCIS case pF1) mutations [35]. A p.H798L mutation in FLCIS (case pF2) has not been previously characterized but is located near the ATP binding site in the kinase domain (Fig. 2, Supplementary Fig. S2B). Two of the three ERBB3 mutations co-occurred with hotspot ERBB2 mutations. Together, ERBB2 and/or ERBB3 alterations were present in 60% (6/10) PLCIS and 50% (3/6) FLCIS.
Mutations in the forkhead transcription factor FOXA1 were identified in 40% (4/10) PLCIS and 17% (1/6) FLCIS. FOXA1 mutations in PLCIS were truncating (p.C268*), known hotspots (p.D226N, p.S250F), or missense mutations in the forkhead domain (p.S174L) [33, 34]. In the FLCIS case (F4), two nearby FOXA1 missense mutations in the forkhead domain (p.C258W and p.C268W) were present on separate alleles.
TP53 mutations were identified in 30% (3/10) PLCIS but not in FLCIS. Conversely, CBFB mutations were identified in FLCIS (2/6) but not in PLCIS. Other recurrently altered genes included CCND1 (amplification in 2/10 PLCIS and 1/6 FLCIS), GATA3 (missense mutations in 1/10 PLCIS and 1/6 FLCIS), and ARHGAP35 (truncating mutations in 1/10 PLCIS and 1/6 FLCIS). One patient with PLCIS (P3) and ERBB2 amplification was a germline pathogenic BRCA2 carrier (p.V3365fs), but none of the lesional samples showed somatic loss of heterozygosity at this allele.
Copy number analysis revealed 16q loss in all cases and 1q gain in 73% (n = 15), with a mean of 11.3 ± 9.3 CNA in PLCIS (n = 9) compared with 4.7 ± 2.9 in FLCIS (n = 6) (p = 0.116, two-tailed t test). Additional recurrent CNA included loss of distal or whole-arm 8p in 44% (4/9) PLCIS and 33% (2/6) FLCIS; gain of 8q or whole-chromosome 8 in 33% (3/9) PLCIS and 33% (2/6) FLCIS; loss of distal or whole-arm 11q in 44% (4/9) PLCIS and 33% (2/6) FLCIS; loss of 13q or whole-chromosome 13 in 44% (4/9) PLCIS and 17% (1/6) FLCIS; gain of distal or whole-arm 16p in 22% (2/9) PLCIS and 33% (2/6) FLCIS; loss of distal or whole-arm 17p in 56% (5/9) PLCIS and 17% (1/6) FLCIS; gain of interstitial or whole-arm 17q in 33% (3/9) PLCIS and 17% (1/6) FLCIS, and loss of distal or whole-arm 18q or whole-chromosome 18 in 44% (4/9) PLCIS and 17% (1/6) FLCIS (Supplementary Table S4).
The mutational profiles of PLCIS or FLCIS and the total number of predicted pathogenic mutations in UCSF500 panel genes per case were similar between LCIS variants associated with invasive carcinoma (range 3–8, n = 11) and pure LCIS variants without invasive carcinoma (range 3–5, n = 5) and included ERBB2 and ERBB3 mutations in both groups (Fig. 2). Although mean CNA were similar between LCIS variants associated with invasive carcinoma (10.8 ± 9.1) and those that were pure (4.4 ± 1.8; p = 0.15, two-tailed t test), wider CNA variation was observed in the former group (range 2–27 vs. 2–7), which closely paralleled associated ILC (p = 0.051, paired two-tailed t test) (Fig. 3 and Supplementary Table S4). Recurrent CNA in PLCIS associated with invasive carcinoma that were absent in pure PLCIS included losses in distal or whole-arm 11q (4/7 vs. 0/3), 13q or whole-chromosome 13 (4/7 vs. 0/3), and distal or whole-arm 17p (5/7 vs. 0/3). No recurrent CNA distinguished FLCIS associated with invasive carcinoma from pure FLCIS; however, this comparison was limited by small sample size.

aPLCIS indicates apocrine PLCIS, CLCIS classic LCIS, FLCIS florid LCIS, ILC invasive lobular carcinoma, LCIS lobular carcinoma in situ, PILC pleomorphic ILC, PLCIS pleomorphic LCIS. Asterisk indicates sequenced LCIS variant component of mixed PLCIS/FLCIS case.
Genetic relationships of pleomorphic and florid lobular carcinoma in situ with synchronous classic lobular carcinoma in situ and invasive lobular carcinoma
For the eleven cases with synchronous invasive carcinoma, sequencing results were compared across paired synchronous components (PLCIS or FLCIS, ILC, and CLCIS, when available) to identify clonal relationships and genetic features associated with progression. Paired predicted pathogenic and/or recurrent alterations are shown in Fig. 3. A list of all identified nonsynonymous mutations are provided in Supplementary Table S3, and all CNA are provided in Supplementary Table S4. In 8 of 9 (89%) cases in which synchronous CLCIS was sequenced in addition to the LCIS variant (5/6 PLCIS, 3/3 FLCIS), the paired components were clonally related to one another and shared pathogenic aberrations and CNA. A clonally distinct CLCIS component was identified in two cases (PLCIS case P7 and FLCIS case F3), which in both harbored different CDH1 frameshift mutations from the respective paired LCIS variant and ILC. In one of these (case F3), a second focus of CLCIS was additionally sequenced and found to be clonally related to paired FLCIS/ILC. In all 11 cases in which synchronous ILC was sequenced in addition to the LCIS variant (seven PLCIS, four FLCIS), the paired components were clonally related to one another and harbored shared pathogenic mutations and CNA.
Loss of 16q and CDH1 mutations were shared between CLCIS, PLCIS or FLCIS, and ILC in all clonally related cases. In three of the four cases with paired CLCIS and hotspot PIK3CA mutations, the mutations were clonal and shared between CLCIS and the LCIS variant (two PLCIS, one FLCIS), consistent with other studies that PIK3CA mutations tend to occur early in lobular carcinogenesis and are present at the CLCIS stage [15, 36].
Pleomorphic lobular neoplasia progression
Compared with paired CLCIS, PLCIS was associated with additional clonal pathogenic mutations in 4/5 (80%) cases and with additional CNA in 2/3 (67%) cases. Paired analysis revealed more nonsynonymous coding mutations in UCSF500 panel genes per case in PLCIS (mean 6.6 ± 3.3) compared with CLCIS (mean 4.4 ± 2.6) (p = 0.03, paired two-tailed t test, n = 6). Progression from CLCIS to PLCIS was associated with TP53 mutation in both evaluable cases with TP53 mutations. Whereas one of these (P5) had an identical truncating TP53 mutation in paired PLCIS and ILC, the other (P3) had different TP53 mutations in PLCIS and ILC, indicative of driver-level genetic divergence at this stage. Indeed, 2/5 CLCIS and 3/7 PLCIS had private pathogenic mutations not present in their respective paired components, and 2/6 PLCIS had private CNA not present in paired ILC, consistent with clonal heterogeneity at early and later stages of in situ lobular carcinogenesis. In one case (P6), progression to PLCIS was associated with concomitant pathogenic ERBB2 and ERBB3 mutations, and progression to ILC was associated with two additional private ERBB3 mutations. However, two other cases shared either ERBB2 mutation (P5) or ERBB2 amplification (P3) among all three components (CLCIS, PLCIS, and ILC), indicating that ERBB2 aberrations can occur either early or late in the pleomorphic lobular neoplasia pathway. Similarly, CCND1 amplification was associated with progression from CLCIS to PLCIS/ILC in one case (P5) but was present in all three components in another case (P4).
Progression from PLCIS to ILC was not associated with a significant increase in the total number of nonsynonymous coding mutations (mean 6.6 ± 3.3 in PLCIS vs. 7.3 ± 4.3 in ILC) (p = 0.518, paired two-tailed t test, n = 7) or CNA (mean 14.8 ± 9.6 in PLCIS vs. 17.2 ± 8.4 in ILC) (p = 0.376, paired two-tailed t test, n = 6). PLCIS harbored similar or identical CNA to synchronous ILC (Fig. 4), including two PLCIS with more CNA than associated ILC (Supplementary Fig. S3A–C). In contrast, paired CLCIS harbored fewer nonsynonymous coding mutations (mean 4.4 ± 2.6) than ILC (p = 0.027, paired two-tailed t test, n = 5). Mean CNA in the three evaluable cases of CLCIS (6.7 ± 3.4, n = 3) was also less than paired ILC, although the sample size of paired lesions was too small for meaningful statistical analysis (Supplementary Table S4). Progression from PLCIS to ILC was associated with additional clonal pathogenic mutations in 3/7 (43%) cases, including NF1, WRN, and HIST1H3B in one case (P1), CDKN1B and KMT2A in another (P3), and ERBB3 in the third (P6). Progression from PLCIS to ILC was also associated with additional CNA in 4/6 (67%) cases, including recurrent losses in 4p or whole-chromosome 4 in two cases (P3 and P6). No other recurrent alterations were associated with progression to invasion.

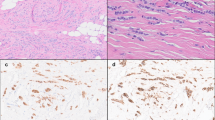
a–d Representative hematoxylin and eosin images of the areas sequenced from paired CLCIS (a), PLCIS (b), and ILC (c) in case P4. The three components shared most copy number alterations, including CCND1 amplification, with three additional changes shared only by PLCIS and ILC (d). e–h Representative hematoxylin and eosin images of the areas sequenced from paired CLCIS (e), PLCIS (f), and pleomorphic ILC (g) in case P5. PLCIS and pleomorphic ILC shared numerous copy number alterations, whereas synchronous CLCIS shared only three of these with PLCIS and ILC (h). Chromosomes are organized from 1 (left) to X (right). Thick arrows indicate large-scale (chromosome or whole-chromosome arm level) copy number alterations; thin arrows, smaller-scale (sub-chromosomal arm) alterations; dotted arrows, subclonal loss or copy neutral loss of heterozygosity; arrowheads, amplification. Black indicates copy number alterations shared among CLCIS, the LCIS variant, and ILC; red, copy number alterations shared between the LCIS variant and ILC; blue, copy number alterations private to a single component.
Florid lobular neoplasia progression
Progression from CLCIS to FLCIS was associated with additional clonal pathogenic mutations in only 1/3 cases (F4), which included PIK3R1, FOXA1, and KMT2C (Fig. 3). However, paired FLCIS and ILC were genetically divergent in this case, with different clonal FOXA1 mutations in both components, and FLCIS also had other private clonal and subclonal pathogenic mutations compared with CLCIS and ILC. Indeed, all four cases with FLCIS harbored private pathogenic mutations in the FLCIS component, again suggestive of driver-level genetic divergence at this level of lobular carcinogenesis.
Overall, the mean number of nonsynonymous coding mutations was 5.3 ± 1.5 in ILC compared with 7.8 ± 2.1 in FLCIS and 4.3 ± 2.6 in CLCIS, but the sample size of paired lesions was too small for meaningful statistical analysis. Mean CNA per case were 11.3 ± 8.5 in ILC compared with 4.8 ± 3.8 in FLCIS (p = 0.07, paired two-tailed t test, n = 4), and all four cases had additional private CNA in ILC compared with FLCIS. In the two evaluable cases with CLCIS, no additional CNA were identified in paired FLCIS. In one case (F4), FLCIS had only two CNA (1q gain and 16 loss), identical to synchronous CLCIS and fewer than associated ILC (Supplementary Fig. S3D–G). Interestingly, ILC in the other case (F2) shared identical CNA with paired CLCIS and more than FLCIS, suggesting direct progression to ILC from CLCIS and not FLCIS. In the remaining three cases, progression to ILC was associated with clonal pathogenic aberrations in MCL1 and RB1 (F1), ASXL2 (F3), and FOXA1 (F4). No recurrent alterations associated with progression to invasion were identified.
Comparative genetics of pleomorphic and florid lobular neoplasia with other invasive lobular carcinomas
The genomic profiles of PLCIS, FLCIS, and associated ILC in our study were compared with publicly available sequencing data of ILC (n = 127) previously analyzed in The Cancer Genome Atlas (TCGA) (Fig. 5) [33, 34, 37]. PLCIS, FLCIS, and LCIS variant-associated ILC were more likely to harbor ERBB2 mutations than ILC in the TCGA cohort (40%, 33%, and 27%, compared with 4% in TCGA, p = 0.002, p = 0.032, and p = 0.017, respectively) or to have either ERBB2 or ERBB3 alterations (60%, 50%, and 45%, compared with 11% in TCGA, p < 0.001, p = 0.006, and p = 0.008, respectively). ERBB3 mutations were more common in FLCIS (33%) than TCGA ILC (1%) (p = 0.005). FOXA1 mutations were more frequent in PLCIS (40%) than ILC in the TCGA cohort (7%) (p = 0.008), and this trended toward significance in ILC associated with LCIS variants (27%, p = 0.056). ILC associated with PLCIS harbored TP53 mutations more frequently than ILC in the TCGA cohort (43% vs. 8%, respectively; p = 0.02) and was also more likely to harbor mutations in HIST1H3B (29% vs. 2%, p = 0.02). In contrast to PLCIS, FLCIS, and/or associated ILC, there were no significant differences between CLCIS associated with these LCIS variants and ILC in the TCGA cohort.

All data except TCGA ILC are from cases in this study. *p < 0.05.
Discussion
In this study, we used capture-based next-generation sequencing to analyze the genetics of PLCIS and FLCIS, including those present as pure lesions and those associated with invasive carcinoma. Our findings demonstrate a similar spectrum of aberrations in both groups, with notable enrichment for pathogenic alterations in the PI-3K pathway, ERBB2/ERBB3, and FOXA1. The results also reveal clonal relationships between CLCIS, LCIS variants, and ILC and demonstrate that PLCIS is a genetically advanced direct precursor to ILC.
Frequent association of PLCIS and FLCIS with CLCIS, as well as shared biomarker profiles, loss of E-cadherin expression, and 1q gain and 16q loss has suggested that these LCIS variants evolve along the lobular lineage; on the other hand, increased CNA in the LCIS variants has suggested that they are genetically more advanced than CLCIS [1, 4, 14]. However, previous genetic studies of PLCIS and FLCIS have been primarily limited to aCGH analysis of nonpaired PLCIS, FLCIS, and CLCIS and could not establish clonal relationships between these lesions [1, 4]. Only a rare PLCIS/PILC pair analyzed by aCGH has suggested a genetic relationship [38]. Here, we provide direct evidence that PLCIS and FLCIS are descended from synchronous paired CLCIS precursors and also show that PLCIS in particular is a genetically more advanced lesion. PLCIS shared truncal mutations (such as CDH1, PIK3CA, and PTEN, among others) with paired CLCIS but also harbored comparatively more nonsynonymous coding mutations in UCSF500 panel genes, additional pathogenic aberrations in genes such as TP53, ERBB2, ERBB3, and CCND1, and increased CNA. In contrast, there were no significant differences in the mean number of total nonsynonymous mutations or CNA between paired PLCIS and ILC, together suggesting that PLCIS is genetically more advanced than CLCIS and similar to ILC. On the other hand, although the altered genomic fraction appeared to be similar overall between PLCIS and ILC, the acquisition of pathogenic mutations such as NF1, WRN, ERBB3, and FOXA1 in some ILC suggests a role for these genes in invasion in individual cases. Although cross-platform comparisons are not accurate, this study and our prior work using aCGH [1] suggest fewer genetic alterations in PLCIS than published studies of high-grade DCIS, despite the histologic similarities between these lesions [39, 40]. Rather, our study supports the concept that PLCIS is a genetically advanced precursor in the lobular lineage that more closely resembles ILC.
In contrast to PLCIS, FLCIS as a group were not obviously more advanced genetically than paired CLCIS. FLCIS harbored more clonal pathogenic mutations than paired CLCIS in only one case (F4), but these were not shared with ILC, which was instead genetically divergent from FLCIS. ILC in another case (F2) was genetically more closely related to paired CLCIS than FLCIS, whereas the third evaluable case was not informative in this respect. Although analysis is limited by the small number of paired CLCIS/FLCIS/ILC samples, the findings suggest that FLCIS-associated ILC does not necessarily develop from the synchronous FLCIS in these cases but can progress directly from the CLCIS precursor. This is in contrast to PLCIS-associated ILC, which more often progressed from CLCIS to PLCIS to ILC. In this respect, we found evidence for subclonal or clonal heterogeneity and/or genetic divergence in all cases of FLCIS and in smaller subsets of PLCIS and CLCIS, including identification of synchronous CLCIS clones that were unrelated to paired LCIS variants or ILC. This in situ heterogeneity likely provides the soil for selection and neoplastic progression to PLCIS/FLCIS or ILC [15]. Interestingly, PIK3CA and PIK3R1 mutations were commonly private to FLCIS (and less frequently PLCIS), suggesting that these alterations are not always among the earliest in situ genetic events and are also not necessarily required for progression to invasion. Although our data do not obviously implicate FLCIS in the progression from CLCIS to ILC in paired cases, the presence of frequent ERBB2/ERBB3 alterations in FLCIS and the frequent association with ILC [3, 14] nevertheless indicate that this variant has aggressive potential. If not a direct precursor, one could hypothesize that FLCIS may instead serve as a surrogate marker for the presence of LCIS that is more aggressive than typical CLCIS. Additional studies with larger numbers of FLCIS paired with CLCIS and ILC would be illustrative.
The enrichment of ERBB2 and ERBB3 alterations in PLCIS and FLCIS, which are shared in associated ILC, may have prognostic and/or management implications. Sequencing of a large cohort of ILC previously demonstrated a higher prevalence of ERBB2 and ERBB3 mutations in ILC than in invasive breast cancers overall, with ERBB2/ERBB3 mutations or ERBB2 amplification present in 14.5% of ILC and an even higher prevalence (23.1%) in the mixed, nonclassic histologic subtype [41]. Other studies have observed a high frequency of ERBB2 mutations in solid or pleomorphic variants of ILC [42, 43]. Furthermore, ERBB2 mutations have been identified at higher frequency (18%) in relapsed ILC and have been associated with worse outcomes in multiple studies [44,45,46,47]. A recently described prognostic molecular signature (LobSig) for ILC revealed a high prevalence of ERBB2 (20%) and ERBB3 (14.3%) mutations in tumors associated with poor outcomes (LobSig high) [46]. In comparison to these studies and to TCGA data [37], our results demonstrate an even higher frequency of ERBB2/ERBB3 alterations in PLCIS (60%), FLCIS (50%), and ILC associated with these variants (45%), including ERBB2 mutations in 40% PLCIS and 33% FLCIS. Our results indicate that ERBB2 and ERBB3 mutations are early events in the progression of these tumors and provide further evidence for the aggressive biological potential of these LCIS variants, supporting their excision [6,7,8,9,10,11,12,13,14]. Interestingly in this regard, a small subset of CLCIS associated with PLCIS and FLCIS in our study also harbored ERBB2 mutations, and one PLCIS-associated CLCIS had ERBB2 amplification, suggesting that these aberrations can potentially occur very early in lobular carcinogenesis, prior to recognized morphologic changes of the LCIS variants. In contrast, the vast majority of CLCIS have been shown to lack ERBB2 or ERBB3 aberrations [15, 48]. Whether ERBB2/ERRB3-altered or other subsets of CLCIS have more aggressive precursor potential than other CLCIS will require further study.
Identification of ERBB2/ERBB3 mutations in tumors lacking ERBB2 amplification may have therapeutic value. Breast cancers with ERBB2 mutations have demonstrated sensitivity to small molecule tyrosine kinase inhibitors in preclinical models and clinical trials [31, 32]. The most highly recurrent ERBB2 mutation in our study, p.L755S, confers resistance to the reversable tyrosine kinase inhibitor lapatinib [30, 32] but shows sensitivity to the irreversible tyrosine kinase inhibitor neratinib [32]. In a recently published basket trial of cancers with ERRB2 and ERBB3 mutations, tumors with the common ERBB2 kinase domain mutations reported here (p.L755S, p.G778_P780dup) showed best responses to neratinib therapy [31]. The occurrence and functional relevance of ERRB3 mutations is less well studied, although functional studies in cell lines and animal models have demonstrated that ERBB3 mutations promote ligand-independent oncogenic signaling that is effectively blocked by anti-ERBB antibodies and small molecule inhibitors [35, 49]. These studies and case reports [50,51,52] suggest that patients with ERBB2- and ERBB3-mutated breast cancers may benefit from existing HER2-targeted drugs. The high frequency of these alterations in our cohort supports evaluating the clinical benefit of mutation testing in PLCIS- and FLCIS-associated invasive carcinoma as an important area for further study and also opens a discussion of potential preventative therapeutic intervention for patients with high risk LCIS variants.
In conclusion, our results demonstrate clonal relationships between PLCIS/FLCIS and synchronous CLCIS and ILC, delineating genetic progression in the lobular neoplasia pathway, and implicate PLCIS as a direct precursor to ILC. Frequent ERBB2 and ERBB3 activating mutations or amplifications in particular are consistent with more aggressive behavior of PLCIS, FLCIS, and associated ILC and may have treatment implications. The genetic similarities between pure LCIS variants and those associated with ILC suggest that pure LCIS variants may also have aggressive potential. Larger-scale molecular studies and long-term clinical follow-up data are indicated to better stratify classification and management of these lesions.
References
Chen YY, Hwang ES, Roy R, DeVries S, Anderson J, Wa C, et al. Genetic and phenotypic characteristics of pleomorphic lobular carcinoma in situ of the breast. Am J Surg Pathol. 2009;33:1683–94.
Sneige N, Wang J, Baker BA, Krishnamurthy S, Middleton LP. Clinical, histopathologic, and biologic features of pleomorphic lobular (ductal-lobular) carcinoma in situ of the breast: a report of 24 cases. Mod Pathol. 2002;15:1044–50.
Bagaria SP, Shamonki J, Kinnaird M, Ray PS, Giuliano AE. The florid subtype of lobular carcinoma in situ: marker or precursor for invasive lobular carcinoma? Ann Surg Oncol. 2011;18:1845–51.
Shin SJ, Lal A, De Vries S, Suzuki J, Roy R, Hwang ES, et al. Florid lobular carcinoma in situ: molecular profiling and comparison to classic lobular carcinoma in situ and pleomorphic lobular carcinoma in situ. Hum Pathol. 2013;44:1998–2009.
Alvarado-Cabrero I, Picon Coronel G, Valencia Cedillo R, Canedo N, Tavassoli FA. Florid lobular intraepithelial neoplasia with signet ring cells, central necrosis and calcifications: a clinicopathological and immunohistochemical analysis of ten cases associated with invasive lobular carcinoma. Arch Med Res. 2010;41:436–41.
Chivukula M, Haynik DM, Brufsky A, Carter G, Dabbs DJ. Pleomorphic lobular carcinoma in situ (PLCIS) on breast core needle biopsies: clinical significance and immunoprofile. Am J Surg Pathol. 2008;32:1721–6.
De Brot M, Koslow Mautner S, Muhsen S, Andrade VP, Mamtani A, Murray M, et al. Pleomorphic lobular carcinoma in situ of the breast: a single institution experience with clinical follow-up and centralized pathology review. Breast Cancer Res Treat. 2017;165:411–20.
Flanagan MR, Rendi MH, Calhoun KE, Anderson BO, Javid SH. Pleomorphic lobular carcinoma in situ: radiologic-pathologic features and clinical management. Ann Surg Oncol. 2015;22:4263–9.
Guo T, Wang Y, Shapiro N, Fineberg S. Pleomorphic lobular carcinoma in situ diagnosed by breast core biopsy: clinicopathologic features and correlation with subsequent excision. Clin Breast Cancer. 2018;18:e449–54.
Fasola CE, Chen JJ, Jensen KC, Allison KH, Horst KC. Characteristics and clinical outcomes of pleomorphic lobular carcinoma in situ of the breast. Breast J. 2018;24:66–9.
Susnik B, Day D, Abeln E, Bowman T, Krueger J, Swenson KK, et al. Surgical outcomes of lobular neoplasia diagnosed in core biopsy: prospective study of 316 cases. Clin Breast Cancer. 2016;16:507–13.
Carder PJ, Shaaban A, Alizadeh Y, Kumarasuwamy V, Liston JC, Sharma N. Screen-detected pleomorphic lobular carcinoma in situ (PLCIS): risk of concurrent invasive malignancy following a core biopsy diagnosis. Histopathology. 2010;57:472–8.
Lavoue V, Graesslin O, Classe JM, Fondrinier E, Angibeau H, Leveque J. Management of lobular neoplasia diagnosed by core needle biopsy: study of 52 biopsies with follow-up surgical excision. Breast. 2007;16:533–9.
Shamir ER, Chen YY, Chu T, Pekmezci M, Rabban JT, Krings G. Pleomorphic and florid lobular carcinoma in situ variants of the breast: a clinicopathologic study of 85 cases with and without invasive carcinoma from a single academic center. Am J Surg Pathol. 2019;43:399–408.
Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA, et al. Lobular carcinomas in situ display intralesion genetic heterogeneity and clonal evolution in the progression to invasive lobular carcinoma. Clin Cancer Res. 2019;25:674–86.
Harrison BT, Hwang ES, Partridge AH, Thompson AM, Schnitt SJ. Variability in diagnostic threshold for comedo necrosis among breast pathologists: implications for patient eligibility for active surveillance trials of ductal carcinoma in situ. Mod Pathol. 2019;32:1257–62.
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Pathol Lab Med. 2010;134:e48–72.
Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch Pathol Lab Med. 2014;138:241–56.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform. 2013;43:11.10.1–33.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71.
Picard: a set of tools (in Java) for working with next generation sequencing data in the BAM: Broad Institute. http://broadinstitute.github.io/picard.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv 2012;1207.3907 [q-bio.GN].
Kancha RK, von Bubnoff N, Bartosch N, Peschel C, Engh RA, Duyster J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE. 2011;6:e26760.
Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. 2018;554:189–94.
Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–37.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23:603–17.
Shah V, Nowinski S, Levi D, Shinomiya I, Kebaier Ep Chaabouni N, Gillett C, et al. PIK3CA mutations are common in lobular carcinoma in situ, but are not a biomarker of progression. Breast Cancer Res. 2017;19:7.
Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast. Cancer Cell. 2015;163:506–19.
Reis-Filho JS, Simpson PT, Jones C, Steele D, Mackay A, Iravani M, et al. Pleomorphic lobular carcinoma of the breast: role of comprehensive molecular pathology in characterization of an entity. J Pathol. 2005;207:1–13.
Iakovlev VV, Arneson NC, Wong V, Wang C, Leung S, Iakovleva G, et al. Genomic differences between pure ductal carcinoma in situ of the breast and that associated with invasive disease: a calibrated aCGH study. Clin Cancer Res. 2008;14:4446–54.
Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, et al. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015;6:7597–607.
Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, et al. Genomic characterization of primary invasive lobular breast cancer. J Clin Oncol. 2016;34:1872–81.
Deniziaut G, Tille JC, Bidard FC, Vacher S, Schnitzler A, Chemlali W, et al. ERBB2 mutations associated with solid variant of high-grade invasive lobular breast carcinomas. Oncotarget. 2016;7:73337–46.
Rosa-Rosa JM, Caniego-Casas T, Leskela S, Cristobal E, Gonzalez-Martinez S, Moreno-Moreno E, et al. High frequency of ERBB2 activating mutations in invasive lobular breast carcinoma with pleomorphic features. Cancers (Basel). 2019;11. pii: E74. https://doi.org/10.3390/cancers11010074.
Ross JS, Wang K, Sheehan CE, Boguniewicz AB, Otto G, Downing SR, et al. Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res. 2013;19:2668–76.
Ping Z, Siegal GP, Harada S, Eltoum IE, Youssef M, Shen T, et al. ERBB2 mutation is associated with a worse prognosis in patients with CDH1 altered invasive lobular cancer of the breast. Oncotarget. 2016;7:80655–63.
McCart Reed AE, Lal S, Kutasovic JR, Wockner L, Robertson A, de Luca XM, et al. LobSig is a multigene predictor of outcome in invasive lobular carcinoma. NPJ Breast Cancer. 2019;5:18.
Ross JS, Gay LM, Wang K, Ali SM, Chumsri S, Elvin JA, et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: an emerging opportunity for anti-HER2 targeted therapies. Cancer. 2016;122:2654–62.
Sakr RA, Schizas M, Carniello JV, Ng CK, Piscuoglio S, Giri D, et al. Targeted capture massively parallel sequencing analysis of LCIS and invasive lobular cancer: repertoire of somatic genetic alterations and clonal relationships. Mol Oncol. 2016;10:360–70.
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23:603–17.
Bidard FC, Ng CK, Cottu P, Piscuoglio S, Escalup L, Sakr RA, et al. Response to dual HER2 blockade in a patient with HER3-mutant metastatic breast cancer. Ann Oncol. 2015;26:1704–9.
Grellety T, Soubeyran I, Robert J, Bonnefoi H, Italiano A. A clinical case of invasive lobular breast carcinoma with ERBB2 and CDH1 mutations presenting a dramatic response to anti-HER2-directed therapy. Ann Oncol. 2016;27:199–200.
Ben-Baruch NE, Bose R, Kavuri SM, Ma CX, Ellis MJ. HER2-mutated breast cancer responds to treatment with single-agent neratinib, a second-generation HER2/EGFR tyrosine kinase inhibitor. J Natl Compr Canc Netw. 2015;13:1061–4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shamir, E.R., Chen, YY. & Krings, G. Genetic analysis of pleomorphic and florid lobular carcinoma in situ variants: frequent ERBB2/ERBB3 alterations and clonal relationship to classic lobular carcinoma in situ and invasive lobular carcinoma. Mod Pathol 33, 1078–1091 (2020). https://doi.org/10.1038/s41379-019-0449-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0449-8
This article is cited by
-
The morphologic spectrum of lobular carcinoma in situ (LCIS) observations on clinical significance, management implications and diagnostic pitfalls of classic, florid and pleomorphic LCIS
Virchows Archiv (2022)
-
Invasive lobular carcinoma of the breast: the increasing importance of this special subtype
Breast Cancer Research (2021)
-
Morphologic subtypes of lobular carcinoma in situ diagnosed on core needle biopsy: clinicopathologic features and findings at follow-up excision
Modern Pathology (2021)
-
Why is LCIS Important—Pathological Review
Current Breast Cancer Reports (2021)
