
Abstract
ZFP42 zinc finger protein (REX1), a pluripotency marker in mouse pluripotent stem cells, has been identified as a tumor suppressor in several human cancers. However, the function of REX1 in cervical cancer remains unknown. Both IHC and western blot assays demonstrated that the expression of REX1 protein in cervical cancer tissue was much higher than that in normal cervical tissue. A xenograft assay showed that REX1 overexpression in SiHa and HeLa cells facilitated distant metastasis but did not significantly affect tumor formation in vivo. In addition, in vitro cell migration and invasion capabilities were also promoted by REX1. Mechanistically, REX1 overexpression induced epithelial-to-mesenchymal transition (EMT) by upregulating VIMENTIN and downregulating E-CADHERIN. Furthermore, the JAK2/STAT3-signaling pathway was activated in REX1-overexpressing cells, which also exhibited increased levels of p-STAT3 and p-JAK2, as well as downregulated expression of SOCS1, which is an inhibitor of the JAK2/STAT3-signaling pathway, at both the transcriptional and translational levels. A dual-luciferase reporter assay and qChIP assays confirmed that REX1 trans-suppressed the expression of SOCS1 by binding to two specific regions of the SOCS1 promoter. Therefore, all our data suggest that REX1 overexpression could play a crucial role in the metastasis and invasion of cervical cancer by upregulating the activity of the JAK2/STAT3 pathway by trans-suppressing SOCS1 expression.
Similar content being viewed by others


Introduction
Cervical cancer is the fourth most commonly diagnosed cancer worldwide and the second leading cause of cancer-related death among women in developing countries [1]. Epidemiological case series have shown that 99.7% of cervical carcinoma cases are positive for human papillomavirus (HPV) [2]. HPV16 and HPV18 are the high-risk, carcinogenic HPV genotypes that account for ~55–60% and 10–15% of all cervical cancers, respectively [2,3,4]. Although high-risk HPV is the most important carcinogenic factor for cervical cancer development [2], including the activation of oncogenes and inactivation of tumor suppressors, are also necessary for tumorigenesis and the development of cervical cancer. For example, p53 protein overexpression during cervical tumorigenesis could play a pivotal role in cervical cancer progression as a late event [5]. In contrast, promoter methylation and the loss of the expression of PTEN, which acts as an antioncogene, occur frequently in carcinomas of the uterine cervix [6]. Our laboratory also clarified that some stem cell-related genes are associated with the carcinogenesis of cervical cancer. For example, EZH2 and DAX1 can promote WNT/β-CATENIN signaling-dependent cell expansion in cervical carcinoma [7, 8]. Moreover, SLUG, SOX9, and KLF4 have been demonstrated to suppress cell proliferation in vitro and tumor growth in vivo by different mechanisms [9,10,11].
REX1 (reduced expression-1), also known as ZFP42 (zinc finger protein-42), is a member of the zinc finger protein family of transcription factors, and the expression of REX1 is rapidly reduced by retinoic acid in F9 teratocarcinoma cells. The REX1 gene encodes a protein containing four Cys-His-type zinc finger domains with similarities to the Yin Yang-1 (YY1) transcription factor [12, 13]. REX1 is a widely used genetic marker of pluripotency in human stem cells, and the expression of REX1 is positively linked to increased pluripotency in human and mouse embryonic stem cells [14, 15]. Furthermore, REX1 plays different roles in different cancers. On the one hand, REX1 can act as an antioncogene in prostate cancer, clear cell renal cell carcinoma, and esophageal squamous cell carcinoma (ESCC) [16,17,18]. On the other hand, REX1 can act as an oncogene in malignant glioblastoma multiforme patients by inducing p38/JNK and AKT/PI3K/GSK3β signaling and by leading to apoptosis [19]. As far as we known, there is no report on the function of REX1 in cervical carcinoma.
In the present study, we are the first to reveal that REX1 could facilitate the migration and invasion of cervical cancer cells in vitro and distant tumor metastasis in vivo by activating the JAK2/STAT3-signaling pathway in cervical cancer through trans-suppressing the expression of suppressor of cytokine signaling 1 (SOCS1).
Results
The expression of REX1 protein in normal cervix and cervical carcinoma samples
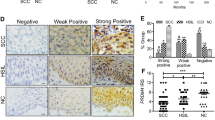
To explore the role of REX1 in cervical carcinogenesis, REX1 protein expression was detected in normal human cervix (NC, N = 30) and invasive cervical squamous cell carcinoma (SCC, N = 48) samples using immunohistochemistry (Fig. 1a). REX1 staining was classified into three categories: negative, weak positive, and strong positive. The percentage of strongly positive REX1 staining was 10.00% in NC tissue (3/30) and 47.9% in SCC tissue (23/48). The percentage of weakly positive REX1 staining was 36.7% in NC tissue (11/30) and 29.2% in SCC tissue (14/48). The percentage of negative REX1 staining was 53.3% in NC tissue (16/30) and 22.9% in SCC tissue (11/48). The positive rate (including weak positive and strong positive) of REX1 staining in SCC was significantly higher than that in NC (77.1% vs 46.7%, Fig. 1b, Table S1, p < 0.01). Moreover, the immunoreactivity score (IRS) of REX1 staining also increased from 3.567 ± 0.438 in NC samples to 6.354 ± 0.486 in SCC samples (Fig. 1c, p < 0.001). In addition, the expression level of REX1 was detected by western blotting in seven NC tissue samples and ten SCC samples, all of which were selected randomly (Fig. 1d). The expression level of REX1 relative to that of GAPDH was much higher in SCC tissue than in NC tissue (Fig. 1e, p < 0.001).

The expression of REX1 in normal cervix and cervical carcinomas samples. a Immunohistochemical (IHC) detection of REX1 in normal cervical samples (NC) and cervical squamous cell carcinoma (SCC) samples. REX1 staining was classified into three levels according to staining intensity (negative, weak positive, and strong positive), original magnification, ×400. b The bar chart showed the percentage of REX1-positive staining containing weak positive and strong positive in NC (n = 30) and SCC (n = 48). c The scatter plots showed the IHC scores obtained for the staining of REX1 in NC and SCC (points represent the IHC score per specimen, Student’s t-test is performed). d The expression of REX1 in NC (n = 7) and SCC (n = 10) samples was detected using western blotting. e The relative expression of REX1 in each tissue was shown. The data shown were the ratios of the REX1/GAPDH of each specimen and the means ± standard error of the NC and SCC groups. f Cervical cancer patients were classified into two groups (29 patients with distant metastasis and 275 patients with no distant metastasis) according to the TCGA clinical data, and the mRNA expression of REX1 of two groups in the TCGA database by RNAseq was shown. This dataset shows the gene-level transcription estimates of REX1, by RSEM normalized count, percentile ranked within each sample. Data were statistically analyzed with Student’s t-test and values are shown as the mean ± SD. g The relationship between survival probability of CESC patients (n = 304) and the expression level of REX1 in their tumors was shown by Kaplan–Meier estimator in the TCGA database. **p < 0.01, ***p < 0.001
Furthermore, we analyzed REX1 expression in 304 patients with cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) including 260 SCC, 22 endocervical type of adenocarcinoma, 15 mucinous adenocarcinoma of endocervical type, and 7 adenosquamous in The Cancer Genome Atlas (TCGA) RNAseq database. The patients were divided into two groups according to their distant metastasis status listed in the clinical information of the TCGA clinical database, including 29 patients with distant metastasis and 275 patients with no distant metastasis. The mRNA expression level of REX1 in the patients with distant metastasis was found to be significantly higher than that of the patients without distant metastasis (Fig. 1f, p < 0.001). Moreover, survival analysis of the correlation between REX1 expression and overall survival probability of CESC patients, as assessed by Kaplan–Meier estimator, showed that as REX1 expression increased, the probability of CESC patient survival decreased (Fig. 1g, p = 0.011). All of these results suggest that REX1 might function as a promoter of cervical carcinogenesis and development.
REX1 promotes distant metastasis in cervical cancer in vivo
Immunocytochemical assays and western blot analyses were performed in cervical cancer cell lines. It is revealed that REX1 expression was found in cervical cancer cell lines (Fig. S1a, b). To identify the effect of REX1 on cervical cancer in vivo, a total of 1 × 106 REX1-overexpressing SiHa or HeLa cells and the respective control cells were injected subcutaneously into female nude mice at the same time to assess tumor formation. As shown in Fig. S2, the volume and weight of the tumors formed by SiHa-REX1 and HeLa-REX1 cells were not significantly different from the same parameters of the tumors formed by the respective control cells (Fig. S2a–f). Surprisingly, macroscopic metastatic focuses, in which a large number of tumor cells infiltrated with inflammatory cells could be observed by staining with hematoxylin and eosin (H&E-stained), were observed in the liver and lung tissues of the xenograft mice injected with REX1-overexpressing cells (Fig. 2a, c). The average number of metastatic lesions observed under microscopy in a liver section from the SiHa-REX1 group was 3.5 ± 1.643, threefold more than that from the SiHa-GFP group (1.0 ± 0.6325, Fig. 2b, p < 0.01). Similarly, metastatic lesions were found in the lung tissue, with an average number of 4.0 ± 1.789 lesions in the HeLa-REX1 group, more than threefold higher than the 1.333 ± 0.8165 lesions in the HeLa-GFP group (Fig. 2d, p < 0.01).

REX1 overexpression promotes SiHa and HeLa cell metastasis in vivo. a–d Female BALB/c nude mice were injected subcutaneously with the SiHa and HeLa cells that overexpress REX1 (labeled SiHa-REX1 and HeLa-REX1) and the respective control cells (labeled SiHa-GFP and HeLa-GFP), n = 6 mice per group. Representative livers/lungs and hematoxylin and eosin-stained images presented to show tumor lesions in the livers/lungs (scale bar, 500 μm and 200 μm). The scatter plots showed the number of lesions in the livers/lungs as mean ± SD (n = 6). a, b SiHa-REX1 and SiHa-GFP cells. c, d HeLa-REX1 and HeLa-GFP cells. e–h Female BALB/c nude mice were injected via tail vein with the SiHa and HeLa cells that overexpress REX1 and the respective control cells, n = 6 mice per group. Representative lungs and hematoxylin and eosin-stained images presented to show metastatic nodules in the lungs (scale bar, 500 μm and 200 μm). The scatter plots showed the number of metastatic nodules in the lungs as mean ± SD (n = 6). e, f SiHa-REX1 and SiHa-GFP cells. g, h HeLa-REX1 and HeLa-GFP cells. For each sample, all micrometastases were counted under a light microscope at ×10 magnifications. Three sections were counted per mouse sample at 50 μm intervals and average the results. Data were statistically analyzed with Student’s t-test and values are shown as mean ± SD. **p < 0.01, ***p < 0.001
To further study the effect of REX1 on distant metastasis in vivo, 5 × 105 REX1-overexpressing SiHa or HeLa cells and the control cells were injected respectively into female nude mice via the tail vein. The organ metastases in nude mice were observed after one and a half months. The metastatic tumor lesions of the REX1-overexpressing group and the control group were counted under microscopy by H&E staining (Fig. 2e, g). The average number of metastatic lesions in one section of lung tissue (Fig. 2f) from the SiHa-REX1 group was 20.67 ± 6.713, threefold greater than that from the SiHa-GFP group (6.167 ± 2.85, p < 0.001). Similarly, the average number of metastatic lesions in one section of lung tissue (Fig. 2h) from the HeLa-REX1 group was 7.667 ± 3.011, fourfold more than that from the HeLa-GFP group (1.833 ± 1.835, p < 0.01). All of these results demonstrate that the REX1 protein promotes the distance metastasis of cervical cancer in vivo.
REX1 promotes the migration and invasion of cervical cancer cell lines in vitro
To explore the mechanism by which REX1 promotes the migration and invasion of cervical cancer, we evaluated the ability of REX1 overexpression to influence the motility of SiHa and HeLa cells using a wound-healing assay and a Transwell assay in vitro. In the wound-healing assay, SiHa-REX1 and HeLa-REX1 cells closed the wound to a greater extent in either direction than the SiHa-GFP and HeLa-GFP cells, respectively. A significant increase in wound closure was found with SiHa-REX1 cells after 48 h (p < 0.05) and 72 h (p < 0.001) and with HeLa-REX1 cells after 24 h (p < 0.001), 48 h (p < 0.001), and 72 h (p < 0.001) compared with wound closure by the respective control cells (Fig. 3a–d). Similarly, in a Transwell migration assay, after incubating for 48 h, the numbers of SiHa-REX1 and HeLa-REX1 cells (797.7 ± 187.5 and 972.9 ± 344.2, respectively) migrating across the uncoated membrane were threefold greater than those of SiHa-GFP and HeLa-GFP cells (246.1 ± 66.12 and 321.4 ± 183.4, respectively, p < 0.001) (Fig. 3e–g). All of these results demonstrate that REX1 overexpression significantly increases the migration capacity of SiHa and HeLa cells in vitro.

REX1 enhances migration and invasion ability of SiHa and HeLa cells in vitro. a, b The migratory potential of REX1-overexpressing SiHa or HeLa cells and the respective control cells was analyzed in a wound-healing assay for 0, 24, 48, and 72 h. Scale bar, 200 μm. c, d Scratch area of REX1-overexpressing SiHa or HeLa cells and the respective control cells was shown as mean ± SD from three independent experiments using triplicate measurements and statistically analyzed with Student’s t-test in each experiment. e, f The migratory potential of REX1-overexpressing SiHa or HeLa cells and the respective control cells was analyzed by the transwell cell migration assay. Scale bar, 100 μm. g Number of migratory cells was shown as mean ± SD from three independent experiments using triplicate measurements and statistically analyzed with Student’s t-test in each experiment. h, i The invasive potential of REX1-overexpressing SiHa or HeLa cells and the respective control cells was analyzed by the transwell cell invasion assay. Scale bar, 100 μm. g Number of invasive cells was shown as mean ± SD from three independent experiments using triplicate measurements and statistically analyzed with Student’s t-test in each experiment. *p < 0.05, ***p < 0.001
Furthermore, the effect of REX1 on the invasion capability of SiHa and HeLa cells was detected in a Transwell invasion assay where the cells invaded an artificial basement membrane made of Matrigel. The numbers of SiHa-REX1 and HeLa-REX1 cells (704.4 ± 169.1 and 393.5 ± 127.2, respectively) invading across the Matrigel membrane were threefold greater than those of SiHa-GFP and HeLa-GFP cells (249.2 ± 138.2 and 131.5 ± 19.44, respectively, p < 0.001), respectively, after incubating for 48 h (Fig. 3h–j). All these results demonstrate that REX1 overexpression enhances the migration and invasion capacities of SiHa and HeLa cells in vitro.
REX1 facilitates the epithelial–mesenchymal transition by regulating EMT-related proteins
To explore whether REX1 promotes the migration and invasion of cervical cancer, a transcriptome sequencing analysis of three HeLa-REX1 monoclonal cell lines and three HeLa-GFP monoclonal cell lines was performed (Fig. 4a). Gene Ontology (GO) enrichment analysis identified 33 epithelial-to-mesenchymal transition (EMT)-related genes, which are involved in the extracellular matrix, collagen trimer, cell junctions, or cytoskeleton, whose expression significantly changed between the two groups (Fig. 4b).

REX1 facilitates the epithelial–mesenchymal transition (EMT) via regulating the expression of EMT-related proteins. a Heatmap of all differentially expressed genes between REX1-overexpressing HeLa cells (three HeLa-REX1 lines, labeled HeLa-REX1-1, 2, 3) and the control cells (three HeLa-GFP lines, labeled HeLa-GFP-1, 2, 3) using transcriptome sequencing. b Heatmap visualization of the differentially expressed genes identified by transcriptome sequencing between three HeLa-REX1 lines and three HeLa-GFP lines enriched in the extracellular matrix, collagen trimer, cell junction, and cytoskeleton of the GO enrichment analysis. Data were log10 normalized. c The mRNA levels of EMT-related genes in SiHa-REX1 and SiHa-GFP cells detected by Real-time PCR analysis. d The mRNA levels of EMT-related genes in HeLa-REX1 and HeLa-GFP cells detected by Real-time PCR analysis. e The expression of EMT-related proteins in REX1-overexpressing SiHa or HeLa cells and the respective control cells was determined by western blot and f the quantitative analysis was shown. g The expression level of VIMENTIN and E-CADHERIN in REX1-overexpressing SiHa or HeLa cells and the respective control cells detected by Immunocytochemistry. Scale bar, 50 μm. Data represent mean ± SD of three independent experiments and statistical analyzed with Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001
Four classic EMT-related genes of the 33 genes, E-CADHERIN, VIMENTIN, MMP2, and MMP9, and other important modulators of EMT, SNAIL1, SLUG, ZEB1, ZEB2, and TWIST were chosen for validation. Real-time PCR showed that the mRNA expression levels of E-CADHERIN (using β-ACTIN as a reference gene) were significantly decreased, and the levels of MMP2, MMP9, and VIMENTIN (using β-ACTIN as a reference gene) were significantly increased (p < 0.05) in the REX1-overexpressing cells (the expression level of MMP2 was not significantly increased in HeLa-REX1 compared with that of the HeLa-GFP cells) compared with the respective control cells (Fig. 4c, d). The mRNA level of SNAIL1 was increased in SiHa-REX1 cells but decreased in HeLa-REX1 cells. The mRNA levels of SLUG, ZEB1, ZEB2, and TWIST (using β-ACTIN as a reference gene) were not altered significantly after REX1 overexpression in HeLa and SiHa cells compared with those in the respective control cells (Fig. 4c, d). Moreover, western blotting analysis showed that the protein expression level of E-CADHERIN was lower, and the protein expression levels of MMP9 and VIMENTIN were much higher in the REX1-overexpressing cells than the respective control cells, which was consistent with the mRNA results (Fig. 4e, f). Furthermore, an immunocytochemistry analysis showed a decrease in E-CADHERIN expression and an increase in VIMENTIN expression in SiHa-REX1 and HeLa-REX1 cells compared with the respective control cells (Fig. 4g). Immunohistochemical staining was also used to detect the expression of E-CADHERIN and VIMENTIN in lung tumor tissue from nude mice. E-CADHERIN expression was downregulated and VIMENTIN expression was upregulated in the lung metastatic foci of mice that received REX1-overexpressing cells compared with those of the mice that received the respective control cells (Fig. S3a, IRS shown in Fig. S3b). All of these results indicate that REX1 overexpression promotes the migration and invasion, as well as the metastasis of cervical cancer cells, possibly by regulating classical EMT-related genes, including MMP9, VIMENTIN, and E-CADHERIN.
REX1 activates the JAK2/STAT3 pathway by downregulating SOCS1 expression in SiHa and HeLa cells
To the best of our knowledge, these classical EMT-related genes are downstream targets of the JAK2/STAT3-signaling pathway, which is negatively regulated by the SOCS family [20,21,22,23,24,25,26,27]. Therefore, we investigated the expression of the key proteins of the JAK2/STAT3 pathway in the present study. As shown in Fig. 5, the protein levels of p-STAT3 (Tyr705), p-JAK2 (Tyr1007/1008), and JAK2 were much higher in the SiHa-REX1 and HeLa-REX1 cells than in the respective control cells (Fig. 5a, b, p < 0.05). However, the JAK2 mRNA expression level was lower in HeLa-REX1 cells than in HeLa-GFP cells, and there were no significant differences in JAK2 mRNA expression between SiHa-REX1 and SiHa-GFP cells (Fig. S3c), suggesting that JAK2 expression could be regulated at the protein level by upstream genes. Therefore, the negative regulators of the JAK2/STAT3 pathway, SOCS1 and SOCS3, were selected as target genes for our further research.

REX1 overexpression downregulates SOCS1 expression and activates JAK2/STAT3-signaling pathway. a The expression of REX1, SOCS1, SOCS3, JAK2, p-JAK2, STAT3, p-STAT3, and GAPDH in REX1-overexpressing SiHa or HeLa cells and the respective control cells were detected by western blotting and b the quantitative analysis was shown. c The mRNA expression levels of SOCS1 and SOCS3 in REX1-overexpressing SiHa or HeLa cells and the respective control cells detected by Real-time PCR analysis. d Immunohistochemical staining of SOCS1 and JAK2 in the lung metastatic nodules of mice that received REX1-overexpressing SiHa or HeLa cells and the respective control cells injected via tail vein and e the Immunoreactivity scores of SOCS1 and JAK2 were shown. Scale bar, 50 μm. f, g Fragments of the SOCS1 promoter were constructed to generate promoter reporter. The luciferase activity relative to Renilla control was measured in REX1-overexpressing SiHa or HeLa cells and the respective control cells. Data represent mean ± SD of three independent experiments and statistical analyzed with Student’s t-test. h A schematic diagram of the four possible specific binding regions of SOCS1 promoter by REX1. i, j The qChIP assay was shown in REX1-overexpressing SiHa or HeLa cells and the respective control cells and Immunoprecipitation by REX1 antibody and IgG antibody (as the negative control). Data represent mean ± SD of three independent experiments and statistical analyzed with Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001, ns: no significant
Western blotting and RT-PCR analysis showed that SOCS1 expression, at both the protein and mRNA levels, was lower in SiHa-REX1 and HeLa-REX1 cells than in the respective control cells (Fig. 5a–c, p < 0.05). However, the expression of SOCS3 at both the mRNA and protein levels was not significantly changed after REX1 overexpression in SiHa and HeLa cells (Fig. 5a–c). Therefore, we hypothesized that REX1 activates the JAK2/STAT3-signaling pathway by inhibiting the expression of SOCS1 rather than SOCS3. Moreover, SOCS1 protein expression was decreased, and JAK2 protein expression was increased in the lung metastatic foci of mice that probably received REX1-overexpressing cells compared with those of the mice that received the respective control cells (Fig. 5d, e, p < 0.001). To confirm the kinetic impact of REX1 on SOCS1 expression, 4 µg of recombinant REX1 plasmids or control plasmids were transiently transfected into 293T cells, SiHa and HeLa cells. The mRNA expression levels of SOCS1 and REX1 were detected by the RT-PCR assay every 12 h. With the time extension after REX1 plasmids transfection, SOCS1 mRNA was significantly decreased at 48 h in 293T cells, 60h in SiHa cells, and 72 h in HeLa cells (Fig. S4a–c, p < 0.05), suggesting that the expression of SOCS1 is regulated by REX1 in a time-dependent manner. To further study the regulation of the SOCS1/JAK2/STAT3-signaling pathway by REX1, different doses of recombinant REX1 plasmids or control plasmids were transiently transfected into 293T cells. As the concentration of the transfected REX1 plasmids increased, the expression of REX1 increased gradually; concomitantly, noticeable downregulation of SOCS1 and upregulation of JAK2, p-JAK2, and p-STAT3 proteins occurred (Fig. S4d, e, p < 0.05), suggesting that the regulation of the SOCS1/JAK2/STAT3-signaling pathway by REX1 is dose dependent in 293T cells. All these results suggest that REX1 overexpression might activate the JAK2/STAT3 pathway and regulate the expression of EMT-related genes by inhibiting the mRNA and protein expression of SOCS1.
To assess whether REX1 activates the JAK2/STAT3 pathway by repressing the promoter of SOCS1, a dual-luciferase reporter assay was performed to study the activity of the SOCS1 promoter. Reporter constructs containing the full-length SOCS1 promoter region, from −2024 to +504 (the SOCS1 transcription start site is at 0), and other truncated fragments were transiently transfected into REX1-overexpressing cells and the respective control cells. The luciferase activity of the full-length SOCS1 promoter in both HeLa-REX1 and SiHa-REX1 cells was significantly lower than that in the respective control cells (Fig. 5f, g, p < 0.001). However, there was no significant difference in the luciferase activity of the reporters containing other truncated fragments between the REX1-overexpressing cells and the respective control cells, suggesting that REX1 trans-suppresses SOCS1 expression by binding to the −2024 to −1596 region of the SOCS1 promoter in SiHa and HeLa cells.
Furthermore, a quantitative chromatin immunoprecipitation (qChIP) assay was performed to determine whether REX1 protein binds to the −2024 and −1596 regions of the SOCS1 promoter. Four pairs of primers were designed to amplify the four P1–P4 fragments of the −2024 and −1596 SOCS1 promoter regions (Fig. 5h and Table S2). As shown in Fig. 5i, significantly more P1 and P3 promoter fragments were amplified by RT-PCR in SiHa-REX1 cells than in SiHa-GFP cells after immunoprecipitation with the REX1 antibody (p < 0.001). However, there was no significant difference in the amplification of the P2 and P4 promoter fragments by RT-PCR between the SiHa-REX1 cells and SiHa-GFP cells after immunoprecipitation with the REX1 antibody. Furthermore, there was no significant difference in the amplification of any of the four P1–P4 promoter fragments by RT-PCR between the SiHa-REX1 cells and SiHa-GFP cells after immunoprecipitation with the IgG control antibody. Similar results were also acquired with HeLa-REX1 and HeLa-GFP cells (Fig. 5j). All of these results indicate that REX1 binds to the P1 and P3 fragments of the SOCS1 promoter to trans-suppress SOCS1 in SiHa and HeLa cells.
SOCS1 overexpression or treatment with the JAK2/STAT3 inhibitor WP1066 inhibits REX1-induced SiHa and HeLa cell migration and invasion
To further confirm that REX1 promotes the migration and invasion of SiHa and HeLa cells by trans-suppressing SOCS1 and activating the JAK2/STAT3-signaling pathway, recombinant SOCS1 (GV492-SOCS1-3Flag-GFP) and control plasmids were transiently transfected into SiHa-REX1 and HeLa-REX1 cells, respectively. As shown in Fig. 6, the JAK2, p-JAK2 (Tyr1007/1008), p-STAT3 (Tyr705), and VIMENTIN protein levels were much lower in the SiHa-REX1 and HeLa-REX1 cells transfected with the SOCS1 plasmids than the cells transfected with control plasmids (Fig. 6a, b, p < 0.05). In addition, the migration and invasion of the SiHa-REX1 and HeLa-REX1 cells transfected with the recombinant SOCS1 plasmids were much weaker than those of the cells transfected with control plasmids (Fig. 6c, d, p < 0.01).

SOCS1 overexpression or treatment with the JAK2/STAT3 inhibitor WP1066 inhibits REX1-induced SiHa and HeLa cell migration and invasion. a The expression levels of SOCS1, JAK2, p-JAK2, STAT3, p-STAT3, VIMENTIN, and GAPDH protein in REX1-overexpressing SiHa or HeLa cells transiently transfected with recombinant SOCS1 plasmids or control plasmids were detected by western blotting and b the quantitative analysis was shown. c, d The migratory and invasive potential of REX1-overexpressing SiHa or HeLa cells transiently transfected with recombinant SOCS1 plasmids or control plasmids were analyzed by the transwell cell migration and invasion assay. Scale bar, 100 μm. Number of migratory and invasive cells was shown as mean ± SD from three independent experiments using triplicate measurements and statistically analyzed with Student’s t-test in each experiment. e The expression levels of JAK2, p-JAK2, STAT3, p-STAT3, VIMENTIN, and GAPDH protein in REX1-overexpressing SiHa or HeLa cells treated with WP1066 or DMSO were detected by western blotting and f the quantitative analysis was shown. g, h The migratory and invasive potential of REX1-overexpressing SiHa or HeLa cells treated with WP1066 or DMSO were analyzed by the transwell cell migration and invasion assay. Scale bar, 100 μm. Number of migratory and invasive cells was shown as mean ± SD from three independent experiments using triplicate measurements and statistically analyzed with Student’s t-test in each experiment. i Proposed model of the REX1-mediated transcription inhibition of SOCS1 and JAK2/STAT3 signaling activated in cervical cancer cells. *p < 0.05, **p < 0.01, ***p < 0.001
Furthermore, SiHa and HeLa cells were treated with a STAT3-specific activator, Colivelin. As shown in Fig. S5, SiHa and HeLa cells treated with Colivelin for 8 h and 24 h at concentrations of 1 μmol/mL and 2 μmol/mL, respectively. The levels of p-STAT3(Tyr705) and VIMENTIN protein were increased compared with those in the untreated control cells (Fig. S5a, b, p < 0.05). In addition, after Colivelin treatment, SiHa and HeLa cells acquired much stronger abilities in the cell migration and invasion (Fig. S5c, p < 0.05). Therefore, the activation of STAT3 pathway promotes the migration and invasion of cervical cancer cells. Moreover, the JAK2/STAT3-specific inhibitor WP1066 was used to further assess whether the activation of the JAK2/STAT3-signaling pathway is indispensable for the migration and invasion of cervical cancer cells affected by REX1. WP1066 has been shown to inhibit the activity of p-JAK and p-STAT3 and reduce the level of JAK2 protein. Therefore, as shown in Fig. 6e, f, in SiHa-REX1 and HeLa-REX1 cells treated with WP1066 for 24 h at concentrations of 2 µmol/mL and 4 µmol/mL, respectively, the level of VIMENTIN protein as well as the levels of JAK2, p-JAK2 (Tyr1007/1008), and p-STAT3 (Tyr705) protein was decreased compared with those in the corresponding untreated cells (p < 0.01). In addition, the migration and invasion of SiHa-REX1 and HeLa-REX1 cells were reduced by treatment with the inhibitor WP1066 (Fig. 6g, h, p < 0.001).
All of these results further confirmed that REX1 promotes the migration and invasion capabilities of cervical cancer cells by trans-suppressing SOCS1 and activating the JAK2/STAT3-signaling pathway (Fig. 6i).
Discussion
It has been shown that the expression of REX1 is related to increased pluripotency in many pluripotent self-renewing cells, including mES cells as well as human ES and iPS cells [15, 28,29,30,31]. The function of REX1 in cancer is still controversial. REX1 in prostate cancer, renal carcinoma and ESCC migh be a tumor suppressor gene, but in glioblastoma multiforme REX1 is an oncogene [16,17,18,19, 32]. As we known, REX1 is a zinc finger protein of YY1 family. YY1 could promote or inhibit tumor growth. For example, YY1 could inhibit the growth of pancreatic ductal adenocarcinoma [33], but stimulate the growth of melanoma cells [34]. The molecular mechanisms underlying the conflicting effects of YY1 might depend on the type of cancer cells. Therefore, the function of REX1, which might be similar to that of YY1, was also depend on the type of cancer cells. As far as we know, there are no reports about the relationship between REX1 and cancer metastasis, and there are also no reports about the role of REX1 in cervical cancer. The present study is the first report to study REX1 in cervical cancer and the first to potentially link REX1 overexpression with cancer metastasis.
The rates of cervical cancer diagnosis in developed countries have decreased dramatically because of cytologic screening and DNA testing for high-risk HPVs [35]. However, throughout the world, cervical cancer remains a considerable problem with 250,000 deaths annually attributed to cervical cancer [36]. Although early-stage and locally advanced cancers may be cured with radical surgery and chemoradiotherapy, patients with metastatic cancer have limited treatment options and extremely low survival rates [36]. The 5-year survival rate of metastatic cervical cancer is just 16.5% [37]. Therefore, it is essential to study the mechanism of cervical cancer metastasis. In the present study, the expression of REX1 in cervical carcinoma tissue was found to be higher than that in NC tissue. The TCGA database showed that the expression of REX1 was higher in cervical cancer patients with distant metastasis than in patients without metastasis. Furthermore, the Kaplan–Meier estimator survival analysis showed that as REX1 expression increased, the probability of CESC patient survival decreased (Fig. 1).
As REX1, which contains four C2H2 zinc finger motifs, is considered as a transcription factor, REX1 staining might be expected to be predominantly nuclear, as opposed to cytoplasmic. However, that was not the case. In recent studies, REX1 has been detected in the cytoplasm of cells in normal human renal tissue [32] and in both the nucleus and cytoplasm of all cells in the mouse blastocyst [38]. Our results showed that REX1 protein was detected in both the nucleus and cytoplasm of cervical cancer cell lines especially in the ICC assay (Fig. S1). Palko et al. [39] have demonstrated that the subcellular distribution pattern of YY1 alters during the cell cycle; YY1 is predominantly cytoplasmic in the G1 phase, primarily nuclear during the early and middle S phase and subsequently returns to the cytoplasm later in the S phase. We hypothesize that the expression of REX1 might be similar to that of YY1, the cellular localization of which is related to the cell cycle, but this hypothesis needs to be confirmed in a further study.
Moreover, we found that REX1 overexpression might promote distant metastasis of cervical cancer in vivo and promote the migration and invasion of cervical cancer cell lines in vitro (Figs. 2 and 3). It has been reported that EMT is a primary process that involves increased cervical cancer progression, invasion, and metastasis with a loss of epithelial markers, such as E-CADHERIN, and a gain of mesenchymal markers, such as VIMENTIN [40]. Our study found that overexpression of REX1 induced significant expression changes in several EMT-related genes, including downregulated expression of the epithelial marker E-CADHERIN, and upregulated expression of the mesenchymal marker VIMENTIN (Fig. 4). Our results suggested that REX1 could promote the metastasis of cervical cancer. This is the first report to link REX1 to the metastasis of cervical cancer, possibly through the EMT process.
It has been reported that activated JAK/STAT signaling is involved in metastasis in cervical carcinoma. In a study, positive p-STAT3 staining was observed in 56.8% of cervical SCC patients and was significantly correlated with lymph node metastasis and lymphovascular invasion [41]. STAT3 is reported to increase invasion and metastasis by inducing matrix metalloproteinase (MMP) expression [22]. STAT3 can also bind to an antisilencer element in the VIMENTIN promoter to enhance VIMENTIN gene expression and can directly or indirectly target certain transcriptional regulators of E-CADHERIN, such as HIF-1 and SLUG, to regulate EMT progression in cancer [20, 21]. Consistent with the above findings, we found that the levels of JAK2, p-JAK2 (Tyr1007/1008), and p-STAT3 (Tyr705) were upregulated in REX1-overexpressing cervical cancer cells (Fig. 5a, b, d, e), suggesting that REX1 promotes the metastasis of cervical cancer, probably through the activation of the JAK2/STAT3-signaling pathway.
It has been reported that SOCS1, a known JAK kinase-binding protein, can inhibit JAK/STAT3 activation directly through binding to the JAK2 activation loop with its SH2 domain and targeting JAK2 for ubiquitination and proteasomal degradation with its COOH terminal homology domain [40]. The downregulation of SOCS1 expression can activate the JAK2/STAT3-signaling pathway and elevate the expression levels of MMP9 in melanoma [42]. In the present study, both the mRNA and protein expression levels of SOCS1 were decreased after REX1 overexpression (Fig. 5a–e). The SOCS1 staining in SiHa-GFP cells was much higher than that in HeLa-GFP cells in the IHC assay, which was inconsistent with the western bolt results shown in Fig. 5a. The SOCS1 expression in SiHa-GFP and HeLa-GFP cells was not assessed under the same exposure conditions or on the same western blot membrane, but the SOCS1 expression in the IHC assay was analyzed under the same conditions and DAB chromogenic reaction time. Therefore, the expression of SOCS1 was detected on the same membrane by the western blot assay (shown in Fig. S6) and found to be consistent with the SOCS1 staining in the IHC assay in Fig. 5d.
Furthermore, the levels of JAK2, p-JAK2 (Tyr1007/1008), p-STAT3 (Tyr705), and VIMENTIN protein were reduced in REX1-overexpressing cells transiently transfected with recombinant SOCS1 plasmids or treated with the JAK2/STAT3 inhibitor WP1066, and the migration and invasion of the treated cells were reduced (Fig. 6a–h). Therefore, REX1 promotes the migration and invasion of cervical cancer cells by increasing the activity of the JAK2/STAT3-signaling pathway, probably by trans-suppressing SOCS1.
There was no statistical significance of the correlation between REX1 and SOCS1 gene expression in the TCGA database, whether in all 304 patients (CESC) or in the 29 patients with distant metastasis. There might be three explanations for the lack of correlation between REX1 and SOCS1 mRNA expression levels in the TCGA database. First, the gene expression of patients is influenced by many factors, such as pathological type, pathological stage, age, drug treatment, and basal gene expression level, which affect the mRNA expression of genes. Second, the pathological tissues of patients we obtained might include a variety of cell types, such as cervical cancer cells, stromal cells, and immune cells, which could influence the correlation of two genes in cancer cells. Third, the TCGA data are the mRNA levels of genes, but the mRNA expression level is not completely consistent with that of protein expression. In our study, we detected decreased expression of SOCS1 at the mRNA and protein levels in REX1-overexpressing cells, and confirmed by the dual-luciferase reporter assay and qChIP assays that REX1 trans-suppresses the expression of SOCS1 by binding to the SOCS1 promoter, which makes our results more convincing than the TCGA correlation results.
Sobti et al. [43] found that the mRNA and protein expression levels of SOCS1 in cervical cancer tissues were undetectable or reduced, and aberrant promoter methylation of SOCS1 was also found; thus, the transcriptional inactivation of the SOCS1 gene might be due to SOCS1 promoter hypermethylation. Kim et al. [44] demonstrated that histone deacetylation might be another regulatory mechanism involved in the regulation of SOCS1 expression in cervical cancer. In the present study, using the dual-luciferase reporter assay, we observed that the SOCS1 promoter was trans-suppressed in REX1-overexpressing cervical cancer cells (Fig. 5f–g) and qChIP assays confirmed that REX1 bound to two specific fragments of the SOCS1 promoter (Fig. 5h–j). These data indicate that REX1 probably acts as a transcriptional repressor to trans-suppress SOCS1 expression. Kim et al. [45] demonstrated that YY1 likely plays a role in the de novo DNA methylation of the differentially methylated regions of Peg3 and Xist during oogenesis. Other studies have found that YY1 negatively regulates the transcription of target genes by recruiting histone deacetylase and inducing the deacetylation of associated histones [46, 47]. Therefore, we wonder whether the mechanism of REX1-mediated SOCS1 transcriptional regulation in cervical cancer is similar to that of YY1, involving the promotion of target methylation or the recruitment of histone deacetylase to play a negative regulatory role. More in-depth research might be required to further define the molecular mechanism of the REX1-mediated reduction in SOCS1 expression in cervical cancer.
In conclusion, our study is the first to demonstrate that REX1 activates the JAK2/STAT3-signaling pathway by binding to the SOCS1 promoter and trans-suppressing SOCS1, promotes the EMT process by upregulating VIMENTIN and downregulating E-CADHERIN, finally promotes the metastasis of cervical cancer in vivo and the migration and invasion of cervical cancer cells in vitro (Fig. 6i).
Materials and methods
Human tissue specimens
A total of 30 NC and 48 cervical SCC samples were collected from the First Affiliated Hospital of Xi’an Jiaotong University from 2009 to 2016. All the procedures followed approved medical ethics practices. None of the patients had received chemotherapy, immunotherapy, or radiotherapy before the specimens were collected. The histological classifications and clinical staging were done in accordance with the International Federation of Gynecology and Obstetrics classification system. The institutional review board named as Ethics Committee of Medical School of Xi’an Jiaotong University in Shannxi, China approved the population study, while all the patients provided their informed consents prior to specimen collection.
Immunohistochemistry and immunocytochemistry
The immunohistochemical staining procedure was performed as previously described [7]. According to the percentage and staining intensity of REX1 positive cells in basal cells of NC tissues, or in tumor cell nests of cervical cancer tissues which defined as clustered tumor cells surrounded by tumor stroma [48, 49], REX1 staining was classified into three categories: negative, weak positive, and strong positive. The percentage of positive cells was divided into five score ranks: <10% (0), 10–25% (1), 25–50% (2), 50–75% (3), and >75% (4). The intensity of staining was divided into four score ranks: no staining (0), light brown (1), brown (2), and dark brown (3). The staining positivity was determined by the following formula: IRS = intensity score × quantity score. The overall score of ≤3 was defined as negative, >3 but ≤6 as weak positive, and >6 as strong positive. Two different pathologists evaluated all the specimens in a blinded manner. The antibodies used were as follows: anti-REX1 (1:100 dilution, #377095, Santa Cruz, USA), anti-VIMENTIN (1:100 dilution, #sc-6260), anti-E-CADHERIN (1:150 dilution, #sc-8426), anti-STAT3 (1:100 dilution, #9139, Cell Signaling Technology, USA), anti-JAK2 (1:100 dilution, #sc-390539), and anti-SOCS1 (1:100 dilution, #A7754, ABclonal, China).
For the immunocytochemistry experiments, cells were cultured on cover slips, fixed with 4% paraformaldehyde for 30 min at room temperature, permeabilized with 0.2% Triton X-100 for 15 min at room temperature, and then incubated with the primary antibodies described above.
Cell lines and cell culture
The human cervical cancer cell lines (HeLa, SiHa, C-33 A, CaSki, and HT-3) were purchased from American Type Culture Collection (ATCC; Manassas, VA) and were tested using RT-PCR for mycoplasma contamination. The cell lines purchased from ATCC have been authenticated by STR profiling, so authentications were not performed. The HeLa, SiHa, C-33 A cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich, St Louis, MO, USA); CaSki and HT-3 cells were cultured in RPMI-1640 (Sigma-Aldrich) and McCoy’s 5A medium (Sigma-Aldrich), respectively, supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone, Thermo Scientific, Waltham, MA, USA). All cell lines were maintained at 37 °C with 5% CO2.
Vector construction and transfection
Human full-length REX1 (NM_174900.4) cDNA was amplified by reverse transcription polymerase chain reaction using mRNA extracted from HeLa cells. The primer sequences were designed as follows:
REX1-F: 5-′ CCGGTCGACGAAAACATGAGCCAGCAACT -3′;
REX1-R: 5-′ CGCGGATCCGTTGGAGGACTACTTTCCCTC -3′.
The REX1 DNA fragment was subsequently cloned into the Sal I and BamH I (TaKaRa, Tokyo, Japan) sites of an internal ribosome entry site vector, pIRES2-AcGFP1-Neo (Clontech, Mountain View, CA), to generate the recombinant pIRES2-AcGFP1-Neo-REX1 plasmid.
The empty plasmid pIRES2-AcGFP1-Neo and overexpression plasmid pIRES2-AcGFP1-Neo-REX1 were transfected into HeLa and SiHa cells by liposome transfection using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Successful transfection was verified as green fluorescence under fluorescence microscope. Then, G418 with a concentration of 1 g/L was added for screening. Stable transfected HeLa and SiHa cells were treated with G418 (Calbiochem, La Jolla, CA, USA) for ~3 weeks, the clones expressing neomycin resistance steadily were obtained, expanded, and the expression of REX1 protein was identified using western blotting. REX1-overexpressing cell clones (transfected pIRES2-AcGFP1-Neo-REX1, labeled SiHa-REX1, and HeLa-REX1) and control cells clones (transfected pIRES2-AcGFP1-Neo, labeled SiHa-GFP, and HeLa-GFP) were maintained as monoclonal cells treated with G418 with a concentration of 1 g/L all the time. After pIRES2-AcGFP1-Neo-REX1 or pIRES2-AcGFP1-Neo plasmids been transient transfected into 293T cell lines, protein was collected after being transfected 48 h. After pIRES2-AcGFP1-Neo-REX1 or pIRES2-AcGFP1-Neo plasmids been transient transfected, mRNA was collected after being transfected 24, 36, and 48 h in 293T cells and 36, 48, 60, and 72 h in SiHa and HeLa cells.
Murine experimental metastasis experiments
Four- to six-week-old female BALB/c nude mice (obtained form Shanghai Slac Laboratory animal co. ltd, China) were housed in a specific pathogen free room with constant temperature (22–25 °C) and humidity (40–50%).
For the spontaneous metastasis assay, twelve mice were randomly divided into two groups, with six mice in each group. REX1-overexpressed SiHa or HeLa cells and the respective control cells (1 × 106/mouse) in the exponential growth phase were harvested and injected (100 μL per site) into the subcutis on the dorsum of each mouse, respectively. Tumors were measured in two dimensions by using manual calipers. Tumor volume was calculated using the following formula: V = 0.5 × length × width2. Tumor volume was measured every 2–3 days. About 8 weeks later, the mice were executed; the livers and lungs were removed and subjected to histologic examination.
For experimental lung metastasis assay, twelve mice were randomly divided into two groups, with six mice in each group. REX1-overexpressed SiHa or HeLa cells and the respective control cells (5 × 105/mouse) in the exponential growth phase were harvested and injected into tail veins of each mouse. About 6 weeks after injection, mice were executed; the livers and lungs were removed and subjected to histologic examination.
Livers and lungs were fixed in 4% formalin and embedded in paraffin, and 5 μm sections were stained with H&E. For each sample, all micrometastases were counted under a light microscope at ×10 magnifications. Three sections were counted per mouse sample at 50 μm intervals and average the results.
All animal studies (including the mice euthanasia procedure) were done in compliance with the regulations and guidelines of the Xi’an Jiaotong University Institutional Animal Care and conducted according to the AAALAC and the IACUC guidelines.
Wound repair assays
Cells were plated in 24-well plates at 106 cells/well in 1 mL of culture medium. After 2 days, a wound was scratched in the adherent cell monolayers with an Eppendorf tip, and the medium was changed to DMEM supplemented with 1% FBS (Invitrogen). The wells were examined every day, and photomicrographs were taken on a Nikon Eclipse Ti as described above. Wound width was measured on the photomicrographs, using the same area of the well for each measurement. The experiment was repeated in three independent experiments.
Migration and invasion assays
Transwell chambers (Corning, Corning, NY, USA) equipped with 8-μm pore insets were used for the migration and invasion assays. For the migration assay, 8 × 104 REX1-overexpressing SiHa or HeLa cells and the respective control cells in serum-free medium were plated on uncoated insets and incubated for 48 h. For the invasion assay, the insets were coated with 70 μL of 1:8-diluted Matrigel (BD Biosciences), and 1 × 105 cells were plated in the serum-free medium described above for an incubation period of 48 h. Quantities of 600 μL of culture medium containing 20% FBS (Invitrogen) were added to the lower chamber. Noninvaded cells were removed, and the cells that were attached to the bottom of the membrane were fixed with 4% paraformaldehyde, stained with 5% crystal violet (Sigma-Aldrich), and counted at 200-fold magnification. The experiment was repeated in three independent experiments.
RNA preparation and transcriptome resequencing
Three HeLa-GFP monoclonal cells and three HeLa-REX1 monoclonal cells were used for transcriptome resequencing. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). A total of six samples were measured using the BGISEQ-500 platform, and the average output of each sample was 11.03 Gb. The average ratio of sample to genome was 94.15%, and the ratio of comparison to gene set was 75.40%. The experiment analysis used the NOISeq method, which is a novel nonparametric approach for the identification of differentially expressed genes (DEGs) based on log2 fold change > 1 and a probability ≥ 0.80 [50]. According to the results of GO annotation and official classification, we classify the DEGs and analysis by using the Phyper function in R software [51, 52]. Then FDR was corrected for p value, and the function of FDR ≤ 0.01 was usually seen as significant enrichment.
Western blotting
Cells and clinical tissues were lysed for 30–45 min on ice in lysis buffer containing freshly added protease inhibitor cocktail (Roche Diagnostics, USA). After BCA quantification (Pierce, USA), the protein was added to 5× loading buffer and boiled at 95 °C for 10 min. Equal amounts of protein were separated by SDS-PAGE and blotted onto activated polyvinylidene difluoride membranes (Millipore, USA). After blocking, the membranes were incubated with primary antibodies overnight at 4 °C. The antibodies used were as follows: anti-REX1 (1:1000 dilution, #GTX60780, Genetex, USA), anti-GAPDH (1:1000 dilution, #sc-47724, Santa Cruz, CA, USA), anti-VIMENTIN (1:1000 dilution, #sc-6260), anti-E-CADHERIN (1:500 dilution, #sc-8426), anti-MMP9 (1:500 dilution, #sc-21733), anti-STAT3 (1:1000 dilution, #9139, Cell Signaling Technology, USA), anti-p-STAT3 (Tyr705) (1:1000 dilution, #9131, Cell Signaling Technology), anti-JAK2 (1:500 dilution, #WL02188, Wanleibio, China), anti-p-JAK2 (Tyr1007/1008) (1:500 dilution, #WL02997, Wanleibio), anti-SOCS1 (1:1000 dilution, #A7754, ABclonal, China), and anti-SOCS3 (1:500 dilution, #WL01364, Wanleibio). The secondary incubation antibodies used a horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Thermo Fisher Scientific, New York, NY, USA). The signals of antigen–antibody complexes were detected by ECL enhanced chemiluminescence solution (Millipore, Billerica, MA, USA). The signal intensity was quantified using the protein imprinting imaging system (Tanon 5200, China). To clearly demonstrate the difference, the relative gray-scale value of target protein versus GAPDH of the control group was set as 1. Results shown were representative of three independent experiments.
Dual-luciferase reporter assay
Fragments of the SOCS1 promoter (predicted from position −2024 bp to +504 bp), were cloned into the pGL3-Basic Vector (Promega, Madison, WI, USA) to generate promoter reporter constructs, respectively. All constructs were verified by sequencing. Cells were seeded in 24-well plates and transiently transfected with plasmids containing firefly luciferase reporters and recombinant promoter reporter constructs. The luciferase activity was measured after incubation for 48 h using the dual-luciferase assay kit (Promega). All experiments were performed as three independent experiments. The transfection efficiency was normalized with Renilla luciferase activity. The specific promoter activity was presented as the change in the experimental group versus the control group. The primers and oligonucleotides are listed in Table S2. The specific activity was shown as the fold change of the experimental group versus the control group.
Quantitative chromatin immunoprecipitation
qChIP assays were performed according to the manufacturer’s protocol for the EZ-Magna ChIP Assay kit (Millipore, Darmstadt, Germany). Chromatin–protein complexes were immunoprecipitated with 5 μg of anti-REX1 antibodies (sc-377095, Santa Cruz, CA, USA) and 20 μL of fully resuspended protein A/G magnetic beads. For the negative control, 1 μg of normal mouse IgG was used. Real-time PCR was performed to amplify the regions of interest or internal negative control regions. Each sample was assayed in triplicate, and the amount of precipitated DNA was calculated as a percentage of the input sample. The primers used in quantitative ChIP assays are listed in Supplementary Table 2.
TCGA data acquisition
RNAseq and clinical data were acquired using TCGA by cervical squamous cell CESC patients (n = 304, including 260 SCC, 22 endocervical type of adenocarcinoma, 15 mucinous adenocarcinoma of endocervical type, and 7 adenosquamous). According to their distant metastasis status listed in the clinical information, patients can be divided into two groups (n = 29 for patients with distant metastasis, n = 275 for patients with no distant metastasis). Moreover, survival analyses were performed with Kaplan–Meier estimator from the Kaplan–Meier plotter (http://kmplot.com/analysis/index.php?p=background).
The corresponding clinicopathological information is from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp). The personal information of the patients was anonymized, and the patients were not identified. According to TCGA publication guidelines (http://cancergenome.nih.gov/publications/publicationguidelines), these mRNA sequencing data have no restrictions on publication, and no additional approval by an ethics committee was required to publish the use of the data.
Statistical analysis
Statistical analysis was performed with SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Measurement data were analyzed with mean ± standard deviation; The two-tailed χ2 test or Fisher’s exact test was used to determine the significance of the differences between the covariates. A univariate analysis was analyzed by Student’s t-test (two tailed) and the Mann–Whitney U-test. For comparison among groups, the χ2 test or one-way ANOVA was performed. A p value < 0.05 was considered statistically significant.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9.
de Sanjose S, Quint WG, Alemany L, Geraets DT, Klaustermeier JE, Lloveras B, et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol. 2010;11:1048–56.
Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. New Engl J Med. 2003;348:518–27.
Astudillo H, Perez M, Silva J, Silva M, Castillo S, Benitez L, et al. P53, Bcl-2, PCNA expression and apoptotic rates during cervical tumorigenesis. J Clin Oncol. 2004;22:5068.
Rizvi MM, Alam MS, Ali A, Mehdi SJ, Batra S, Mandal AK. Aberrant promoter methylation and inactivation of PTEN gene in cervical carcinoma from Indian population. J Cancer Res Clin Oncol. 2011;137:1255–62.
Chen Q, Zheng PS, Yang WT. EZH2-mediated repression of GSK-3beta and TP53 promotes Wnt/beta-catenin signaling-dependent cell expansion in cervical carcinoma. Oncotarget. 2016;7:36115–29.
Liu XF, Li XY, Zheng PS, Yang WT. DAX1 promotes cervical cancer cell growth and tumorigenicity through activation of Wnt/beta-catenin pathway via GSK3beta. Cell Death Dis. 2018;9:339.
Cui N, Yang WT, Zheng PS. Slug inhibits the proliferation and tumor formation of human cervical cancer cells by up-regulating the p21/p27 proteins and down-regulating the activity of the Wnt/beta-catenin signaling pathway via the trans-suppression Akt1/p-Akt1 expression. Oncotarget. 2016;7:26152–67.
Wang HY, Lian P, Zheng PS. SOX9, a potential tumor suppressor in cervical cancer, transactivates p21WAF1/CIP1 and suppresses cervical tumor growth. Oncotarget. 2015;6:20711–22.
Yang WT, Zheng PS. Kruppel-like factor 4 functions as a tumor suppressor in cervical carcinoma. Cancer. 2012;118:3691–702.
Mongan NP, Martin KM, Gudas LJ. The putative human stem cell marker, Rex-1 (Zfp42): structural classification and expression in normal human epithelial and carcinoma cell cultures. Mol Carcinog. 2006;45:887–900.
Thomas MJ, Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236:197–208.
Henderson JK, Draper JS, Baillie HS, Fishel S, Thomson JA, Moore H, et al. Preimplantation human embryos and embryonic stem cells show comparable expression of stage-specific embryonic antigens. Stem Cells. 2002;20:329–37.
Toyooka Y, Shimosato D, Murakami K, Takahashi K, Niwa H. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development. 2008;135:909–18.
Arai E, Chiku S, Mori T, Gotoh M, Nakagawa T, Fujimoto H, et al. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis. 2012;33:1487–93.
Lee MY, Lu A, Gudas LJ. Transcriptional regulation of Rex1 (zfp42) in normal prostate epithelial cells and prostate cancer cells. J Cell Physiol. 2010;224:17–27.
Oka D, Yamashita S, Tomioka T, Nakanishi Y, Kato H, Kaminishi M, et al. The presence of aberrant DNA methylation in noncancerous esophageal mucosae in association with smoking history: a target for risk diagnosis and prevention of esophageal cancers. Cancer. 2009;115:3412–26.
Kim BS, Kang KS, Choi JI, Jung JS, Im YB, Kang SK. Knockdown of the potential cancer stem-like cell marker Rex-1 improves chemotherapeutic effects in gliomas. Hum Gene Ther. 2011;22:1551–62.
Anzai E, Hirata K, Shibazaki M, Yamada C, Morii M, Honda T, et al. FOXA1 induces E-cadherin expression at the protein level via suppression of slug in epithelial breast cancer cells. Biol Pharm Bull. 2017;40:1483–9.
Cui Y, Li YY, Li J, Zhang HY, Wang F, Bai X, et al. STAT3 regulates hypoxia-induced epithelial mesenchymal transition in oesophageal squamous cell cancer. Oncol Rep. 2016;36:108–16.
Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res. 2007;13:1362–6.
Kamizono S, Hanada T, Yasukawa H, Minoguchi S, Kato R, Minoguchi M, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. 2001;276:12530–8.
Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci USA. 2012;109:5004–9.
Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. 2001;193:471–81.
Wu Y, Diab I, Zhang X, Izmailova ES, Zehner ZE. Stat3 enhances vimentin gene expression by binding to the antisilencer element and interacting with the repressor protein, ZBP-89. Oncogene. 2004;23:168–78.
Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999;18:1309–20.
Brivanlou AH, Gage FH, Jaenisch R, Jessell T, Melton D, Rossant J. Stem cells. Setting standards for human embryonic stem cells. Science. 2003;300:913–6.
Chan EM, Ratanasirintrawoot S, Park IH, Manos PD, Loh YH, Huo H, et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol. 2009;27:1033–7.
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Raman JD, Mongan NP, Liu L, Tickoo SK, Nanus DM, Scherr DS, et al. Decreased expression of the human stem cell marker, Rex-1 (zfp-42), in renal cell carcinoma. Carcinogenesis. 2006;27:499–507.
Zhang JJ, Zhu Y, Xie KL, Peng YP, Tao JQ, Tang J, et al. Yin Yang-1 suppresses invasion and metastasis of pancreatic ductal adenocarcinoma by downregulating MMP10 in a MUC4/ErbB2/p38/MEF2C-dependent mechanism. Mol Cancer. 2014;13:130.
Zhao G, Li Q, Wang A, Jiao J. YY1 regulates melanoma tumorigenesis through a miR-9 ~ RYBP axis. J Exp Clin Cancer Res. 2015;34:66.
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30.
Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, et al. Improved survival with bevacizumab in advanced cervical cancer. New Engl J Med. 2014;370:734–43.
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–403.
Garcia-Tuñon I, Guallar D, Alonso-Martin S, Benito AA, Benítez-Lázaro A, Pérez-Palacios R, et al. Association of Rex-1 to target genes supports its interaction with polycomb function. Stem Cell Res. 2011;7:1–16.
Palko L, Bass HW, Beyrouthy MJ, Hurt MM. The Yin Yang-1 (YY1) protein undergoes a DNA-replication-associated switch in localization from the cytoplasm to the nucleus at the onset of S phase. J Cell Sci. 2004;117:465–76.
Qureshi R, Arora H, Rizvi MA. EMT in cervical cancer: its role in tumour progression and response to therapy. Cancer Lett. 2015;356:321–31.
Takemoto S, Ushijima K, Kawano K, Yamaguchi T, Terada A, Fujiyoshi N, et al. Expression of activated signal transducer and activator of transcription-3 predicts poor prognosis in cervical squamous-cell carcinoma. Br J Cancer. 2009;101:967–72.
Zhou X, Yan T, Huang C, Xu Z, Wang L, Jiang E, et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J Exp Clin Cancer Res. 2018;37:242.
Sobti RC, Singh N, Hussain S, Suri V, Nijhawan R, Bharti AC, et al. Aberrant promoter methylation and loss of suppressor of cytokine signalling-1 gene expression in the development of uterine cervical carcinogenesis. Cell Oncol. 2011;34:533–43.
Kim MH, Kim MS, Kim W, Kang MA, Cacalano NA, Kang SB, et al. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PLoS One. 2015;10:e0123133.
Kim JD, Kang K, Kim J. YY1’s role in DNA methylation of Peg3 and Xist. Nucleic Acids Res. 2009;37:5656–64.
Aoyama T, Okamoto T, Fukiage K, Otsuka S, Furu M, Ito K, et al. Histone modifiers, YY1 andp300, regulate the expression of cartilage-specific gene, chondromodulin-I, in mesenchymal stem cells. J Biol Chem. 2010;285:29842–50.
Yang WM, Inouye C, Zeng Y, Bearss D, Seto E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc Natl Acad Sci USA. 1996;93:12845–50.
Boxberg M, Kuhn PH, Reiser M, Erb A, Steiger K, Pickhard A, et al. Tumor budding and cell nest size are highly prognostic in laryngeal and hypopharyngeal squamous cell carcinoma: further evidence for a unified histopathologic grading system for squamous cell carcinomas of the upper aerodigestive tract. Am J Surg Pathol. 2019;43:303–13.
Karpathiou G, Vieville M, Gavid M, Camy F, Dumollard JM, Magne N, et al. Prognostic significance of tumor budding, tumor-stroma ratio, cell nests size, and stroma type in laryngeal and pharyngeal squamous cell carcinomas. Head Neck. 2019;41:1918–27.
Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21:2213–23.
Berkopec A. HyperQuick algorithm for discrete hypergeometric distribution. J Discret Algorithms. 2007;5:341–7.
Kumar L, Matthias EF. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7.
Acknowledgements
This work was supported by a to grant Dr Xiao-Fang Liu from the National Natural Science Foundation of China (No. 81802946) and Prof. Peng-Sheng Zheng from the National Natural Science Foundation of China (Nos. 81472728 and 81672910).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, YT., Liu, XF., Yang, WT. et al. REX1 promotes EMT-induced cell metastasis by activating the JAK2/STAT3-signaling pathway by targeting SOCS1 in cervical cancer. Oncogene 38, 6940–6957 (2019). https://doi.org/10.1038/s41388-019-0906-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-0906-3
This article is cited by
-
METTL3 facilitates stemness properties and tumorigenicity of cancer stem cells in hepatocellular carcinoma through the SOCS3/JAK2/STAT3 signaling pathway
Cancer Gene Therapy (2024)
-
Stress-induced phosphoprotein 1: how does this co-chaperone influence the metastasis steps?
Clinical & Experimental Metastasis (2024)
-
Detection of the DNA methylation of seven genes contribute to the early diagnosis of lung cancer
Journal of Cancer Research and Clinical Oncology (2024)
-
LncRNA KIAA0087 suppresses the progression of osteosarcoma by mediating the SOCS1/JAK2/STAT3 signaling pathway
Experimental & Molecular Medicine (2023)
-
Computational analysis of synergism in small networks with different logic
Journal of Biological Physics (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}