
Abstract
Calcifying nested stromal–epithelial tumor (CNSET) is a rare hepatic tumor that occurs in children and young adults. With <40 cases in the literature, the mechanism for tumorigenesis and the biological behavior of CNSET remain uncertain. Here, we studied the clinicopathologic and molecular genetic features of eight CNSETs. Six patients (75%) were female, and the median age at presentation was 22.5 years (range 14–34 years). The median tumor size was 14 cm (range 2.7–18 cm). All tumors had fibrous stroma that contained organoid nests of epithelioid to spindled tumor cells with moderate amounts of palely eosinophilic cytoplasm and ovoid, vesicular nuclei. Five tumors showed calcifications, and one showed lymphovascular invasion. Necrosis was absent in all. Immunohistochemistry demonstrated nuclear β-catenin expression in five of five tested tumors and focal to diffuse nuclear WT-1 positivity in five of seven. Hepatocellular markers (HepPar-1, arginase-1, and albumin in situ hybridization) and neuroendocrine markers (synaptophysin, chromogranin, and INSM1) were uniformly negative. Next-generation sequencing demonstrated CTNNB1 alterations in all seven sequenced tumors. Sanger sequencing demonstrated TERT promoter mutations in all six sequenced tumors. Clinical follow-up was available for seven patients (median duration 4.4 years; range 1.2–6.2 years): four (57%) developed metastatic disease; all four developed lung metastases; and two also had abdominal metastases. All four patients with metastatic disease also had persistent or recurrent liver tumors. Three patients with metastases were alive with disease at the most recent follow-up and one died of disease. The other three patients with available follow-up did not develop metastasis or recurrence. One tumor treated with neoadjuvant chemotherapy showed no response, and another showed 90% tumor fibrosis; the latter patient remained disease-free at 6.2 years of follow-up. Our series demonstrates the presence of TERT promoter mutations and CTNNB1 alterations in all sequenced tumors and suggests that CNSET might perhaps be more aggressive than previously reported.
Similar content being viewed by others
Introduction
Calcifying nested stromal–epithelial tumor (CNSET) is a rare primary hepatic tumor of uncertain lineage that predominantly occurs in young patients, with a reported age range from 22 months to 32 years [1]. CNSET was initially described in 2001 in a series of three tumors [2], and as of now, there are ~40 reported cases in the literature [3]. Many CNSETs are found incidentally on imaging, although some patients present with abdominal pain or Cushing syndrome [3]. Grossly, CNSET is an unencapsulated, well-defined, large, and multilobulated mass. Calcification and ossification are often identified by both imaging studies and histologic evaluation. Microscopically, in addition to calcification, CNSET shows characteristic nests of epithelioid cells with palely eosinophilic cytoplasm and round to ovoid nuclei with inconspicuous or small nucleoli, surrounded by a myofibroblastic proliferation with abundant fibrosis. Immunohistochemically, CNSET shows nuclear immunoreactivity for β-catenin and WT-1. To date, most tumors have been reported to follow a benign course, although there are rare reports of multifocal liver recurrence and distant metastasis [3, 4].
Genetic data are limited to a single report in which in-frame CTNNB1 exon 3 deletions were found in two tumors that underwent directed CTNNB1 sequencing [5]; to date, there are no published next-generation sequencing studies of this tumor type. Here, we report a comprehensive clinicopathologic evaluation of a series of eight CNSETs. We also performed next-generation DNA sequencing and additional targeted TERT promoter sequencing, with the aim of gaining better insight into the molecular pathogenesis of CNSET.
Materials and methods
Eight tumors diagnosed as CNSET were obtained: five from the consultation files of one of the authors (C.D.M.F.) and one each from three institutions (X.Z., R.F., and O.B.). Any available hematoxylin and eosin slides and immunohistochemistry were reviewed for each case, and clinical follow-up data were obtained from referring pathologists or clinicians if possible (see “Acknowledgements”). This study was approved by institutional review.
Immunohistochemistry at Brigham and Women’s Hospital was performed on tissue sections prepared from formalin-fixed, paraffin-embedded (FFPE) sections using citrate-buffered pressure cooker antigen retrieval with the following antibodies and conditions: β-catenin (BD Biosciences, San Jose, CA; dilution 1:1000) on four tumors; WT-1 (Cell Marque, Rocklin, CA; clone: 6F-H2; dilution: 1:100) on five tumors; HepPar-1 (Dako products, Agilent Technologies, Santa Clara, CA; clone: OCH1E5; dilution: 1:600) on five tumors; and arginase-1 (Sigma-Aldrich, St. Louis, MO; rabbit polyclonal antibody; dilution: 1:1500) on six tumors. Albumin RNA in situ hybridization (Advanced Cell Diagnostics Inc., Newark, CA; albumin probe: RS7752) was performed on four tumors by one of the authors (X.Z.) at Yale, using a Leica RNAscope detection system (Leica Biosystems, Wetzlar, Germany). Other stains in this study were performed by coauthors (X.Z., R.F., and O.B.) at their respective institutions during clinical workup.
One tumor was sequenced as part of its clinical workup, using UW-OncoPlex testing performed at the University of Washington [6]. In the present study, next-generation DNA sequencing was performed for six tumors not already sequenced for clinical purposes, using methods described previously in detail [7]. In brief, tissue sections cut from FFPE tissue were macrodissected to enrich tumor content. DNA was isolated from macrodissected tumor and at least 50 ng input DNA was used to prepare libraries with Illumina TruSeq LT reagents (Illumina Inc., San Diego, CA). Hybrid capture sequencing was performed using a custom RNA bait set (Agilent Technologies, Santa Clara, CA) for the entire coding regions of 447 genes and for intronic regions of 60 genes for rearrangement detection (“OncoPanel” version 3; Supplemental Table 1). Massively parallel sequencing was performed using an Illumina HiSeq 2500 (Illumina Inc., San Diego, CA).
TERT promoter mutations were detected via OncoPanel sequencing, although the coverage of the TERT promoter was insufficient to be certain of the significance of these findings. Thus, due to the suggestive but incomplete OncoPanel results, additional bi-directional Sanger sequencing of the TERT promoter was performed on isolated DNA at NeoGenomics (Fort Myers, FL).
Results
Clinical findings
The tumors occurred in six females and two males (Table 1), with an age at presentation ranging from 14 to 34 years (median 22.5 years). Four patients had multiple liver tumors at presentation; in three of four, the largest tumor involved segments 5–6 of the right lobe. All three tumors that were unifocal at presentation occurred in the right lobe.
The reported presenting symptoms were abdominal pain (four patients), weight loss (one), and fatigue (one). Clinical diagnoses were hemangioma (two patients), metastatic tumor (two), focal nodular hyperplasia (one), hepatocellular carcinoma (one), and soft tissue sarcoma (one). The median preoperative duration was 8 months (range 1 month–2 years).
Clinical follow-up data were available for seven of eight patients (88%), with a median follow-up duration of 4.4 years (range 1.2–6.2 years) for patients alive with disease at the most recent follow-up. All seven patients underwent partial hepatectomies; two resections had positive margins. Four patients (57%) had both local recurrence and metastases to the lung (4/4) and peritoneum or abdominal lymph nodes (2/4). Two of these patients had positive margins in the initial resection, while the other two had negative margins. Two of these four patients had metastatic disease at presentation. One of these patients developed local recurrence and lung metastases 2 months after initial resection and died of disease 4 months later (11 months after initial diagnosis). Lastly, one of these patients developed local recurrence 1 year after initial resection, followed by six additional local recurrences treated with resections or radiofrequency ablations, and peritoneal and lung metastases 5 years after the initial surgery. No other patients developed recurrent or metastatic disease. Overall, one patient died of disease (patient 1) and the other three patients with metastases were alive with disease at 1.2, 4.4, and 5.3 years of follow-up (patients 2–4; Table 1).
One patient with residual disease after surgery (patient 3; Table 1) received adjuvant chemotherapy with etoposide and cisplatin, with no change in tumor size on follow-up imaging. Three patients received neoadjuvant chemotherapy (patients 1, 4, and 5; Table 1). One patient (patient 5) with the multifocal disease received neoadjuvant chemotherapy with 13 cycles of vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide and received no adjuvant chemotherapy. This patient’s tumor exhibited 90% fibrosis, consistent with the treatment effect, and the patient did not develop recurrence or metastasis in 6.2 years of follow-up. Another patient with lung and abdominal metastases at presentation (patient 4) received neoadjuvant vincristine–irinotecan, with a reported mild reduction in tumor size on imaging but with no apparent treatment response in the resection.
Gross and microscopic findings
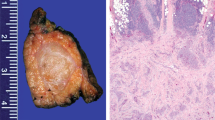
Gross descriptions were available for five tumors, which were described as tan/white (four tumors), yellow-white (one), firm (one), and lobulated (three); one tumor was noted to have calcifications on gross inspection, and another resection was noted to have satellite tumor nodules. Size ranged from 2.7 to 18 cm (median 14 cm). Among tumors sent for diagnostic consultation, suggested diagnoses from referring pathologists included poorly differentiated carcinoma (three tumors), CNSET (two), biphasic synovial sarcoma (one), large cell neuroendocrine carcinoma (one), interdigitating dendritic cell neoplasm (one), fibrolamellar carcinoma (one), and undifferentiated embryonal sarcoma of the liver (one).
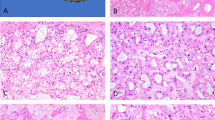
Histologically, all CNSETs were composed of organoid nests of epithelioid cells with moderate amounts of pale cytoplasm and round to ovoid, vesicular nuclei (Figs. 1–3). Tumors showed variably cellular myofibroblastic proliferation with myxoid to collagenous stroma between organoid nests of tumor cells. Three tumors lacked calcifications. Lymphovascular invasion was identified in one tumor. No necrosis was identified in any tumor. Most tumors had prominent associated ductular proliferation: in four, the ductular proliferation concentrically surrounded nests of tumor cells, while in three other tumors there was haphazardly distributed ductular proliferation within the dense stroma between tumor nests (Fig. 4). Three tumors exhibited focal clear-cell features (Fig. 5) and one showed focal pseudo-glandular architecture (Fig. 6). Mitotic indices ranged from 2 to 22 mitoses per 10 high-powered fields (2.38 mm2), and higher mitotic indices tended to be associated with multifocal disease and/or distant metastases (Table 1).

Calcifying nested stromal–epithelial tumors exhibited organoid tumor nests in a dense fibrous stroma.

A Tumor cells were epithelioid and monomorphic, with ovoid nuclei exhibiting minimal atypia. B Mitotic indices ranged from 2 to 22 mitoses per high-powered field (HPF). This example (from patient 1; Table 1) had 22 mitoses per 10 HPF and was multifocal at presentation.

The neoplastic cells in this tumor showed somewhat spindled cytomorphology.

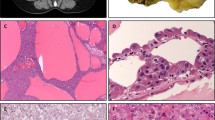
A Four tumors had ductular proliferations arranged concentrically around tumor nests. B Three other tumors had prominent ductular proliferations that were more haphazardly distributed between nests of tumor cells.

Clear-cell features were present focally in three tumors.

One tumor had focal pseudo-glandular architecture.
Immunohistochemical and in situ hybridization findings
Immunohistochemistry for β-catenin demonstrated nuclear positivity in five of five tested tumors, and WT-1 was positive in five of seven (Fig. 7A, B). Broad-spectrum keratins were positive in five of six tumors, typically more weakly than in adjacent ductular proliferations (Fig. 7C); one tumor treated with neoadjuvant chemotherapy was negative for keratins. Epithelial membrane antigen, hepatocellular markers (HepPar-1, arginase-1, and albumin in situ hybridization), and neuroendocrine markers were uniformly negative in all tested tumors (Fig. 7D). Immunohistochemistry and in situ hybridization results are summarized in Table 2.

A Five of five tested tumors exhibited nuclear β-catenin positivity. B Five of seven tumors exhibited nuclear WT-1 positivity. C CAM5.2 was positive in five of six tumors, as was pan-K. As shown here, broad-spectrum keratins were more strongly positive in adjacent ductular proliferations than in tumor cells. D Arginase-1 was negative in all six tested tumors; this image highlights the contrast between negative tumor cells and positive adjacent hepatocytes. Other consistently negative stains included HepPar-1, albumin (in situ hybridization), synaptophysin, chromogranin, and INSM1. Immunohistochemistry findings are summarized in Table 2.
Molecular findings
One tumor sequenced during clinical workup was found to harbor an unspecified CTNNB1 mutation. Six other tumors were sequenced as part of this study and were found to harbor CTNNB1 deletions or activating hotspot mutations. The deletions included two involving exon 3—CTNNB1 c.94_189del (p.D32_K63del) and CTNNB1 c.78_182del (p.Q27_Q61del)—and one involving exons 3 and 4—CTNNB1 c.108_290del (p.S37_A97del). The point mutations were T41A (two tumors) and S33F (one tumor). TERT promoter sequencing was performed for the same six tumors, which were all found to harbor TERT promoter hotspot mutations: −146C > T (three tumors) and −124C > T (three tumors). Two sequenced tumors were found to have multiple chromosomal copy number gains, including in chromosomes 8 and 12 in both tumors. Full molecular findings for each tumor are reported in Supplemental Table 2.
Discussion
CNSET is a rare primary hepatic tumor that occurs predominantly in young women, including children and young adults. Compared to previous reports, our series contained a higher proportion of tumors occurring in adults. CNSET has been reported to sometimes give rise to Cushing syndrome, with adrenocorticotropic hormone production [8]. No patients in our series were reported to have these symptoms, and instead, they most often presented with abdominal pain. An association between CNSET and Beckwith–Wiedemann syndrome has been reported, although it was not identified in our series [9].
In the ~40 reported CNSETs in the literature, there are only three reports of local recurrence and one report of a patient developing lung metastases [4, 10,11,12,13]. In contrast, over half of the patients in our series (four of seven for whom follow-up was available) developed lung metastases, and two of these four patients also developed peritoneal or abdominal lymph node metastases. Three of these four patients had multifocal liver disease and/or metastatic disease at presentation. The mitotic index seemed to correlate with the development of metastases; however, given the small number of tumors in this series, it is difficult to assess how accurately the mitotic rate can predict aggressive behavior. Based on these data, multifocal disease at presentation seems to be a poor prognostic factor. Given that most tumors in this series were reviewed in diagnostic consultation, the high metastatic rate could reflect an inherent bias in diagnostic consultations, which are more likely to have unusual or aggressive clinical presentations.
CTNNB1 alterations and activating TERT promoter mutations were present in all tested cases, making them remarkably consistent findings in this tumor type. CTNNB1 alterations included an even mixture of in-frame deletions and hotspot mutations. CTNNB1 exon 3 deletions and point mutations are present in many tumor types, including other liver tumors such as hepatocellular carcinoma, combined hepatocellular–cholangiocarcinoma, and hepatoblastoma [14,15,16]. In our series, all tumors with CTNNB1 deletions were multifocal at presentation and gave rise to distant metastases; in contrast, only one tumor with CTNNB1 point mutation was multifocal at presentation, and none gave rise to distant metastases. While these findings suggest that CTNNB1 deletions might portend a worse prognosis in CNSET, the significance of this association remains uncertain due to the overall low number of sequenced tumors. One patient with multifocal disease at presentation died of disease 11 months after initial diagnosis, representing an unusually aggressive clinical course compared to other tumors in this series and in the literature. In addition to a CTNNB1 exon 3 deletion, this patient’s tumor also harbored several chromosomal copy number alterations, which was an uncommon finding; we speculate that these additional changes might have contributed to the unusually aggressive behavior of this tumor.
TERT promoter mutations are associated with aggressive behavior in other tumor types, including papillary thyroid carcinoma and solitary fibrous tumor [17, 18]. However, in our series, TERT promoter mutations were present in all tumors and therefore were not predictive of aggressive behavior. TERT promoter mutations are frequently present in hepatocellular carcinoma [19], where they are often associated with CTNNB1 alterations [20]. β-Catenin has been shown to regulate TERT expression and telomere length in both non-neoplastic and neoplastic tissue [21]. Given the frequent co-occurrence of TERT and CTNNB1 alterations in CNSET and hepatocellular carcinoma, but infrequent co-occurrence in other neoplasms, it is possible that concurrent alterations are an important aspect of tumorigenesis in the liver.
Clinical follow-up data were available for three patients treated with neoadjuvant chemotherapy; one had metastatic disease at presentation, while the other two had multifocal liver disease but no distant metastases at presentation. The patient with metastatic disease at presentation received vincristine–irinotecan for 6 months preoperatively; there was a mild decrease in tumor size on imaging, but there was no significant histologic response in the resection. One patient with only multifocal local disease was treated with 13 cycles of vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide, with a 90% histologic tumor response in the resection. The other patient with only multifocal local disease at presentation was treated with four cycles of doxorubicin, with no information available regarding tumor response; this patient developed liver recurrence and lung metastases 2 months after surgery and died of disease 4 months later (11 months after initial diagnosis). Thus, while neoadjuvant chemotherapy might provide benefit and could be considered in patients presenting with multifocal liver disease, factors that might predict favorable clinical outcomes remain unclear.
The main differential diagnostic consideration for CNSET is hepatoblastoma, which occurs in young patients and frequently expresses β-catenin [22]. In particular, the fetal subtype exhibits similar architecture, with nests of primitive tumor cells. However, in contrast to CNSET, the fetal subtype of hepatoblastoma does not contain dense fibrous stroma, and it consistently expresses hepatocellular markers [23].
The differential diagnosis also includes hepatocellular carcinoma, which generally exhibits more cytologic atypia than CNSET and expresses hepatocellular markers such as HepPar-1, arginase-1, and albumin (as detected via in situ hybridization); these stains were negative in all tested cases of CNSET. The scirrhous variant of hepatocellular carcinoma can exhibit similarly nested growth with abundant fibrous stroma. However, scirrhous hepatocellular carcinoma occurs in older men, often demonstrates higher histologic grade, and maintains some immunoreactivity for hepatocellular markers.
CNSET is a rare, distinctive primary hepatic neoplasm that occurs in children and young adults. CTNNB1 and TERT promoter mutations were found to co-occur in all sequenced tumors, and no other recurrent mutations were found on next-generation sequencing. The behavior of the tumors in this series suggests that CNSET might perhaps be more aggressive than previously appreciated and that close clinical follow-up is warranted after resection.
Data availability
The data generated and/or analyzed for this study are available from the corresponding author upon reasonable request.
References
Geramizadeh B. Nested stromal-epithelial tumor of the liver: a review. Gastrointest Tumors. 2019;6:1–10.
Ishak KG, Goodman ZD, Stocker JT. Miscellaneous malignant tumors (Chapter 11). In: Rosai J, Sobin L, editors. Tumors of the liver and intrahepatic bile ducts. Washington: Armed Forces Institute of Pathology; 2001. p. 276–8.
Benedict M, Zhang X. Calcifying nested stromal-epithelial tumor of the liver an update and literature review. Arch Pathol Lab Med. 2019;143:264–8.
Heerema-McKenney A, Leuschner I, Smith N, Sennesh J, Finegold MJ. Nested stromal epithelial tumor of the liver: Six cases of a distinctive pediatric neoplasm with frequent calcifications and association with Cushing syndrome. Am J Surg Pathol. 2005;29:10–20.
Assmann G, Kappler R, Zeindl-Eberhart E, Schmid I, Häberle B, Graeb C, et al. β-Catenin mutations in 2 nested stromal epithelial tumors of the liver - a neoplasia with defective mesenchymal-epithelial transition. Hum Pathol. 2012;43:1815–27.
Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16:56–67.
Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of oncopanel a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–8.
Weeda VB, De Reuver PR, Bras H, Zsíros J, Lamers WH, Aronson DC. Cushing syndrome as presenting symptom of calcifying nested stromal-epithelial tumor of the liver in an adolescent boy: a case report. J Med Case Rep. 2016;10:4–7.
Khoshnam N, Robinson H, Clay MR, Schaffer LR, Gillespie SE, Shehata BM. Calcifying nested stromal-epithelial tumor (CNSET) of the liver in Beckwith-Wiedemann syndrome. Eur J Med Genet. 2017;60:136–9.
Brodsky SV, Sandoval C, Sharma N, Yusuf Y, Facciuto ME, Humphrey M, et al. Recurrent nested stromal epithelial tumor of the liver with extrahepatic metastasis: case report and review of literature. Pediatr Dev Pathol. 2008;11:469–73.
Heywood G, Burgart LJ, Nagorney DM. Ossifying malignant mixed epithelial and stromal tumor of the liver: a case report of a previously undescribed tumor. Cancer. 2002;94:1018–22.
Makhlouf HR, Abdul-Al HM, Wang G, Goodman ZD. Calcifying nested stromal-epithelial tumors of the liver. Am J Surg Pathol. 2009;33:976–83.
Hommann M, Kaemmerer D, Daffner W, Prasad V, Baum RP, Petrovitch A, et al. Nested stromal epithelial tumor of the liver-liver transplantation and follow-up. J Gastrointest Cancer. 2011;42:292–5.
Yamaoka H, Ohtsu K, Sueda T, Yokoyama T, Hiyama E. Diagnostic and prognostic impact of β-catenin alterations in pediatric liver tumors. Oncol Rep. 2006;15:551–6.
Perugorria MJ, Olaizola P, Labiano I, Esparza-Baquer A, Marzioni M, Marin JJG, et al. Wnt–β-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol. 2019;16:121–36.
Joseph NM, Tsokos CG, Umetsu SE, Shain AH, Kelley RK, Onodera C, et al. Genomic profiling of combined hepatocellular-cholangiocarcinoma reveals similar genetics to hepatocellular carcinoma. J Pathol. 2019;248:164–78.
Wong KS, Higgins SE, Marqusee E, Nehs MA, Angell T, Barletta JA. Tall cell variant of papillary thyroid carcinoma: impact of change in WHO definition and molecular analysis. Endocr Pathol. 2019;30:43–8.
Bahrami A, Lee S, Schaefer I-M, Boland JM, Patton KT, Pounds S, et al. TERT promoter mutations and prognosis in solitary fibrous tumor. Mod Pathol. 2016;29:1511–22.
Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:1–6.
Nault JC, Martin Y, Caruso S, Hirsch TZ, Bayard Q, Calderaro J, et al. Clinical impact of genomic diversity from early to advanced hepatocellular carcinoma. Hepatology. 2020;71:164–82.
Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, et al. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549–54.
Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014;147:690–701.
López-Terrada D, Alaggio R, De Dávila MT, Czauderna P, Hiyama E, Katzenstein H, et al. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol. 2014;27:472–91.
Acknowledgements
We thank the following pathologists and clinicians who kindly provided case material and clinical follow-up information when available: Prof. A.P. Dei Tos (Padova, Italy), Dr. N. Donner (Amsterdam, The Netherlands), Dr. W.R. Jeck (Durham, NC), Dr. P. Puspanathan (Alor Setar, Malaysia), Dr. J.E. Rostedt (Tacoma, WA), Dr. C.D. Savci Heijink (Amsterdam, The Netherlands), Dr. S.A. Whitworth (Denver, CO), and Dr. X. Zhang (Cleveland, OH).
Funding
No external funding was used for this project, which was supported through intradepartmental funds.
Author information
Authors and Affiliations
Contributions
D.J.P., C.D.M.F., and L.Z. conceived of and designed this study. All authors (D.J.P., F.D., X.Z., R.K., O.B., C.D.M.F., and L.Z.) performed data acquisition, analysis, and/or interpretation. D.J.P., C.D.M.F., and L.Z. wrote the manuscript. All authors revised the manuscript, and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval/consent to participate
This retrospective study was approved by the Institutional Review Board of Mass General Brigham (protocol #2019P002519). No human subjects were recruited for the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Papke Jr., D.J., Dong, F., Zhang, X. et al. Calcifying nested stromal–epithelial tumor: a clinicopathologic and molecular genetic study of eight cases highlighting metastatic potential and recurrent CTNNB1 and TERT promoter alterations. Mod Pathol 34, 1696–1703 (2021). https://doi.org/10.1038/s41379-021-00822-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00822-w
