
Abstract
Medulloblastoma (MB) is the most common malignant pediatric brain tumor. Since surviving patients experience severe neurocognitive disabilities, better and more effective treatments are needed to enhance their quality of life. Casein kinase 2 (CK2) is known to regulate cell growth and survival in multiple cancers; however, the role of CK2 in MB is currently being studied. In this study, we verified the importance of CK2 in MB tumorigenesis and discovered that inhibition of CK2 using the small molecule inhibitor, CX-4945, can sensitize MB cells to a well-known and tolerated chemotherapeutic, temozolomide (TMZ). To study the role of CK2 in MB we modulated CK2 expression in multiple MB cells. Exogenous expression of CK2 enhanced cell growth and tumor growth in mice, while depletion or inhibition of CK2 expression decreased MB tumorigenesis. Treatment with CX-4945 reduced MB growth and increased apoptosis. We conducted a high-throughput screen where 4000 small molecule compounds were analyzed to identify compounds that increased the anti-tumorigenic properties of CX-4945. TMZ was found to work synergistically with CX-4945 to decrease cell survival and increase apoptosis in MB cells. O-6-methylguanine-DNA methyltransferase (MGMT) activity is directly correlated to TMZ sensitivity. We found that loss of CK2 activity reduced β-catenin expression, a known MGMT regulator, which in turn led to a decrease in MGMT expression and an increased sensitivity to TMZ. Our findings show that CK2 is important for MB maintenance and that treatment with CX-4945 can sensitize MB cells to TMZ treatment.
Similar content being viewed by others


Introduction
Medulloblastoma (MB) is the most common malignant brain tumor in children and accounts for ~20% of all pediatric central nervous system (CNS) tumors [1]. Current multimodal treatment has led to a 70–90% 5-year overall survival rate [2]. However, the 5-year disease-free survival rate remains at 30%, and the prognosis for patients with tumor dissemination and recurrent MB remains poor [3]. Additionally, the majority of survivors exhibit long-term neurocognitive and neuroendocrine complications as a result of the cytotoxic drugs and high-dose radiation. Consequently, more effective and less toxic treatments are necessary in order to raise the quality of life for these young patients.
Recently, we and other researchers have identified a pleiotropic kinase called casein kinase 2 (CK2) as a potentially interesting target for cancer therapies. CK2 is a highly ubiquitous and conserved serine/threonine kinase that forms a tetramer consisting of a catalytic subunit (CK2α) and regulatory subunit (CK2β) [4]. CK2 expression and activity are increased in many cancers and its growth-related functions have been supported by its involvement in many tumorigenic pathways [5]. Interestingly, upregulation of CK2 correlates with poor cancer prognosis in numerous cancers, including cervical, gastric, liver, head and neck cancers, and adult gliomas [6, 7]. In our recent findings we showed through a developmental phosphoproteomics study that CK2 is a driver of hundreds of phosphorylation events during MB growth [8].
To further study the role of CK2 in MB tumorigenesis, we modulated CK2 expression in multiple MB cell lines, Daoy and Med1-MB. We initially discovered that exogenous CK2 expression increased cell growth and anchorage independent growth, while knocking out CK2 isoforms decreased growth and colony formation. Inhibition of CK2, using small molecule inhibitor CX-4945, decreased MB growth in a dose-dependent manner. To further explore the ability of CK2 inhibition to treat MB, we conducted a high-throughput assay to identify small molecule inhibitors that enhanced the efficacy of CX-4945. Through multiple screens we identified temozolomide (TMZ) as a molecule that worked synergistically with CX-4945 to reduce MB growth and induce apoptosis. We discovered that inhibition or knockout of CK2 reduced expression of O-6-methylguanine-DNA methyltransferase (MGMT) through β-catenin. Our findings demonstrate for the first time that inhibition of CK2 using CX-4945 sensitized MB cells to TMZ treatment.
Results
MB patients with high expression of CK2 have a worse prognosis
Using a web-based genomics analysis and visualization application called R2 platform (http://r2.amc.nl), we correlated MB patient survival to expression of CK2 in the patients’ tumors. We analyzed a cohort of 763 primary MB samples from the Medulloblastoma Advanced Genomics International Consortium and determined that MB patients with high-mRNA expression of CK2α or CK2β have a significantly worse prognosis compared to their low-expressing counterparts (p < 0.05) (Fig. 1a) [9]. To further investigate if CK2 expression is dysregulated in MB, we examined three different datasets that analyzed the mRNA expression of primary MB patients [10, 11] (MAS5.0—u133p2) and compared them to an mRNA array of normal brain tissue [12]. Each MB mRNA array contained ~60 to 80 primary MB samples and the normal brain tissue array had 172 samples. Using one-way analysis of variance (ANOVA) we determined that CK2α expression was significantly elevated in all three datasets compared to the normal brain tissue array (p < 0.05), while there was no increase in CK2β expression in any of the three MB datasets (Fig. 1b). Since CK2β expression was not elevated compared to normal brain tissue we analyzed the expression of CK2α in a tissue microarray (TMA). We conducted a TMA that contained 46 primary MB samples and six control tissue samples (cerebellar tonsil, and placenta). The array was scored 0 to 3 in a blinded study (Supplementary Fig. S1). Consistent with our previous finding, CK2α expression was significantly elevated in the MB patients’ samples compared to the control tissues (p < 0.0001) (Fig. 1c). Since MB is widely categorized into four different molecular subgroups, we analyzed CK2α expression across each group [13]. We discovered that CK2α expression was increased in each MB molecular subgroup compared to control tissue (p < 0.05), but there was no difference between the subgroups (Fig. 1c). Altogether these findings suggest that CK2 expression is enhanced in MB patients and that it correlates with a worse prognosis.

CK2 expression in primary MB patient samples. a Left—Kaplan–Meyer survival curve for MB patients with either high or low CK2α expression. Right—Kaplan–Meyer survival curve for MB patients with either high or low CK2β expression. N = 612. b Left—The relative mRNA expression level of CK2α in three MB microarray patient samples compared to a normal brain microarray. Right—The relative mRNA expression level of CK2β. Boxes represent the interquartile range; lines represent the location of the first quartile, median, and third quartile. The log-rank test was used to calculate p-value. *p < 0.05, sample compared to normal brain samples. c Left—Tissue Micro Array (TMA) of primary MB patient samples for CK2α expression. Tonsil and placenta were used as normal tissue control (n = 6). Right—TMA of primary MB samples separated by molecular subgroup (WNT n = 8, SHH n = 14, Group 3 n = 12, Group 4 n = 17). Bars represent mean ± SD, two-tailed t-test
Enhancing expression of CK2 increases MB tumorigenesis
To verify that CK2 is involved in MB tumorigenesis we modulated CK2 expression in two MB cell lines, Daoy and Med1-MB. We stably transduced YFP-tagged CK2α or CK2β using retroviral infections and verified expression of each CK2 isoform (Fig. 2a). The cells transduced with YFP-CK2α and YFP-CK2β exhibited increased cell growth and anchorage independent growth compared to the YFP-transduced control cells in both Daoy and Med1-MB cells (Fig. 2b, c).

Exogenous expression of CK2 enhanced MB tumorigenesis. a Daoy and Med1-MB cells were stably transduced with YFP, YFP-CK2α, or YFP-CK2β. b Left—Cell growth of Daoy cell lines transduced with YFP-CK2α or YFP-CK2β. Right—Cell growth of transduced Med1-MB cells. Bars represent mean of three technical or experimental replicates, mean ± SD, student t-test. c Soft-agar analysis of Daoy and Med1-MB-expressing YFP-CK2α or YFP-CK2β. Bars represent mean of three technical or experimental replicates, mean ± SD, student t-test. d Survival curve of mice intracranially injected with transduced Daoy cell lines. N = 5 mice per treatment and experiment was done in duplicate. Log-rank was used to determine p-value. e Tumors from mice counted for number of Ki67-positive cells. Bars represent mean of three technical or experimental replicates, mean ± SD, student t-test. *p < 0.05, sample compared to control cells
We extended our study by testing the effects of enhanced CK2 expression in tumor formation in intracranial xenografts (n = 5). Mice injected with Daoy YFP-CK2α had significantly worse survival compared to the mice injected with the control Daoy YFP cells, with the median survival time after tumor implantation decreasing from 95 days to 70 days (p = 0.014) (Fig. 2d). The mice injected with Daoy YFP-CK2β did have a small decrease in survival, but it was not statistically significant (p = 0.27). Tumors were stained for the proliferative marker Ki67 and we saw a significant increase in the number of proliferative cells (p = 0.03) in mice bearing the YFP-CK2α tumors compared to the YFP control tumors, while YFP-CK2β tumors only had a trend of increased proliferation (p = 0.06) (Fig. 2e). To determine if the exogenous expression of YFP-CK2α or YFP-CK2β had increased CK2 activity, we stained for phosphorylated STAT3 (p-STAT3), a well-known CK2 substrate. Tumors transduced with YFP-CK2α or YFP-CK2β tumors had increased p-STAT3-positive cells compared to the YFP tumors, but there was no statistical difference (Supplementary Fig. S2A).
Knocking out or inhibiting CK2 decreases MB tumorigenesis
To assess whether inhibition of CK2 can decrease MB growth, we used the CRISPR-Cas9 technology to generate MB cell lines lacking each CK2 subunit. We generated Daoy and Med1-MB cells that used the CRISPR/Cas9 system where single-guide RNA (sgRNA) targeting sequences were directed to CSNK2a1 (sgCK2α), CSNK2b (sgCK2β), or GFP (sgControl). We verified using western blot analysis that the sgCK2α and sgCK2b cells had a significant and specific decrease in CK2 expression in Daoy and Med1-MB cells (Fig. 3a). Both cell lines lacking either CK2α or CK2β expression consistently had a significant decrease in cell growth and colony formation compared with the sgControl cells (Fig. 3b, c). To confirm that decreasing CK2α expression decreased MB growth, we used lentivirus to stably transduce Daoy or Med1-MB cells with two different short-hairpin RNAs to CSNK2a1 (shCK2α v1 and shCK2α v2). Consistent with the CRISPR/Cas9 cell lines, reduction of CK2α expression reduced cell growth and colony formation in both Daoy and Med1-MB cells in both shCK2α v1 and shCK2α v2 cell lines (Supplementary Fig. S3A–C).

Decreasing CK2 expression or activity inhibits MB growth. a Knockout of CK2α (sgCK2α) or CK2β (sgCK2β) in MB cell lines using CRISPR-Cas9 technology. The fold change is indicated below each blot normalized to sgControl sample. b Left—Cell growth of Daoy lacking CK2α or CK2β compared to the control cells transduced with sgRNA to GFP (sgControl). Right—Cell growth of Med1-MB lacking CK2α or CK2β. Each data point represents mean of three technical or experimental replicates, mean ± SD, student t-test. c Soft-agar analysis of Daoy and Med1-MB lacking CK2α or CK2β expression. d Left—Growth curves of Daoy cells treated with CX-4945. Right—Growth curves of Med1-MB cells treated with CX-4945. e Soft-agar analysis of Daoy and Med1-MB treated with CX-4945. f Growth of MB55 or MB56 cells treated with CX-4945. Bars represent mean of three technical or experimental replicates, mean ± SD, student t-test. g Proliferative growth of mouse xenograft treated with CX-4945. Log-rank test was used to calculate p-value. *p < 0.05, sample compared to control cells. N = 5 mice per treatment and experiment
Since knocking out CK2 expression decreased MB tumorigenesis, we determined if treatment with a CK2 small molecule inhibitor could phenocopy this effect. Daoy or Med1-MB cells treated with the CK2 inhibitor, CX-4945, reduced cell growth and colony formation in a dose-dependent manner (Fig. 3d, e). Since Daoy and Med1-MB cells are immortalized cell lines, we also analyzed two mouse-derived, serum-free, MB cell lines, MB55 and MB56. These MB lines retain characteristics of primary MB tumors that arise in a mutant Patched 1 (Ptch) mouse model [8]. Treatment with CX-4945 significantly reduced MB55 and MB56 growth in a dose-dependent manner (Fig. 3f).
To expand on these in vitro findings, we analyzed the number of proliferative cells in MB tumor xenografts treated with 75 mg/kg per day of CX-4945. We discovered that treatment with CX-4945 significantly reduced the number of Ki67-positive cells (p = 0.02) compared to the vehicle (Fig. 3g). We also discovered that there was a trend in which the mice treated with CX-4945 had decreased phosphorylated STAT3-positive tumor cells compared to the mice treated with the vehicle control, but there was no statistical difference (Supplementary Fig. S2B).
Two additional CK2 inhibitors, TBB and TBBz, were also tested to verify that the reduction in MB growth was due to CK2 inhibition and not off-target effects. We found that both TBB and TBBz reduced MB growth in a dose-dependent manner similar to CX-4945 treatment (Supplementary Fig. S4A–C). These findings suggest that a small molecule inhibitor of CK2 could be a potentially new therapeutic for MB patients.
High-throughput screen to identify potential synergistic compounds with CX-4945
While single target inhibitor treatments can deliver dramatic responses, cancer cells inevitably acquire drug resistance and additional treatments are needed. One method to combat this resistance is to undergo combinational treatments that enhance targeting of specific target pathways or administer multiple drugs that work by different molecular mechanisms. Therefore, we developed a multiple drug screening strategy to identify, characterize, and categorize FDA-approved drugs that specifically enhance CX-4945 treatment. We conducted a high-throughput screen analyzing 4000 FDA-approved drugs at the Stanford High-Throughput Bioscience Center (HTBC). In order to identify compounds that further reduce MB cell growth together with CX-4945 treatment, we initially determined the percent each compound alone inhibited Daoy growth. We determined that 5 μM of CX-4945 treatment inhibited growth 51.4%, while treatment with compounds from the library inhibited growth 0 to ~60% (Table 1). We identified three compounds that, together with CX-4945, increased inhibition by an additional 25%, compared to CX-4945 alone (Table 1). These three compounds were TMZ, flubendazole, and ralititrexed (Table 1). Two compounds, pitavastatin and pterostilbene, were found to be “weak” inhibitors since they only increased inhibition an additional 10–15%. We repeated the screen and discovered that only TMZ and flubendazole repeated as strong inhibitors of Daoy cell growth (Table 1). We analyzed the effects of flubendazole and ralititrexed on Daoy and Med1-MB growth and compared to prior studies, we did not observe a significant change in cell growth (Supplementary Fig. S5) [14, 15]. As TMZ is the most commonly used chemotherapy in adult brain tumors and because it is an oral medication with usually tolerable toxicity, we further studied the combination of CX-4945 and TMZ.
Combinatorial treatments with TMZ and CX-4945 decrease MB tumorigenesis
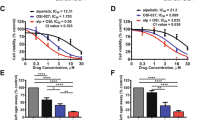
We verified the findings from the high-throughput screen using an in vitro combinatorial study with CX-4945 and TMZ. The Chou–Talalay method was used to determine if combining CX-4945 and TMZ had a synergistic or additive effect by calculating the combination index (CI). We discovered that each drug combination in Daoy cells had a CI < 0.3 indicating a synergistic response (Fig. 4a). Interestingly, the combinatorial treatments in Med1-MB cells had a CI > 1, indicating an antagonistic or no change in cell response (Fig. 4a). We verified the synergistic response by analyzing both Daoy and Med1-MB cells treated with 1 μM CX-4945 and 100 μM or 500 μM TMZ (Supplementary Fig. S6A, B). To confirm the synergy with TMZ and CX-4945 we analyzed the MB55 and MB56 cell lines. Treatment with 500 μM TMZ and 10 μM CX-4945 resulted in a synergistic response in MB55 and MB56 cells (CI < 1) (Fig. 4b). We verified these findings by using two additional CK2 inhibitors, TBB or TBBz. Consistent with our previous findings, inhibition of CK2 using either TBB or TBBz combined with TMZ had a synergistic effect in Daoy cells, but not in the Med1-MB cell line (Supplementary Fig. S6C, D).

Combinatorial treatments with TMZ and CX-4945 decreases MB tumorigenesis. a Left—Combinatorial treatment of Daoy cells with CX-4945 and TMZ. Bars represent mean of three technical or experimental replicates, mean ± SD. Combination index (CI) was determined using Chou–Talalay method, #CI = 0.13 ± 0.06, ##CI = 0.02 ± 0.01. Right—Combinatorial treatment Med1-MB cells with CX-4945 and TMZ. b Left—Combinatorial treatment for MB55 with CX-4945 and TMZ. Bars represent mean of three technical or experimental replicates, mean ± SD. Combination index (CI) was determined using Chou–Talalay method ###CI = 0.07 ± 0.03. Right—Combinatorial treatment for MB56 with CX-4945 and TMZ, bars represent mean of three technical or experimental replicates, mean ± SD. Combination index (CI) was determined using Chou–Talalay method. ####CI = 0.61 ± 0.08. c Left—Growth curve of Daoy cells transduced with CRISPR-Cas9 technology treated with TMZ. Right—Growth curve of Med1-MB cells transduced with CRISPR-Cas9 technology treated with TMZ. Each data point represents mean of three technical or experimental replicates, mean ± SD, student t-test. d Left—Growth curve of Daoy cells transduced with YFP-CK2α or YFP-CK2β treated with TMZ. Right—Growth curve of Med1-MB cells transduced with YFP-CK2α or YFP-CK2β treated with TMZ. Each data point represents mean of three technical or experimental replicates, mean ± SD, student t-test. *p < 0.05, sample compared to control cells
To verify that the combinatorial effect was due to CK2 modulation and not off-target effects of CX-4945, we analyzed our MB knockout cell lines. We discovered that when normalized to the dimethyl sulfoxide (DMSO) control, Daoy sgCK2α or sgCK2β cells showed a significant decrease in their growth curves after treatment with TMZ compared to the sgControl cells, while there was no change in the Med1-MB knockout cells (Fig. 4c). To determine if increasing CK2 expression caused MB cells to become more resistant to TMZ treatment, we conducted a dose-dependent growth assay using increasing amounts of TMZ on Daoy and Med1-MB YFP-CK2α and YFP-CK2β cells. Exogenous expression of CK2α or CK2β allowed a higher percentage of cells to survive compared to the Daoy YFP control cells, while Med1-MB transduced cells had no observable change compared to the control cells (Fig. 4d).
CK2 regulates MGMT expression through β-catenin
In order to elucidate how CX-4945 and TMZ are working synergistically, we analyzed the O6-methylguanine DNA methyltransferase (MGMT) and β-catenin pathway in the MB cell lines. TMZ achieves cytotoxicity by methylation of the O6 position of guanine, which is reversed by MGMT activity. β-catenin was previously shown to regulate MGMT levels in MB cell lines [16], and through our own work we determined that CK2 can control β-catenin activity and levels in adult gliomas [7]. Consequently, we analyzed the expression of both MGMT and β-catenin in our MB cell lines with modulated CK2 activity. We discovered that inhibition of CK2 using CX-4945 or knock-out of CK2α or CK2β caused a threefold reduction in MGMT and β-catenin expression in Daoy cells, while exogenous expression of CK2α or CK2β yielded a three- to fivefold increase in MGMT and β-catenin expression (Fig. 5a–c). Since Med1-MB cells had little to no observable MGMT protein, it was not surprising that modulating CK2 expression did not alter MGMT expression (Fig. 5a–c). We verified that β-catenin played an important role in MGMT expression by inhibiting β-catenin using the compound FH-535. Inhibition of β-catenin in Daoy YFP-CK2α or YFP-CK2β cells restored MGMT expression to levels similar to the Daoy YFP control cells (Supplementary Fig. S7). To determine if β-catenin plays an integral role in maintaining MGMT expression, or if CK2 alone regulates MGMT, we transduced Daoy sgCK2α or Daoy sgCK2β cell lines with exogenous β-catenin (sgCK2 + β-catenin) (Fig. 5c). Adding back β-catenin restored its expression to ~80–90% of its endogenous levels and also increased MGMT expression 7-fold higher than the Daoy sgCK2α or Daoy sgCK2β knockout cells (Fig. 5c). Rescuing with β-catenin also restored growth in the Daoy sgCK2α and sgCK2β cell lines (Fig. 5d, e). To verify that the differential expression of MGMT levels due to CK2 activity were biologically relevant, we treated the β-catenin add-back cell lines with TMZ. Rescuing with β-catenin restored cell growth and anchorage independent growth, and restored TMZ sensitivity to cells lacking CK2 (Fig. 5f).

CK2 regulates MGMT expression through b-catenin. a Daoy and Med1-MB treated with CX-4945. The fold change is indicated below each blot normalized to untreated cells (DMSO). b Daoy and Med1-MB transduced with YFP-CK2α or YFP-CK2β. c Daoy sgCK2 cells transduced with exogenous β-catenin. d Growth curve of Daoy sgCK2 cells transduced with exogenous β-catenin. Bars represent mean of three technical or experimental replicates, mean ± SD, student t-test. e Soft-agar assay of Daoy sgCK2 cells transduced with exogenous β-catenin. Bars represent mean of three technical or experimental replicates, mean ± SD, student t-test. *p < 0.05, sample compared to control cells. f Left—Growth of Daoy sgCK2α cells transduced with exogenous β-catenin treated with TMZ. Right—Growth of Daoy sgCK2β cells transduced with exogenous β-catenin treated with TMZ. Each data point represents mean of three technical or experimental replicates, mean ± SD, student t-test. *p < 0.05, sample compared to control cells
Combining CK2 inhibition and TMZ increases MB apoptosis
Apoptosis plays an important role in the treatment of cancer as it is a popular target of many treatment strategies. We determined if modulating CK2 activity only inhibits MB growth or if it also induced MB apoptosis. To study cellular apoptosis, we monitored caspase 8 activity using a luminescence assay. We treated Daoy cells with 5 μM CX-4945 and 100 μM or 500 μM TMZ, and observed a synergistic effect (CI < 0.2) for both concentrations of TMZ where apoptosis levels were increased two- to threefold compared to the individual treatments (Fig. 6a). Med1-MB cells were also treated with both CX-4945 and TMZ, but there was no observable change in apoptosis in any of the combinatorial treatments (CI > 1.3) (Fig. 6b). Treatments at a lower concentration of CX-4945 was also conducted, and synergy was also seen with 1 μM CX-4945 and 500 μM TMZ (CI < 0.2) (Supplementary Fig. S5B). We also verified that it is the modulation of CK2 activity and not just CX-4945 treatment that can significantly alter MGMT activity. Exogenous expression of CK2α or CK2β decreased Daoy cell apoptosis after TMZ treatment (Fig. 6c), while Daoy cells lacking CK2α or CK2β had a significantly higher level of apoptosis after TMZ treatment compared to the control cell line (p < 0.05) (Fig. 6d). A similar increase in apoptosis after TMZ treatment was seen in the Daoy shCK2α cells, reinforcing the notion that inhibition or removal of CK2 will sensitize Daoy cells to TMZ treatment (Supplementary Fig. S3D–F). Med1-MB cells with modulated CK2 expression did not have any significant changes in apoptosis (Fig. 6e, f). Lastly, we were able to rescue the Daoy cells lacking CK2α or CK2β by reintroducing β-catenin. Adding back β-catenin decreased apoptosis to a similar manner as the sgControl cells (Fig. 6g).

Loss of CK2 activity or expression enhances TMZ-induced apoptosis. a Apoptosis assay for Daoy cells treated with TMZ and CX-4945. Bars represent mean of three technical or experimental replicates, mean ± SD. Combination index (CI) was determined using Chou–Talalay method #CI = 0.11 ± 0.03, ##CI = 0.08 ± 0.01. b Apoptosis assay for Med1-MB cells treated with TMZ and CX-4945. c Apoptosis for Daoy YFP-CK2α and YFP-CK2β cells treated with TMZ. d Apoptosis for Daoy cells transduced with CRISPR-Cas9 technology. e Apoptosis for Med1-MB YFP-CK2α and YFP-CK2β cells treated with TMZ. f Apoptosis for Med1-MB cells transduced with CRISPR-Cas9 technology. g Daoy sgCK2 cells transduced with exogenous β-catenin treated with TMZ. Bars represent mean of three technical or experimental replicates, mean ± SD, student t-test. *p < 0.05, sample compared to control cells
Discussion
In this study, we verified the importance of CK2 in MB tumorigenesis and discovered a novel connection between CK2 and MGMT expression that can lead to new exciting therapeutic advances. Consistent with our previous findings, inhibition of CK2 using CX-4945 reduced MB tumorigenesis [17]. Since single treatment arms rarely work in the clinic, we expanded on this initial finding and conducted a high-throughput assay to identify FDA-approved drugs that work with CX-4945. We found that TMZ works synergistically with CX-4945 to reduce MB growth and increase apoptosis. This is the first time that CK2 has been shown to dictate TMZ efficacy by regulating MGMT expression. Inhibition of CK2 directly led to a decrease in β-catenin levels, which resulted in a loss of MGMT expression (Fig. 7). Our findings are intriguing because it may help revolutionize the way we treat MB patients by combining standard-of-care therapies like TMZ with small molecule inhibitors like CX-4945.

Diagram of the mechanism of CK2 regulation of MB tumorigenesis. a Signaling cascade in which CK2 regulates MB tumorigenesis through β-catenin. b Effects of CK2 inhibition on MB survival and apoptosis
CK2 and MB clinical relevance
Our findings support the idea that CK2 could be a novel prognostic biomarker for MB patients. We analyzed multiple MB microarray datasets and discovered that MB patients with elevated expression of CK2α or CK2β had a significantly worse prognosis than patients with low expression. This is not the first time that CK2 could be viewed as a prognostic marker for cancer. Elevated levels of CK2α corresponded with a worse prognosis in glioblastoma [7], breast [18], ovarian [18], gastric cancer [19, 20], renal clear cell carcinoma [21], AML [20, 22], liver cancer [23, 24], and sarcomas [20]. High expression of CK2β lead to a poor prognosis in lung adenocarcinoma [18], breast [18], gastric cancer [20, 25], and head and neck cancer [20].
Targeting CK2 to inhibit MGMT
Previous clinical studies have analyzed the effectiveness of using TMZ as a treatment for MB patients (NSC# 704865) [26, 27]. While the overall response to TMZ treatment have been promising, numerous reports have shown that sensitivity to TMZ in MB is MGMT-dependent [28]. TMZ exhibited potent cytotoxic activity in MGMT-deficient primary MB cells, while inhibition of MGMT increased the sensitivity of TMZ in MGMT proficient cells [29]. Since there are currently no effective therapeutic MGMT inhibitors and more than half of MB patients have elevated MGMT levels [30], another method is needed to target MGMT. Through our current work, we demonstrated that inhibiting CK2 through CX-4945 can increase TMZ sensitivity to MGMT proficient cells (Daoy) by decreasing β-catenin and subsequently MGMT expression. Consistently, the TMZ sensitivity in MGMT-deficient cells (Med1-MB) are not changed with CX-4945 treatment. These findings indicate a new method to target MGMT using a clinically relevant small molecule inhibitor.
CK2 as a novel therapeutic target for MB
CK2 is an attractive avenue for anti-cancer therapy because it is essential to the survival and maintenance of numerous cancers and it supports multiple oncogenic signaling pathways. Through our report we demonstrated that inhibiting CK2 activity in MB cell lines decreased MB tumorigenesis. CX-4945, also known as silmitasertib, is an especially intriguing treatment option because it is an orally available, small molecule with highly selective inhibition of CK2 that can cross the blood–brain-barrier [31]. CX-4945 is currently undergoing multiple phase I/II clinical trials for patients with advanced solid tumors and it is generally well tolerated with low toxicity [32, 33]. Our findings are consistent with our recent report that showed CX-4945 treatment could increase survival in Ptch+/− mouse that develop MB like tumors [17]. Altogether, our findings indicate that CX-4945 can be an effective and safer treatment for MB patients. These findings provided the basis for the pediatric brain tumor consortium to begin a clinical trial, “A Pediatric Brain Tumor Consortium Phase I and Surgical Study of CX-4945 in Children with Recurrent or Refractory SHH Medulloblastoma (ClinicalTrials.gov PBTC −053)”, which will begin recruiting at the end of 2018.
In this study, we identified CK2 as a novel therapeutic target for MB. Here, we show for the first time that CK2 inhibition works synergistically with TMZ to induce MB apoptosis (Fig. 7). We believe our findings will be invaluable for future preclinical and clinical trials for MB patients with high levels of CK2 and MGMT.
Materials and methods
Dataset mining
Survival analysis within the MB dataset was performed using the MB microarray dataset from Cavalli et al. [9] from the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). Samples within a dataset were sorted into high or low expression of the investigated gene based on a cutoff expression value: CSNK2a1 (1214.900), CSNK2b (2161.300), CSNK2a1 WNT (837.2), CSNK2a1 SHH (1077.7), CSNK2a1 Group 3 (1164.1), CSNK2a1 Group 4 (584.7), CSNK2b WNT (2159.6), CSNK2b SHH (4049.9), CSNK2b Group 3 (3314.1), and CSNK2b Group 4 (2282.8). The log-rank significance of the projected survival was calculated and the best p-value and corresponding cutoff value was selected.
Gene expression
The relative mRNA expression levels were analyzed using the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). The normal brain microarray dataset from Berchtold et al. [12] (n = 172) was compared to the datasets from Gilbertson et al. [11] (n = 76), Delattre (n = 57) (MAS5.0—u133p2), and Kool [10] (n = 62). Boxes represent the interquartile range; lines represent the location of the first quartile, median, and third quartile. One-way analysis of variance (ANOVA) was used to determine the statistical significance between the sample sets by calculating the p-value.
Cell culture
The MB cell lines Daoy (from American Type Culture Collection, Manassas, VA, USA) and Med1-MB (gift from Dr. Gerald Grant) were authenticated and tested for mycoplasma before experiments were conducted. Daoy and Med1-MB were cultured in minimum essential media (MEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. HEK293 cells (from American Type Culture Collection, Manassas, VA, USA) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and 2 mM l-glutamine. FH-535, TBB, TBBz, and CX-4945 (Sigma, St. Louis, MO, USA) were used in inhibitor studies at stated concentrations for the specified time.
Western blot
For protein analysis, protein extracts from cells were harvested and immunoblotted as previously described [34]. The following antibodies were used for immunoblotting: green fluorescent protein (GFP)(CST #2956), CK2α (CST #2656), β-catenin (CST #8480), MGMT (CST #2739), PARP (CST# 9542), Cleaved PARP (CST #5625), glyceraldehyde 3-phosphate dehydrogenase (GAPDH)(CST #5174) (all from Cell Signaling Technology, Danvers, MA, USA), and CK2β (#04–1128, Millipore, Burlington, MA, USA). Chemiluminescence signals were quantitated using NIH Image J (National Institutes of Health).
Retroviral and lentiviral infections
Retroviral infections to exogenously express YFP-CK2α or YFP-CK2β were carried out as previously described [7]. The retroviral shRNA plasmid, pGFP-V-RS, used to express shRNA to the CK2α gene was purchased commercially (CSNK2A1 Human shRNA Plasmid Kit, Locus ID 1457. Origene, Rockville, MD 20850, USA). Retroviral infections were carried out as described above [7]. The Cas-sgRNA, infections were carried out in a similar manner described above [7]. HEK293 cells were transfected with psPAX2, pMD2.G, and pHR-SFFV-Cas9-BFP (# 46910, Addgene, Watertown, MA, USA) to generate virus to introduce Cas9 into the MB cell lines. The BFP-positive cells were subsequently infected with lentivirus generated from HEK293 cells transfected with psPAX2, pMD2.G, and pSLQ1371 sgCK2α or pSLQ1371 sgCK2β. For the sgRNA constructs, 72-bp oligos, including specific sgRNA sequences, were synthesized for PCR amplification with primers for sgCK2α-F (attgatcatgagcacagaa) or sgCK2β-F (gggcagtagagtttcacca) and sgRNA-R (gccgcctaatggatcctagta).
Cell growth analysis
Daoy and Med1-MB-infected cells were plated in six-well plates (5 × 105 cells per well). The number of live cells was counted daily by means of trypan blue exclusion assay. Cell growth was also measured using the CellTiter-blue cell viability assay (#G8080, Promega, Sunnyvale, CA, USA). Colony formation were conducted as previously described [7]. All growth experiments were done in triplicate and results are expressed as mean ± SD.
Xenograft transplants
Six to 8-week-old, female, NOD-scid IL2R gammanull mice were stereotactically injected with 100,000 D425 or DAOY-YFP, YFP-CK2α, or YFP-CK2β overexpressing cells (n = 5) into the cerebella as previously described [35], and monitored for survival. Injected animals that did not develop a primary tumor were excluded from the experiment. Only mice that developed a primary tumor were included in our survival analysis. For drug treatments, the mice (n = 5) were first randomized into two groups (DMSO or CX-4945 treatment) after the intracranial injections. The studies were not blinded. The log-rank (Mantel–Cox) test of Kaplan–Meier survival plots was used to determine the p-value. All animal studies were approved by the Stanford Internal Review board, APLAC-26992.
Immunohistochemistry
A MB TMA from Dr. Cho, Stanford University, was stained using a primary antibody to CK2α (ab70774) with 3,3’-diaminobenzidine (DAB) detection, and finally counterstained with hematoxylin. Brains of tumor-burdened mice were collected and fixed in 4% paraformaldehyde. Six micrometer thick FFPE sections were stained for Ki67 (16667, Abcam, Cambridge, MA, USA) and p-STAT3 (CST #9131) using ImmPRESS HRP reagent kit (anti-Rabbit IgG, Vector Labs. Burlingame, CA, USA), ImmPACT DAB and counterstained with hematoxylin, following heat-induced antigen retrieval. Three 297 × 223 μm sections of each tumor were imaged and quantified. Statistics: Student’s t-test.
High-throughput screen
One thousand Daoy cells were added to Geiner Black walled-clear bottom 384-well plates using the Matrix Wellmate with or without 10 µM CX-4945. Our drug library consisted of ~4000 FDA-approved drugs and known bioactives from the Library of Pharmacologically Active Compounds (LOPAC1280), the NIH Clinical Collection (NIHCC), the Microsource Spectrum, and the Biomol (now Enzo Life Sciences) ICCB Known Bioactives and FDA-Approved Drug Library. Two-hundred nanoliters from the compound libraries were added using the pin tool in the Staccato System (CaliperLS) in the High-Throughput Bioscience Center (HTBC). The plates were incubated for 72 h, and then imaged and analyzed in Molecular Devices ImageXpress Micro. Parameters were set to identify holes 9.67–30 microns in size with a signal intensity 500 units above local background. Hits were identified as compounds that result in a decrease in cell growth with or without CX-4945 as compared to no compound controls.
Apoptosis assay
To measure apoptosis in the MB cell lines, we used the Caspase-Glo® 8 Assay Systems (#G8200, Promega). We plated 1000 cells/well in 200 μL of MEM media and each sample was plated in triplicate. The specified drugs were incubated with the cells for 24 h and then apoptotic levels were measured using a plate reader to determine the luminescence readings according to the manufacturer’s directions.
Statistical analysis
The sample size was chosen based on the power to detect statistically significant effects. For in vitro experiments, at least three technical and experimental replicates from each experiment were compiled. Data represent mean ± standard deviation (SD) with n-values listed in figure legends. Excel was used to plot all bar graphs. Statistical analyses including student’s t-test, two-tailed test, or one-way analysis of variance was used to determine the p-value. p < 0.05 was considered significant. All the data did have a normal distribution and the SD between the groups were similar. For in vivo experiments, our sample size was chosen based on power needed to detect statistical significance. In addition, sample size was further chosen in accordance with the three R’s (replace, refine and reduce). The in vivo studies were done in duplicate. All Kaplan–Meier survival plots were plotted with GraphPad Prism and long-rank (Mantel–Cox) test was performed for comparison of median survival p < 0.05 was considered significant.
Change history
28 October 2019
The original version of this Article contained an error in the spelling of the author David Solow-Cordero, which was incorrectly given as David Solow-Codero. This has now been corrected in both the PDF and HTML versions of the Article.
References
Dhall G. Medulloblastoma. J Child Neurol. 2009;24:1418–30.
Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–20.
Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202–8.
Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15.
Agarwal, M, Nitta RT, Li G. Casein Kinase 2: a novel player in glioblastoma therapy and cancer stem cells. J Mol Genet Med. 2013;8:1–18.
Chua MM, Ortega CE, Sheikh A, Lee M, Abdul-Rassoul H, Hartshorn KL, et al. CK2 in cancer: cellular and biochemical mechanisms and potential therapeutic target. Pharmaceuticals (Basel). 2017;10:1–30.
Nitta RT, Gholamin S, Feroze AH, Agarwal M, Cheshier SH, Mitra SS, et al. Casein kinase 2alpha regulates glioblastoma brain tumor-initiating cell growth through the beta-catenin pathway. Oncogene. 2015;34:3688–99.
Purzner J, Paradiso GO, Cunic D, Saint-Cyr JA, Hoque T, Lozano AM, et al. Involvement of the basal ganglia and cerebellar motor pathways in the preparation of self-initiated and externally triggered movements in humans. J Neurosci. 2007;27:6029–36.
Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31:737–54. e736.
Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE. 2008;3:e3088.
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–8.
Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci USA. 2008;105:15605–10.
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–72.
Guo B, Cao S, Toth K, Azrak RG, Rustum YM. Overexpression of Bax enhances antitumor activity of chemotherapeutic agents in human head and neck squamous cell carcinoma. Clin Cancer Res. 2000;6:718–24.
Michaelis M, Agha B, Rothweiler F, Loschmann N, Voges Y, Mittelbronn M, et al. Identification of flubendazole as potential anti-neuroblastoma compound in a large cell line screen. Sci Rep. 2015;5:8202.
Wickstrom M, Dyberg C, Milosevic J, Einvik C, Calero R, Sveinbjornsson B, et al. Wnt/beta-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat Commun. 2015;6:8904.
Purzner T, Purzner J, Buckstaff T, Cozza G, Gholamin S, Rusert JM, et al. Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma. Sci Signal. 2018;11:1–15.
Ortega CE, Seidner Y, Dominguez I. Mining CK2 in cancer. PLoS ONE. 2014;9:e115609.
Bae JS, Park SH, Kim KM, Kwon KS, Kim CY, Lee HK, et al. CK2alpha phosphorylates DBC1 and is involved in the progression of gastric carcinoma and predicts poor survival of gastric carcinoma patients. Int J Cancer. 2015;136:797–809.
Chua MMJ, Lee M, Dominguez I. Cancer-type dependent expression of CK2 transcripts. PLoS ONE. 2017;12:e0188854.
Rabjerg M, Guerra B, Olivan-Viguera A, Mikkelsen ML, Kohler R, Issinger OG, et al. Nuclear localization of the CK2alpha-subunit correlates with poor prognosis in clear cell renal cell carcinoma. Oncotarget. 2017;8:1613–27.
Kim JS, Eom JI, Cheong JW, Choi AJ, Lee JK, Yang WI, et al. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin Cancer Res. 2007;13:1019–28.
Kim HS, Chang YG, Bae HJ, Eun JW, Shen Q, Park SJ, et al. Oncogenic potential of CK2alpha and its regulatory role in EGF-induced HDAC2 expression in human liver cancer. FEBS J. 2014;281:851–61.
Zhang HX, Jiang SS, Zhang XF, Zhou ZQ, Pan QZ, Chen CL, et al. Protein kinase CK2alpha catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget. 2015;6:34800–17.
Lin KY, Tai C, Hsu JC, Li CF, Fang CL, Lai HC, et al. Overexpression of nuclear protein kinase CK2 alpha catalytic subunit (CK2alpha) as a poor prognosticator in human colorectal cancer. PLoS ONE. 2011;6:e17193.
Aguilera D, Mazewski C, Fangusaro J, MacDonald TJ, McNall-Knapp RY, Hayes LL, et al. Response to bevacizumab, irinotecan, and temozolomide in children with relapsed medulloblastoma: a multi-institutional experience. Childs Nerv Syst. 2013;29:589–96.
Akyuz C, Demir HA, Varan A, Yalcin B, Kutluk T, Buyukpamukcu M. Temozolomide in relapsed pediatric brain tumors: 14 cases from a single center. Childs Nerv Syst. 2012;28:111–5.
Othman RT, Kimishi I, Bradshaw TD, Storer LC, Korshunov A, Pfister SM, et al. Overcoming multiple drug resistance mechanisms in medulloblastoma. Acta Neuropathol Commun. 2014;2:57.
von Bueren AO, Bacolod MD, Hagel C, Heinimann K, Fedier A, Kordes U, et al. Mismatch repair deficiency: a temozolomide resistance factor in medulloblastoma cell lines that is uncommon in primary medulloblastoma tumours. Br J Cancer. 2012;107:1399–408.
Faoro D, von Bueren AO, Shalaby T, Sciuscio D, Hurlimann ML, Arnold L, et al. Expression of O(6)-methylguanine-DNA methyltransferase in childhood medulloblastoma. J Neurooncol. 2011;103:59–69.
Zheng Y, McFarland BC, Drygin D, Yu H, Bellis SL, Kim H, et al. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin Cancer Res. 2013;19:6484–94.
Borad MJ, Hubbard JM, Oh D-Y, Rha SY, Mody K, Lim JKC, et al. A phase IB study of CX-4945 in combination with gemcitabine plus cisplatin in the frontline systemic treatment of patients with advanced cholangiocarcinoma. J Clin Oncol. 2017;35:294–94.
Marschke RF, Borad MJ, McFarland RW, Alvarez RH, Lim JK, Padgett CS, et al. Findings from the phase I clinical trials of CX-4945, an orally available inhibitor of CK2. J Clin Oncol. 2011;29:3087–87.
Nitta RT, Del Vecchio CA, Chu AH, Mitra SS, Godwin AK, Wong AJ. The role of the c-Jun N-terminal kinase 2-alpha-isoform in non-small cell lung carcinoma tumorigenesis. Oncogene. 2011;30:234–44.
Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. 2012;21:601–13.
Acknowledgements
We would like to thank Albert Wong and the Cell Sciences Imaging Facility (CSIF) at Stanford University for use of their equipment. This work was funded by generous donations of the Anatoly Machulskiy family, Keith Tsu and Carmelita Ko, Steven Sommer, and the Yatindra Singh family. This research was also funded by Stanford Cancer Institute Bioscience Screening Award and NIH-KNS085333A and SB was supported by the Swedish Childhood Cancer Foundation.
Author contributions
RTN and GL designed the study. RTN performed majority of in vitro experiments and analyses. SB performed in vivo, in vitro survival studies experiments, and analysis. EL performed in vitro survival studies and WB. DES-C performed the high-throughput assay. PS analyzed and quantified the TMA. TP stained TMA sections. PA conducted the high-throughput analysis. GN assisted in the in vivo experiments. YJC supplied TMA. RTN wrote the manuscript with support from GL and SB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nitta, R.T., Bolin, S., Luo, E. et al. Casein kinase 2 inhibition sensitizes medulloblastoma to temozolomide. Oncogene 38, 6867–6879 (2019). https://doi.org/10.1038/s41388-019-0927-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-0927-y
This article is cited by
-
Cdc2-like kinases: structure, biological function, and therapeutic targets for diseases
Signal Transduction and Targeted Therapy (2023)
-
Protein kinase CK2: a potential therapeutic target for diverse human diseases
Signal Transduction and Targeted Therapy (2021)
-
Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy – potential clinical relevance
Cellular Oncology (2020)
-
Advances in Targeted Therapies for Pediatric Brain Tumors
Current Treatment Options in Neurology (2020)
