
Abstract
Proper follicle development is very important for the production of mature oocytes, which is essential for the maintenance of female fertility. This complex biological process requires precise gene regulation. The most abundant modification of mRNA, N6-methyladenosine (m6A), is involved in many RNA metabolism processes, including RNA splicing, translation, stability, and degradation. Here, we report that m6A plays essential roles during oocyte and follicle development. Oocyte-specific inactivation of the key m6A methyltransferase Mettl3 with Gdf9-Cre caused DNA damage accumulation in oocytes, defective follicle development, and abnormal ovulation. Mechanistically, combined RNA-seq and m6A methylated RNA immunoprecipitation sequencing (MeRIP-seq) data from oocytes revealed, that we found METTL3 targets Itsn2 for m6A modification and then enhances its stability to influence the oocytes meiosis. Taken together, our findings highlight the crucial roles of mRNA m6A modification in follicle development and coordination of RNA stabilization during oocyte growth.
Similar content being viewed by others


Introduction
In mice, a few days after birth, a restricted number of oocytes within primordial follicles serve as the source of mature eggs for reproduction [1]. Through follicle recruitment or activation, resting primordial follicles are recruited into the growing follicle stage featuring oocyte growth and proliferation of somatic granulosa cells [2]. At this stage, oocytes grow rapidly with transcription of a large number of mRNAs that are crucial for subsequent oocyte meiotic maturation and early embryo development. When the oocytes reach the fully-grown germinal vesicle (GV) stage, the maternal transcriptome is completed along with the shutdown of transcription [3]. Throughout the follicle developmental process, oocytes are arrested in the prophase of meiosis I. At puberty, the oocytes resume and complete meiosis I under the stimulation of gonadotropin hormones and are then arrested in metaphase II (MII) until fertilization occurs [4].
Due to the transcriptional silencing of the maternal genome in GV oocytes, normal oocyte meiotic maturation is supported only by early-expressed or stored maternal mRNAs in oocytes. Thus, the maintenance of maternal mRNAs throughout the early developmental stages appears particularly critical. Recent studies have shown that serial epigenetic events participate in process that regulate the composition of oogenesis-associated maternal mRNAs, including DNA methylation [5, 6], histone modifications [7, 8], chromatin remodeling [9, 10], and chromosome stability [11]. Furthermore, the posttranscriptional regulation of poly(A) tail length and RNA modifications has also been validated to play an important role in determining the density of maternal mRNAs [12], suggesting the involvement of a complex regulation mechanism in oocyte development.
In mammals, N6-methyladenosine (m6A) is the most abundant internal modification of mRNAs and is involved in many critical aspects of RNA metabolism, including transcription [13], splicing [14, 15], mRNA stability [16, 17], mRNA decay [18, 19], mRNA export [20,21,22], and translation [23, 24]. The formation of m6A is catalyzed by a multicomponent methyltransferase complex including methyltransferase-like 3 (METTL3) [25], methyltransferase-like 14 (METTL14) [26], Wilms tumor 1 associated protein (WTAP) [27], and other proteins. m6A is a reversible modification that can be erased by the demethylases fat mass and obesity-associated factor (FTO) [28] and AlkB homolog 5 (ALKBH5) [22] and can be recognized by YTH-domain-containing family proteins [19, 29,30,31], insulin-like growth factor 2 (IGF2), mRNA-binding proteins [16, 32], and heterogeneous nuclear ribonucleoproteins (HNRNPs) [33] to regulate different developmental processes. Some m6A-associated proteins have been reported to participate in oocyte and early embryo development by regulating the turnover of maternal mRNAs, including KIAA1429 and YTHDC1, which regulate maternal mRNA splicing in the GV stage [34, 35], and YTHDF2, which regulates maternal mRNA decay in the MII stage [29]. Recently, IGF2BP2 and IGF2BP3 have also been shown to participate in stabilizing maternal mRNAs for early embryo development in mouse and zebrafish, respectively [36, 37]. However, these studies mainly focused on the functions of non-core m6A writer or m6A readers in regulating partial maternal mRNAs, the global function of m6A in follicle development and oocyte maturation still remains elusive. As the core subunit of the m6A methyltransferase complex, METTL3 has usually been used to evaluate the global function of m6A both in vivo and in vitro [38, 39]. Nowadays, METTL3-mediated m6A has been shown to modulate spermatogenesis [40,41,42], postimplantation embryonic development [43], sex determination [14, 15], and human diseases [44]. In addition, by knocking down Mettl3 in GV oocytes from mice, oocyte maturation and early embryo development displayed defects probably due to disrupting mRNA degradation [45]. However, owing to the embryonic lethality of Mettl3 knockout mice, the in vivo function of Mettl3 in follicle development remains unknown.
Here, we investigated how m6A modification specifically regulates follicle development and ovulation. Our results demonstrated that oocyte-specific inactivation of Mettl3 with Gdf9-Cre causes defective follicle development and infertility. Mechanistically, we found that METTL3-mediated m6A modification regulates the stabilization of Itsn2 at the GV stage and then influences the resumption of meiosis during oocyte development.
Results
METTL3 is expressed during follicle development and is required for female fertility
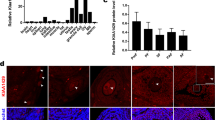
To determine the roles of m6A in follicle development, we first examined whether METTL3 is expressed in mouse ovaries. The immunohistochemistry results showed that METTL3 was expressed at all stages of folliculogenesis and was mainly located in the oocyte nuclei and granulosa cells (Fig. 1A). Further detection at the single-cell level using immunofluorescence showed that the METTL3 protein was indeed located in the oocyte nucleus at postnatal day (PD) 5, PD12, and the GV stage; however, due to meiotic nuclear division, the METTL3 signal was uniformly dispersed in the oocytes at the MII stage (Fig. 1B). The high abundance and relatively dynamic distribution of METTL3 during the whole process of oocyte development suggest that METTL3 might play a role in the regulation of oocyte competence and maturation. To understand the in vivo functions of Mettl3 in female reproduction, we generated Mettl3flox/flox;Gdf9-Cre (referred to as Mettl3Gdf9 cKO) mice by using Gdf9-Cre, which mediates Cre recombinase expression in mouse oocytes at the primordial stage, to knock out Mettl3 specifically in oocytes [46]. qRT-PCR confirmed that oocytes from Mettl3Gdf9 cKO mice had a negligible expression of Mettl3 mRNA as compared to Mettl3flox/flox (referred to as WT) mice (Fig. 1C). Additionally, the immunohistochemistry and immunoblotting results showed that METTL3 protein was nearly undetectable in the oocytes of Mettl3Gdf9 cKO ovaries (Fig. 1D, E), suggesting the successful oocyte-specific knockout of Mettl3.

A Immunohistochemistry of METTL3 in 3-week-old mouse ovaries at different follicle stages with PMSG injection. The arrows indicate oocytes at different follicle stages. Scale bar, 20 μm. B Confocal immunofluorescence images of oocytes from wild-type mice stained with METTL3 antibody (red) and DAPI (blue), as indicated. PD5 postnatal days 5, PD12 postnatal days 12, GV germinal vesicle, MII metaphase II. Scale bar, 20 μm. C qRT-PCR analysis of Mettl3 mRNA levels in oocytes from 3-week-old WT and Mettl3Gdf9 cKO females. The relative mRNA level of Mettl3 in WT oocytes was set to 1.0. ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM (n = 3). D Immunohistochemistry for METTL3 in ovary sections from WT and Mettl3Gdf9 cKO mice. Primordial (PrF) and secondary (SF) follicle stages are indicated. Scale bar, 20 μm. E Immunoblotting analysis of METTL3 protein level in oocytes of WT and Mettl3Gdf9 cKO mice. GAPDH was used as an internal control. One hundred germinal vesicle oocytes were used for each lane of the blots. F Cumulative numbers of pups born from six pairs of WT and Mettl3Gdf9 cKO female mice for 5 months. G Representative images of ovaries from 6-week-old, 8-week-old, and 12-week-old female mice. Scale bar, 200 μm. H Ratio of ovary weight to body weight of 6-week-old, 8-week-old, and 12-week-old female mice. 6-week-old, n = 3; 8-week-old, n = 6; 12-week-old, n = 6. n.s., p > 0.05; ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM.
Next, to detect the effect of Mettl3 knockout on female fertility, we randomly selected six pairs of 6-week-old WT and Mettl3Gdf9 cKO female mice, which were bred to WT male mice for 5 months. Six WT females produced a total of 232 pups, whereas the six Mettl3Gdf9 cKO females produced no offspring (Fig. 1F). Next, we analyzed ovaries obtained at 6 weeks, 8 weeks, and 12 weeks. The sizes of the ovaries and the ovary-to-body weight ratios of Mettl3Gdf9 cKO mice were not significantly different from those of WT mice at 6 weeks, but they were significantly smaller than those of WT mice at 8 weeks. The difference in the ovary-to-body weight ratio between Mettl3Gdf9 cKO and WT mice at 12 weeks was more significant than that at 8 weeks; moreover, the ovary surface was smooth at 12 weeks, with almost no obvious follicle structure (Fig. 1G, H). Together, these results demonstrate that Mettl3 in oocytes is essential for female fertility.
Mettl3 Gdf9 cKO mice display follicle development defects
To investigate the phenotype after Mettl3 knockout during follicle development, we performed hematoxylin-eosin (HE) staining on ovaries from 6-week-old, 8-week-old females and counted the number of follicles at each stage. In 6-week-old females, there was no significant difference in the number of follicles in each stage. However, in 8-week-old females, there were more primary follicles and fewer primordial, secondary, and antral follicles in the Mettl3Gdf9 cKO ovaries than in the WT ovaries (Fig. 2A, B), demonstrating that METTL3 was not necessary for the transition of primordial follicles to the activated growing follicle stage, but mainly functions in the process of growing follicle development.

A Representative ovarian histology of 6-week-old, 8-week-old WT, and Mettl3Gdf9 cKO mice. The primary, secondary, and antral (indicated with PF, SF, and AF, respectively) follicle stages are indicated by the black arrow. Scale bar, 50 μm. B Quantification of the numbers of different types of follicles in ovaries from 6-week-old, 8-week-old WT, and Mettl3Gdf9 cKO mice. Primordial, primary, secondary, and antral follicles (indicated with PrF, PF, SF, and AF, respectively) were counted. n.s., p > 0.05; *p < 0.05; **p < 0.01 by two-tailed Student’s t-test. Data represent the mean ± SEM (n = 3). C Confocal immunofluorescence staining with γH2AX antibody (red) and DAPI (blue) in ovary sections from 6-week-old WT and Mettl3Gdf9 cKO mouse ovaries. Scale bars, 20 μm. D Graph showing the quantification of γ-H2AX staining. ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM (n = 4). E Detection of apoptosis in granulosa cells by TUNEL kit performed in paraffin sections at different stages of follicle development of 6-week-old WT and Mettl3Gdf9 cKO mouse ovaries. Secondary and antral follicles (indicated with SF and AF, respectively) were counted. Scale bars, 20 μm. F Graph showing the quantification of the number of TUNEL positive granulosa cells at different follicle stages. *p < 0.05; **p < 0.01 by two-tailed Student’s t-test. Data represent the mean ± SEM (n = 3). G, H FSH and LH levels of WT and Mettl3Gdf9 cKO mice. Serum samples were collected from female mice of both genotypes (12 to 20 weeks old) and were killed for measurement of FSH and LH levels. FSH: WT, n = 5; Mettl3Gdf9 cKO, n = 9. LH: WT, n = 8; Mettl3Gdf9 cKO, n = 9. **p < 0.01; ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM. I Immunohistochemistry with MVH antibody in ovary sections from 24-week-old WT and Mettl3Gdf9 cKO mice. Scale bar, 50 μm.
The defective follicle development in Mettl3Gdf9 cKO mice suggested that Mettl3-deficient oocytes might undergo apoptosis. As DNA damage is a major inducer of apoptosis, we assessed potential DNA damage in oocytes through immunofluorescence assays for phosphorylated histone H2AX (γ-H2AX), a widely used marker gene for DNA double-strand breaks (DSBs) [47]. The results showed that more DSBs were produced in the secondary follicles of the Mettl3Gdf9 cKO mice than in those of the WT mice (Fig. 2C, D). Moreover, we also performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining to validate this finding. Indeed, Mettl3Gdf9 cKO mice showed a significantly higher apoptosis signal in secondary and antral follicle than WT mice (Fig. 2E, F).
The levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) are two important parameters for the assessment of premature ovarian failure (POF) [48]. An enzyme-linked immunosorbent assay (ELISA) showed that the levels of FSH (Fig. 2G) and LH (Fig. 2H) were higher in the serum of 12- to 20-week-old Mettl3Gdf9 cKO mice than in that of WT mice. Moreover, immunohistochemistry for MVH (an oocyte marker) in 24-week-old ovaries indicated that oocytes were absent in Mettl3Gdf9 cKO ovaries, which lead to POF (Fig. 2I). Thus, METTL3 is indispensable for follicle development because it preserves oocyte survival.
Mettl3 Gdf9 cKO oocytes fail to resume meiosis
To further explore the potential mechanism leading to follicle development defects after Mettl3 deletion, we collected GV oocytes from 6-week-old mice after pregnant mare serum gonadotropin (PMSG) treatment. The experimental results showed that the number of GV oocytes in Mettl3Gdf9 cKO mice was significantly reduced, and the oocyte diameter also became obviously smaller compared with WT mice (Fig. 3A–C).

A Representative images showing GV stage oocyte from 6-week-old WT and Mettl3Gdf9 cKO females. Left column: scale bar, 100 μm; right column: scale bar, 10 μm. B, C Mean number and diameter of GV stage oocytes obtained per mouse after priming with PMSG. WT, n = 72; Mettl3Gdf9 cKO, n = 97. **p < 0.01; ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM. D Confocal immunofluorescence with α-Tubulin antibody (green) and DAPI (blue) for oocytes from 6-week-old WT and Mettl3Gdf9 cKO mice after PMSG and HCG injection. Scale bars, 20 μm. E, F GVBD percentages and first polar body (PB1) rates for oocytes from 6-week-old WT and Mettl3Gdf9 cKO mice after PMSG and HCG injection. WT, n = 73; Mettl3Gdf9 cKO, n = 49. ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM. G Representative images of embryos collected from WT and Mettl3Gdf9 cKO mice at the indicated time points after natural ovulation. Scale bar, 100 μm. H Rates of a zygote, four-cell, and blastocyst formation by ovulated WT and Mettl3Gdf9 cKO oocytes after culture in KSOM medium. WT, n = 45; Mettl3Gdf9 cKO, n = 45. n.s., p > 0.05; **p < 0.01; ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM.
It has been reported that meiotic errors can reduce oocyte developmental competence [49]. We thus investigated the influence of Mettl3 knockout on meiosis and performed immunofluorescence staining for Tubulin to detect spindle formation. We found that most of the oocytes from Mettl3Gdf9 cKO could not undergo germinal vesicle breakdown (GVBD); with only a small number of oocytes reached the end of meiosis I, and almost no first polar body exclusion occurred, suggesting that no mature MII oocytes were produced in Mettl3Gdf9 cKO mice (Fig. 3D–F). These results indicated that the resumption of meiosis in Mettl3Gdf9 cKO oocytes was defective, which influenced the maturation of the oocytes. In addition, the results of subsequent assays examining the developmental competence of ovulated oocytes showed that there was no significant difference in ovulation number between 6-week-old Mettl3Gdf9 cKO and WT mice. However, most of the oocytes from Mettl3Gdf9 cKO mice could not be fertilized to form zygotes or develop beyond the four-cell embryo stage (Fig. 3G, H). But almost no zygotes could be obtained from 8-week-old Mettl3Gdf9 cKO mice compared with WT mice by natural ovulation (Fig. S1A, B). These results suggested that oocyte developmental competence was severely impaired.
METTL3-mediated m6A maintains maternal mRNA stability in oocytes
As the core subunit of the m6A methyltransferase complex, METTL3 mainly mediates m6A formation on mammalian mRNAs. To identify potential maternal mRNA targets regulated by METTL3, we first conducted the m6A methylated RNA immunoprecipitation sequencing (MeRIP-seq) assay on GV oocytes. More than 81% (5368) overlapping m6A peaks were detected in two independent biological replicates (Fig. 4A and Table S1); these peaks corresponded to 3256 (45.8%) maternal mRNAs, indicating that m6A plays a more important role in regulating the density of oocyte maternal mRNAs than previously reported. Consistent with the findings of previous studies, the m6A peaks detected in GV oocytes were significantly enriched in the GGACH motif (H = A/C/U) and were abundant in the coding region (CDS), in the 3′ untranslated region (3′UTR), and near the stop codon (Fig. 4B, C). And 90.1% of the methylated mRNAs contained fewer than four peaks, with an average of two peaks per mRNA (Fig. 4D) in this stage.

A Venn diagram showing the overlay of m6A peaks in transcripts between two independent biological replicates. B Pie plot showing the percentages of m6A peaks on distinct mRNA segments: the TSS, 5′ untranslated region (5′UTR), CDS, stop codon, and 3′ untranslated region (3′UTR). C The m6A motif of GV oocytes enriched with HOMER (inner panel). The distribution pattern displays the position density of m6A across the 5′UTR, CDS, and 3′UTR of mRNAs (outer panel). D Bar plot showing the numbers of transcripts with different numbers of m6A peaks. E Cumulative distribution showing the differences (log2 (fold change) values) in transcript expression between Mettl3Gdf9 cKO and WT oocytes. The transcripts were classified as m6A and non-m6A by MeRIP-seq. The p value was determined with the two-sided Kolmogorov–Smirnov test. F Heatmap showing the significantly enriched KEGG pathways regulated by transcripts with m6A modification.
To further investigate the influence of Mettl3 deficiency on maternal mRNA abundance, we compared the transcriptomes between WT and Mettl3Gdf9 cKO oocytes at the GV stage based on RNA-seq data. We found that the abundance of maternal mRNAs with m6A peaks was significantly decreased than that of maternal mRNAs without m6A peaks upon Mettl3 knockout (Fig. 4E), suggesting that maternal mRNAs with m6A modification are preferentially stabilized in the GV stage. In addition, functional enrichment analysis showed that m6A-modified maternal mRNAs preferentially participate in the regulation of the cell cycle, oocyte meiosis, and RNA degradation (Fig. 4F).
Consistent with the MeRIP-seq data, the RNA-seq data revealed that 2053 maternal transcripts were significantly downregulated upon Mettl3 knockout, approximately two-fold more than those that were upregulated (Fig. 5A and Table S2). Functional enrichment analysis showed that oocyte-specific knockout of Mettl3 globally reduced the abundance of cell cycle-, oocyte maturation-, and meiosis-related maternal mRNAs (Fig. S2A and Table S3). In addition, we identified 1098 (53.5%) downregulated maternal mRNAs with m6A modification, significantly more than the 342 (35.2%) upregulated mRNAs (p < 2.2e-16, Chi-squared test, Fig. 5B). The downregulated maternal mRNAs with m6A modification were also significantly enriched for follicle development and oocyte meiosis-associated biological processes, including the cell cycle and DNA repair (Fig. 4F and Table S3). Overall, these results suggest that METTL3, an m6A methyltransferase, can maintain oocyte development by stabilizing functional maternal mRNAs in the GV stage.

A Volcano plot showing the expression differences for target transcripts under Mettl3Gdf9 cKO. Transcripts with average RPKM values >3 in WT, |log2 fold change| values > log2 (1.2), and p values <0.05 as determined by DESeq2 were regarded as significantly dysregulated transcripts. The numbers of significantly downregulated (blue) and upregulated (red) transcripts are shown. The vertical dashed lines indicate the cutoff of |log2 fold change| = |log2 (1.2)|, and the horizontal dashed lines indicate the cutoff of p = 0.05. B Bar plot showing the ratios of transcripts with m6A modification among downregulated transcripts and upregulated transcripts under Mettl3 knockout. C Bar plot showing the expression of Igf2bp1, Igf2bp2, and Igf2bp3 in GV oocytes, as determined by RNA-seq in this study and in the GSE96638 dataset. The data were shown as the mean ± SEM of two independent experiments for this study and 26 independent experiments for the GSE96638 dataset. D Density plot showing the distance between IGF2BP2/3 peaks identified by eCLIP (GSE78509) in hESCs and m6A peaks identified by MeRIP-seq in this study. The background was obtained by randomly shuffling IGF2BP peaks using BEDTools’ shuffleBed tool. The p value was determined by the two-sided Kolmogorov–Smirnov test. E Venn diagram showing the overlay between downregulated expressed transcripts with m6A peaks and IGF2BP2/3 target transcripts. F Heatmap displaying the transcript abundance in WT and Mettl3Gdf9 cKO oocytes. Transcripts with m6A modifications targeted by IGF2BPs are labeled brown, transcripts involved in meiosis or DNA repair are labeled in green or white, and the expression correlation between transcripts and Mettl3 is shown in red.
IGF2BPs regulate the maternal mRNA abundance during oogenesis
IGF2 mRNA-binding proteins 1, 2, and 3 (IGF2BP1, IGF2BP2, and IGF2BP3, respectively) have been identified as a distinct family of m6A readers mediating mRNA stabilization [16]. Recently, IGF2BP2 and IGF2BP3 have been validated to maintain maternal mRNA abundance for early embryo development in mouse and zebrafish, respectively [36, 37]. As m6A-marked maternal mRNAs in GV stage oocytes are preferentially downregulated upon Mettl3 depletion, we hypothesized that the change in maternal mRNA abundance in GV stage oocytes might be regulated by the IGF2BP family. Thus, we next investigated whether some of the m6A-marked maternal mRNAs could be directly targeted by the IGF2BP family. The expression of only Igf2bp2 and Igf2bp3 could be detected in GV stage oocytes (Fig. 5C), suggesting that Igf2bp2 and Igf2bp3 might be the main participants in the maintenance of maternal mRNA abundance in this stage. To explore the correlation between IGF2BP2/3 and m6A in GV stage oocytes, we downloaded public data on IGF2BP2/3 peaks [50] detected in human embryonic stem cells (hESCs) with eCLIP and converted them to mm10 genomic coordinates using liftOver. The distances between the IGF2BP2/3 and m6A peaks were calculated using BEDTools. The results showed that the distance between peaks was significantly closer than the shuffled background, especially for IGF2BP3 (Fig. 5D), suggesting that the region with m6A modification could be accessible to IGF2BP2 and even more easily accessible to IGF2BP3. Furthermore, we identified 617 (56.2%) downregulated m6A-modified maternal mRNAs targeted by IGF2BP2 or IGF2BP3 (Fig. 5E and Table S4). Among them were a group of transcripts that are responsible for oocyte meiosis and DNA repair (Fig. 5F). Collectively, these data indicate that IGF2BP2 and especially IGF2BP3, as m6A readers, might participate in stabilizing oogenesis-related maternal mRNAs involved in meiosis and DNA repair.
METTL3 participates in oocyte maturation by regulating m6A modification of Itsn2
To elucidate the molecular mechanism of Mettl3 in oocyte development, we compared the transcripts whose abundance positively correlated with Mettl3 in the GV stage with those of 617 IGF2BP2/3 binding targets that contained m6A peaks and were downregulated upon Mettl3 depletion. We found that five oocyte meiosis-associated transcripts (Itsn2, Spire1, Myt1, Pias1, and Cdc42bpa) and five DNA repair-associated transcripts (Brca1, Ercc6l, Palb2, Brca2, and Mcm9) were potentially regulated by METTL3 and IGF2BP2/3 via m6A modification (Fig. 5F and S2B). Among these potential targets, we found the homologs of Itsn2, Spire1, Pias1, Ercc6l, and Brca2 are also targeted by IGF2BP3 using RIP-seq at the sphere stage of zebrafish [36] (Fig. S2C), indicating IGF2BP3 could regulate the fate of these maternal mRNAs at different developmental stages across species. As reported previously, the Itsn2 gene encodes an adapter protein involved in microtubule formation and signal transduction and participates in meiosis during oocyte maturation [51]. Furtherly, we found that the abundance of Itsn2 was significantly positively correlated with that of Mettl3 at the GV stage (Fig. 6A). Next, qRT-PCR was performed, and the results validated the significant downregulation of Itsn2 mRNA upon Mettl3 deletion in the GV stage (Fig. 6B).

A Scatter plot showing the Pearson correlation of RNA abundance between Itsn2 and Mettl3. B qRT-PCR analysis of Itsn2 and Mettl3 mRNA levels in oocytes from 3-week-old WT and Mettl3Gdf9 cKO mice. The relative mRNA levels of Itsn2 and Mettl3 in WT oocytes were set to 1.0. **p < 0.01; ***p < 0.001 by two-tailed Student’s t-test. Data represent the mean ± SEM (n = 3). C qRT-PCR analysis of Itsn2 mRNA stability in growing oocytes of WT and Mettl3Gdf9 cKO mice. Data were presented as means ± SEM, n = 3. D Integrated genomics viewer (IGV) plot showing the m6A peak across Itsn2 mRNA transcript; the green box represents the m6A peak identified by MeRIP-seq. E Confocal immunofluorescence staining for ITSN2 (green) in oocytes. DAPI was used to stain nuclei (blue). Scale bar, 20 μm. F Images of control siRNA (Control) and Itsn2 siRNA microinjected oocytes. Dashed circles indicate oocytes with big polar body or symmetrical division. Scale bar, 100 μm. G, H Rates of PB1 formation and abnormal oocytes after Itsn2 siRNA and control siRNA (Control) microinjection. Control siRNA microinjected oocytes, n = 95; Itsn2 siRNA microinjected oocytes, n = 105. *p < 0.05; **p < 0.01 by two-tailed Student’s t-test. Data represent the mean ± SEM (n = 3). I Confocal immunofluorescence staining with α-Tubulin antibody (green) and DAPI (blue) for Itsn2 siRNA and control siRNA microinjected oocytes. Scale bar, 20 μm.
Next, we examined the stability of Itsn2 mRNA and found a shortened mRNA half-life of Itsn2 in Mettl3Gdf9 cKO growing oocytes compared with WT oocytes (Fig. 6C), indicating that m6A modification may promote Itsn2 mRNA stability. In addition, our MeRIP-seq data and public IGF2BP3 RIP-seq data showed that there was only one METTL3-mediated m6A peak located in the CDS of the Itsn2 transcript that might be recognized by IGF2BP3 (Figs. 6D, 5F, and S2C), indicating that IGF2BP3 is a potential cofactor for Itsn2 abundance maintenance. Moreover, the immunofluorescence results showed that ITSN2 was expressed in the nucleus and cytoplasm of GV oocytes (Fig. 6E). Then, we screened three pairs of Itsn2 candidate siRNAs for knockdown of Itsn2 in oocytes, and we selected the most efficient siRNA, siRNA-1, for subsequent functional verification (Fig. S3A, B and Table S5). After injection of siRNA-1 for Itsn2 knockdown, we found that the rate of polar body exclusion decreased significantly, and some abnormal oocytes showed a large polar body or symmetrical division (Fig. 6F–I). Collectively, the results illustrate that the m6A methyltransferase METTL3 participates in posttranscriptional regulation of Itsn2 stability in oocytes, which is essential for oocyte maturation and meiotic divisions (Fig. 7).

In WT mouse oocytes, Itsn2 mRNA with m6A could be recognized by IGF2BP3 to furtherly enhance its stability and promote oocyte meiotic maturation. However, in Mettl3Gdf9 cKO mouse oocytes, Itsn2 mRNA without m6A could be degraded after losing IGF2BP3 protection, leading to oocyte meiotic maturation failure.
Discussion
METTL3, the core methyltransferase subunit, has been demonstrated to regulate postimplantation development [43], maintenance of the embryonic stem cell pluripotency network [52, 53], and spermatogenesis [38,39,40]. However, because its knockout in mice causes early embryonic lethality [43], the in vivo functions of Mettl3 in female reproduction remain unknown. Our findings indicated that METTL3 was highly expressed during follicle growth and oocyte maturation (Fig. 1A, B). We then generated mice with oocyte-specific knockout of Mettl3 at the primordial follicle stage to explore the potential role of Mettl3 in the female germline. Interestingly, Mettl3 knockout at the primordial follicle stage did not affect the survival of the primordial follicle until 6 weeks (Fig. 2A, B), the phenotype is more manifested in oocyte maturation at this stage (Fig. 3). Until 8 weeks, Mettl3Gdf9 cKO female mice displayed a severe defect in follicle development, which showed more primary follicles and fewer secondary follicles (Fig. 2A). In addition, it has been reported that METTL3 plays an important role in regulating unrepaired DSBs and genome instability by modulating DNA-RNA hybrid accumulation [54]. We found more DSBs are produced in the secondary follicles upon Mettl3Gdf9 cKO (Fig. 2C, D), indicating METTL3-deficient oocytes may also undergo apoptosis. Moreover, it has been further verified by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Fig. 2E, F). Therefore, we speculate that might also be a potential mechanism to regulate oocyte DSBs and apoptosis in the Mettl3Gdf9 cKO mice. Thus, our study shows that METTL3 is not necessary for the transition of resting primordial follicles to activated growing follicles, but participates in follicle and oocyte development and plays an important role in female reproduction.
As reported previously, m6A regulatory enzymes can participate in the meiotic maturation of oocytes by affecting associated RNA metabolic processes. For example, the writer protein KIAA1429 and the reader protein YTHDC1 have been reported to regulate oocyte meiotic maturation by altering RNA alternative splicing [34, 35], and the reader protein YTHDF2 influences oocyte maturation by regulating maternal mRNA degradation [29, 55]. However, the fates of only certain maternal mRNAs can be influenced by these proteins, and the proportion of maternal mRNAs with m6A modification in oocytes is still unclear. Although Mettl3 was knockout from the primordial follicle stage, the phenotype began to appear at later follicle developmental stages (Fig. 3). So in this work, we performed, for the first time, MeRIP-seq using low-input materials from GV oocytes based on a previously described protocol with some modifications [56]. According to the sequencing data, we identified 3256 (45.8%) maternal RNAs expressed in GV oocytes that contained m6A modifications, suggesting that the function of m6A in oocytes has been underestimated. Through a combination of RNA-seq and MeRIP-seq data, we found that m6A-modified transcripts showed significantly lower expression upon Mettl3 deletion than transcripts without m6A (Fig. 4E). We identified 1098 (53.5%) downregulated transcripts with m6A modification that were enriched for meiosis and DNA repair (Figs. 5B, 4F). Consistently, our results showed that the process of oocyte meiotic maturation was defective upon Mettl3 deletion, suggesting that meiosis-associated maternal transcripts with m6A modification are preferentially stabilized in the GV stage (Fig. 4E, F). As reported previously, IGF2BPs have been validated to bind to the maternal mRNA and promote its stability during early embryo development in mouse and zebrafish, respectively [36, 37]. Thus, we hypothesized that IGF2BPs might be the main cofactor to regulate the fate of maternal mRNAs with m6A in follicle and oocyte development. Furtherly, we found 617 (56.2%) downregulated transcripts with m6A modification that were targeted by IGF2BP2 or IGF2BP3 (Fig. 5E), including meiosis-associated genes, such as Itsn2, Cdc42bpa, Spire1, Myt1, and Pias1 (Fig. 5F). Itsn2 has been reported to be an adapter protein that regulates oocyte meiotic resumption through the Cdc42 pathway [51]. Cdc42bpa is a Cdc42 downstream effector that also regulates meiotic oocytes through the Cdc42 signaling pathway [57]. Spire1 has been reported to drive asymmetric oocyte division by cooperating with Formin-2 [58]. Myt1 is a transcription factor that has been identified as a trigger during oocyte meiosis [59]. Pias1 is a member of the SUMO pathway, which is important for the maintenance of centromeric cohesion [60]. In support of these previous findings and based on its significant positive correlation with Mettl3 in the GV stage (Fig. 6A) and that it could be targeted by IGF2BP3 at early embryo development (Fig. S2C), we focused on Itsn2. Specifically, we knocked down Itsn2 by microinjecting siRNA into GV oocytes. Taken together, our results showed that in WT mouse oocytes, m6A modifications on Itsn2 mRNA might be recognized by IGF2BP3, which further enhanced the stability of the mRNA, consequently promoting oocyte meiotic maturation. However, in Mettl3Gdf9 cKO mouse oocytes, Itsn2 mRNA could not be recognized by IGF2BP3; consequently, degradation of the mRNA was promoted, leading to oocyte meiotic maturation failure (Fig. 7).
In conclusion, our results show that m6A serves as a critical regulator to control the stability of oocyte meiotic maturation-related transcripts and that the precise effect of m6A on transcript stability might depend on different reader proteins. In addition to playing a regulatory role in mRNA stability, m6A may also participate in oocyte maturation by affecting other RNA metabolic processes. Exploration of the detailed mechanisms of this possible role will be of great interest in the future.
Materials And methods
Mice
The conditional mutant alleles for Mettl3 (hereafter referred to as Mettl3flox/flox) were kindly provided by Dr. Ming-Han Tong at Shanghai Institute of Biochemistry and Cell Biology [40]. Gdf9-Cre transgenic mice were a gift from Dr. Qing-Yuan Sun at Guangdong Second Provincial General Hospital [61]. All mice described above were maintained on the C57BL/6 J background. Mice lacking Mettl3 in oocytes (referred to as Mettl3Gdf9 cKO) were generated by crossing Mettl3flox/flox mice with Gdf9-Cre mice. The Mettl3flox/flox female mice were used as the control group (referred to as WT). For the fertility test, six pairs of 6 weeks Mettl3flox/flox and Mettl3Gdf9 cKO female mice were randomly selected and continually mated to Mettl3flox/flox male mice which have been confirmed fertility for 5 months. The number of pups and litter size from each female was recorded. Primers for PCR genotyping were listed in Table S6. All animal experiments were done in accordance with the guidelines from the Animal Care and Use Committee of China Agricultural University.
Oocyte and zygote collection
To obtain fully-grown GV oocytes, 6–8 weeks of females were injected with 5 IU PMSG (Ningbo No.2 hormone factory, Zhejiang, China). After 44–48 h, GV oocytes were collected by puncturing the ovarian follicle and released with microcapillary pipettes in M2 media (M-7197, Sigma-Aldrich, Germany). For MII oocyte collection, mice were injected with PMSG as above and after 46–48 h with 5 IU of human chorionic gonadotropin (hCG) (Ningbo No.2 hormone factory, Zhejiang, China). MII oocytes were collected from the oviducts 14–16 h post hCG injection by digestion in M2 medium with hyaluronidase (MR-051-F, Millipore, USA). The detailed procedure to get meiotically incompetent PD5 and PD12 oocytes was described previously [25, 62].
To obtain zygotes, female mice were mated with male mice with known fertility. Successful mating was confirmed by the presence of vaginal plugs. Zygotes were isolated from the oviduct of plugged females. The zygotes were cultured in KSOM medium (MR-107-D, Millipore, USA) and cultured in a 5% CO2 incubator.
Oocyte microinjection
GV oocytes were collected from 6–8 weeks CD-1 mice (Beijing Vital River Laboratory Animal Technology Co., Ltd) follow the same procedure above. For the siRNAs injection experiment, Itsn2 siRNAs were designed and synthesized by GenePharma (Shanghai, China) and were dissolved in RNase-free water to a final concentration of 20 mM. Approximately 5 pl of the siRNAs were microinjected into the cytoplasm of fully-grown GV oocytes in M2 medium serum. After injection, oocytes were cultured for 20–24 h in 200 μM IBMX (I5879, Sigma-Aldrich, Germany) containing medium to maintain GV arrest, and then the oocytes were either collected for assessing the knockdown efficiency by qRT-PCR and western blot analysis or alternatively washed from the IBMX and cultured in maturation medium for 14 h for scoring the progress of meiosis. Sequences of siRNAs were listed in Table S5.
Quantification of ovarian follicles and histological analysis
The ovaries were fixed in 4% paraformaldehyde overnight at 4 °C, dehydrated ethanol series, and embedded in paraffin. To count the numbers of follicles, paraffin-embedded ovaries were serially sectioned at the 5-μm thickness and stained with hematoxylin for histological analyses. Primordial, primary, secondary, and antral follicles were counted in every fifth section of an ovary. In each section, follicles that contained oocytes with clearly visible nuclei were scored and the cumulative number of follicles was multiplied by a correction factor of 5 to represent the estimated number of total follicles in an ovary. A double-blind experiment was performed for the quantification of ovarian follicles.
Immunohistochemistry and immunofluorescence
For immunohistochemistry (IHC) and immunofluorescence on ovary sections, ovaries were fixed, embedded, and sectioned follow the same procedure above. Sections then were boiled in 10 mM sodium citrate buffer (pH 6.0) for 18 min for antigen retrieval, cooled down in ice for 20 min, and washed in PBS with 0.1% Triton X-100. The following steps for IHC were under the instruction from the SP Rabbit or Mouse HRP Kit (ZSGB-BIO, China). The primary antibodies for IHC include Anti-METTL3 antibody (ab195352, 1:500, Abcam, UK), Anti-DDX4/MVH antibody (Ab27591, 1:500, Abcam, UK). The following steps for immunofluorescence on ovary sections was performed as previously described [40]. The primary antibodies for immunofluorescence include the Anti-γH2AX antibody (ab11174, 1:500, Abcam, UK). An apoptotic signal was detected using DeadEnd™ Fluorometric TUNEL System (G3250, Promega, USA) according to the manufacturer’s instructions.
For immunofluorescence on oocytes, oocytes were fixed in 4% paraformaldehyde (PFA) for 30 min at room temperature, and then treated with 0.25% Triton X-100 in PBS for 15 min. After blocking with 1% BSA in PBS for 60 min, oocytes were incubated with primary antibodies: FITC-α-Tubulin antibody (F2168, 1:100, Sigma-Aldrich, USA), anti-intersectin2 antibody (NBP1-71833, 1:1000, NOVUS, USA), anti-METTL3 antibody (ab195352, 1:500, Abcam, UK) at 4 °C overnight, followed by incubation with secondary antibodies for 1 h at room temperature. After washing three times with 0.1% BSA in PBS, oocytes were counterstained with Mounting Medium with DAPI (ab104139, Abcam, UK), and then analyzed by confocal microscopy A1 (Nikon, Japan).
RNA isolation and qRT-PCR
Total RNA was extracted from oocytes samples using Trizol reagent (15596018, Invitrogen, USA), and cDNA was generated using the 5X All-In-One RT Master Mix (G490, Abm, USA). Quantitative real-time PCR using 2× RealStar Green Power Mixture (A311, GenStar, China) was performed using a real-time PCR system (Roche LightCycler 96®, Germany). Primer sequences are listed in Table S7.
RNA stability analysis
Primary (25–35 μm) and secondary (35–65 μm) oocytes from 3-week-old WT and Mettl3Gdf9 cKO ovaries by enzymatic digestion follow the same procedure above [25, 62]. RNA stability analysis was performed as previously described with some modifications [63, 64]. The oocytes were collected at 0, 12, 24 h after in vitro culture with Actinomycin D (10 μg/ml, SBR00013, Sigma, Germany) treatment. Total RNA isolation and qRT-PCR were performed as above and β-actin was used as a loading control for normalization. Plot the relative expression of RNA at each time point relative to t = 0 and then calculate the mRNA decay rate by nonlinear regression curve fitting (one phase decay) using GraphPad Prism (version 8). Primer sequences are listed in Table S7.
Hormone assays
Twelve to 20 weeks old Mettl3Gdf9 cKO and WT female mice were sacrificed in order to measure FSH and LH levels. Serum samples were collected as described previously [48, 61] and stored at −80 °C until measurement. The assays were using ELISA Kit for Follicle-Stimulating Hormone (CEA830Mu, CLOUD-CLONE, China) and ELISA Kit for Luteinizing Hormone (CEA441Mu, CLOUD-CLONE, China) according to the manufacturer’s instructions.
RNA-seq
Ten mouse GV oocytes were collected in lysis buffer and subjected to first-strand cDNA synthesis using SMARTer® PCR Synthesis Kit (634925, Clontech, Japan). The cDNA products were analyzed by Agilent 2100 bioanalyzer and were fragmented with sonication and subjected to library construction using TruSeq® Nano DNA Library Prep kit (FC-121–403, Illumina, USA) according to the manufacturer’s instructions. Sequencing was performed on an Illumina HiSeq X-ten sequencing system.
Western blot
About 100 fully-grown GV oocytes were lysed in 2× Laemmli Sample Buffer (1610737, Bio-Rad, USA) with protease inhibitor. Oocyte lysates were heated at 99.9 °C for 5 min and the denatured protein samples were separated by SDS-PAGE gel and transferred to PVDF membranes (IPVH00010, Millipore, Germany). Membranes were blocked with 5% nonfat milk prepared in Tris-buffer saline-plus 0.1% Tween-20 (TBST) at room temperature for 1 h and then incubated with primary antibodies overnight at 4 °C, followed by the treatment with Goat anti-Rabbit-HRP (BE0106, 1:5000, Easybio, China) for 1 h on the next day. After washing with TBST three times, the blotted membranes were exposed with SuperSignal West Dura Extended Duration Substrate (34075, Thermo, USA). The following primary antibodies were used for western blot: anti-METTL3 antibody (ab195352, 1:500, Abcam, UK), ITSN2 antibody (NBP1-71833, 1:1000, NOVUS, USA), and GAPDH antibody (2118 L, 1:5000, CST, USA).
m6A-MeRIP-seq
m6A-MeRIP using low-input materials was performed based on a previously described protocol [55] with some modifications. Briefly, total RNA from about 2500 mouse GV oocytes from 5 weeks C57BL/6 J mice (Beijing Vital River Laboratory Animal Technology Co., Ltd) was first randomly fragmented to ~200 nt with RNA fragmentation reagents (AM8740, Thermo, USA), and then incubated with the protein A beads (10001D, Thermo, USA) coupled with anti-m6A polyclonal antibody (ABE572, Millipore, USA) in IPP buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 0.1% NP-40, 0.4 U/μl RNasin). After immunoprecipitation, the RNA reaction mixture was washed twice in 1 ml of IP buffer, twice in 1 ml of low-salt IP buffer (50 mM NaCl, 10 mM Tris-HCl, pH 7.4, 0.1% IGEPAL® CA-630 (I8896, Sigma-Aldrich, Germany) in nuclease-free H2O), and twice in 1 ml of high-salt IP buffer (500 mM NaCl, 10 mM Tris-HCl, pH 7.4, 0.1% IGEPAL CA-630 in nuclease-free H2O) for 5 min each at 4 °C. After extensive washing, the m6A-enriched RNA fragments were eluted from the beads by proteinase K digestion followed by phenol-chloroform extraction and ethanol precipitation. The purified RNA was subjected to library construction using the SMARTer Stranded Total RNA-Seq Kit v2 (634413, Clontech, Japan) according to the manufacturer’s instructions. Sequencing was performed on an Illumina HiSeq X-ten sequencing system.
Sequencing data analysis
General preprocessing of reads: the MeRIP-seq of control, the RNA-seq of WT, and Mettl3Gdf9 cKO treatment for GV oocyte were performed using Illumina HiSeq platform with paired-end 150 bp read length. Adapter sequences were trimmed off for all raw reads using the Cutadapt (version 1.18). Especially, for MeRIP-seq that generated using SMARTer Stranded Total RNA-Seq Kit version 2, the first three nucleotides of the second sequencing read which derived from the SMART adapter was trimmed using Cutadapt with parameter “-U 3”. Reads with length less than 35 nt or contained an ambiguous nucleotide were discarded by Trimmomatic (version 0.36). The remaining reads were aligned to the mm10 by HISAT2 (version 2.0.5). To minimize the rate of false positives, only uniquely mapped reads with −q ≥ 20 were kept for the subsequent analysis for each sample.
For MeRIP-seq, the whole-genome m6A-enriched peaks were identified using MACS (version 2.1.4) with the corresponding input sample as a control. MACS was used with parameters “--nomodel, --keep-dup all and -g mm”. Peaks with FDR value <0.05 was annotated based on Ensembl (release 68) gene annotation information by applying BEDTools’ intersectBed (version 2.16.2). Only peaks located in the GV oocyte-expressed gene and annotated as TSS (transcription start sequence), 5′UTR, CDS, stop codon, and 3′UTR were used for the subsequent analysis.
For RNA-seq, the number of reads mapped to each gene was counted using the HTSeq (version 0.6.0), with parameter ‘--mode = union and --stranded = no’. RPKM (reads per kilobase of exon model per million mapped reads) was calculated for each gene using an in-house Rscript. Only transcripts with the average RPKM >3 for WT samples were regarded as expressing in GV oocyte. The R package DESeq2 was used for differential expression analysis (fold change cutoff = 1.2, p value cutoff = 0.05, and the average RPKM >3 for WT). Cumulative distribution analysis of RNA-seq log2 fold changes for expressed transcripts between control and Mettl3Gdf9 cKO treatment was performed in R using the ecdf function. Groups were defined as m6A (MeRIP-seq FDR <0.05) or non-m6A (the remainder of the transcripts). The significance of the difference between the cumulative distribution curves was calculated by the Kolmogorov–Smirnov test.
Motif statistics analysis within m6A peaks
Motif analysis for m6A peaks were identified by HOMER (version 4.7). The sequence of peaks located in TSS, 5′UTR, CDS, stop codon, and 3′UTR for mRNA were extracted as the target sequences and background sequences were obtained by randomly shuffling peaks upon total mRNAs on the genome using BEDTools’ shuffleBed.
m6A reader analysis
IGF2BP2/3 binding peaks identified by eCLIP for hESC were downloaded from GSE78509. Human coordinates were converted to mm10 using UCSC tools’ liftOver. Distance between IGF2BP2/3 peaks by eCLIP and m6A peaks identified in this study was calculated using BEDTools. Background were obtained by randomly shuffling IGF2BP2/3 peaks using BEDTools’ shuffleBed. Expressed transcripts with m6A peaks, targeted by IGF2BP2/3, were obtained using the union genes identified by eCLIP (GSE78509) [50] and RIP-seq (GSE90639) [16].
Gene set enrichment analysis
Functional enrichment analysis of KEGG pathways documented in Gene Ontology were performed using DAVID (https://david.ncifcrf.gov). KEGG terms with p value <0.05 were determined to be statistically significant.
Quantification and statistical analysis
All the experimental data was replicated at least in two independent experiments. Data in figures were expressed as mean ± SEM unless otherwise stated. Replicates in different experiments were stated in corresponding Figure legends. Statistically significant differences between different groups were evaluated by Student’s t-test, two-sided Kolmogorov–Smirnov test, Chisq-test. p < 0.05 were considered as statistically significant. The significance of Pearson correlation (Figs. 3F and 4A) was evaluated by the R program. Statistical significance in other experiments was performed by Graphpad Prism (version 8). For microscope images, n generally refers to the total number of oocytes and embryos.
Data availability
The RNA-seq and MeRIP-seq data have been deposited into National Genomics Data Center (https://bigd.big.ac.cn) with accession number: PRJCA003168.
Code availability
The computer code used during the current study is available from the corresponding authors on reasonable request.
References
Oktem O, Urman B. Understanding follicle growth in vivo. Hum Reprod. 2010;25:2944–54.
Li J, Kawamura K, Cheng Y, Liu S, Klein C, Liu S, et al. Activation of dormant ovarian follicles to generate mature eggs. Proc Natl Acad Sci USA. 2010;107:10280–4.
Pepling ME. Development. Nurs Oocyte Sci. 2016;352:35–36.
Li R, Albertini DF. The road to maturation: somatic cell interaction and self-organization of the mammalian oocyte. Nat Rev Mol Cell Biol. 2013;14:141–52.
Hou G, Sun QY. Maternal ageing causes changes in DNA methylation and gene expression profiles in mouse oocytes. Zygote 2020;28:360–6.
Stewart KR, Veselovska L, Kelsey G. Establishment and functions of DNA methylation in the germline. Epigenomics 2016;8:1399–413.
Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 2017;547:419–24.
Nashun B, Hill PW, Smallwood SA, Dharmalingam G, Amouroux R, Clark SJ, et al. Continuous histone replacement by Hira is essential for normal transcriptional regulation and de novo DNA methylation during mouse oogenesis. Mol Cell. 2015;60:611–25.
Miyara F, Migne C, Dumont-Hassan M, Le Meur A, Cohen-Bacrie P, Aubriot FX, et al. Chromatin configuration and transcriptional control in human and mouse oocytes. Mol Reprod Dev. 2003;64:458–70.
Zhang C, Chen Z, Yin Q, Fu X, Li Y, Stopka T, et al. The chromatin remodeler Snf2h is essential for oocyte meiotic cell cycle progression. Genes Dev. 2020;34:166–78.
De La Fuente R, Baumann C, Viveiros MM. ATRX contributes to epigenetic asymmetry and silencing of major satellite transcripts in the maternal genome of the mouse embryo. Development 2015;142:1806–17.
Morgan M, Much C, DiGiacomo M, Azzi C, Ivanova I, Vitsios DM, et al. mRNA 3’ uridylation and poly(A) tail length sculpt the mammalian maternal transcriptome. Nature 2017;548:347–51.
Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, et al. N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science. 2020;367:580–6.
Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, et al. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–4.
Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, et al. m6A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540:242–7.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626.
Shi HL, Wang X, Lu ZK, Zhao BXS, Ma HH, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 2013;155:793–806.
Roundtree IA, Luo GZ, Zhang ZJ, Wang X, Zhou T, Cui YQ, et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife. 2017;6:e31311.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Zhou J, Wan J, Gao XW, Zhang XQ, Jaffrey SR, Qian SB. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–4.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–47.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Ivanova I, Much C, Di Giacomo M, Azzi C, Morgan M, Moreira PN. et al. The RNA m6A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell. 2017;67:1059–67.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444–7.
Shi H, Zhang X, Weng YL, Lu Z, Liu Y, Lu Z, et al. m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature. 2018;563:249–53.
Zhu S, Wang JZ, Chen, He YT, Meng N, Chen M, et al. An oncopeptide regulates m6A recognition by the m6A reader IGF2BP1 and tumorigenesis. Nat Commun. 2020;11:1685.
Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. 2015;162:1299–308.
Hu Y, Ouyang Z, Sui X, Qi M, Li M, He Y, et al. Oocyte competence is maintained by m6A methyltransferase KIAA1429-mediated RNA metabolism during mouse follicular development. Cell Death Differ. 2020;27:2468–83.
Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 2018;14:e1007412.
Ren F, Lin Q, Gong G, Du X, Dan H, Qin W, et al. Igf2bp3 maintains maternal RNA stability and ensures early embryo development in zebrafish. Commun Biol. 2020;3:94.
Liu HB, Muhammad T, Guo Y, Li MJ, Sha QQ, Zhang CX, et al. RNA-binding protein IGF2BP2/IMP2 is a critical maternal activator in early zygotic genome activation. Adv Sci. 2019;6:1900295.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–9.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24.
Lin Z, Hsu PJ, Xing XD, Fang JH, Lu ZK, Zou Q, et al. Mettl3-/Mettl14-mediated mRNA N6-methyladenosine modulates murine spermatogenesis. Cell Res. 2017;27:1216–30.
Tang C, Xie Y, Yu T, Liu N, Wang Z, Woolsey RJ, et al. m6A-dependent biogenesis of circular RNAs in male germ cells. Cell Res. 2020;30:211–28.
Xu K, Yang Y, Feng GH, Sun BF, Chen JQ, Li YF, et al. Mettl3-mediated m6A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017;27:1100–14.
Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002–6.
Zhang Z, Luo K, Zou Z, Qiu M, Tian J, Sieh L, et al. Genetic analyses support the contribution of mRNA N6-methyladenosine (m6A) modification to human disease heritability. Nat Genet. 2020;52:939–49.
Sui X, Hu Y, Ren C, Cao Q, Zhou S, Cao Y, et al. METTL3-mediated m6A is required for murine oocyte maturation and maternal-to-zygotic transition. Cell Cycle. 2020;19:391–404.
Sun QY, Liu K, Kikuchi K. Oocyte-specific knockout: a novel in vivo approach for studying gene functions during folliculogenesis, oocyte maturation, fertilization, and embryogenesis. Biol Reprod. 2008;79:1014–20.
Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–86.
Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319:611–3.
Nagaoka SI, Hassold TJ, Hunt PA. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet. 2012;13:493–504.
Conway AE, Van Nostrand EL, Pratt GA, Aigner S, Wilbert ML, Sundararaman B, et al. Enhanced CLIP uncovers IMP protein-RNA targets in human pluripotent stem cells important for cell adhesion and survival. Cell Rep. 2016;15:666–79.
Zhang J, Ma R, Li L, Wang L, Hou X, Han L, et al. Intersectin 2 controls actin cap formation and meiotic division in mouse oocytes through the Cdc42 pathway. FASEB J. 2017;31:4277–85.
Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, et al. m6A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16:289–301.
Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–19.
Zhang C, Chen L, Peng D, Jiang A, He Y, Zeng Y, et al. METTL3 and N6-methyladenosine promote homologous recombination-mediated repair of DSBs by modulating DNA-RNA hybrid accumulation. Mol Cell. 2020;79:425–42.
Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A. et al. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017;542:475–8.
Zeng Y, Wang S, Gao S, Soares F, Ahmed M, Guo H, et al. Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol. 2018;16:e2006092.
Ma C, Benink HA, Cheng D, Montplaisir V, Wang L, Xi Y, et al. Cdc42 activation couples spindle positioning to first polar body formation in oocyte maturation. Curr Biol. 2006;16:214–20.
Pfender S, Kuznetsov V, Pleiser S, Kerkhoff E, Schuh M. Spire-type actin nucleators cooperate with Formin-2 to drive asymmetric oocyte division. Curr Biol. 2011;21:955–60.
Hiraoka D, Hosoda E, Chiba K, Kishimoto T. SGK phosphorylates Cdc25 and Myt1 to trigger cyclin B-Cdk1 activation at the meiotic G2/M transition. J Cell Biol. 2019;218:3597–611.
Ding Y, Kaido M, Llano E, Pendas AM, Kitajima TS. The post-anaphase SUMO pathway ensures the maintenance of centromeric cohesion through meiosis I-II transition in mammalian oocytes. Curr Biol. 2018;28:1661–9.
Liang QX, Wang ZB, Lin F, Zhang CH, Sun HM, Zhou L, et al. Ablation of beta subunit of protein kinase CK2 in mouse oocytes causes follicle atresia and premature ovarian failure. Cell Death Dis. 2018;9:508.
Guo J, Zhang T, Guo Y, Sun T, Li H, Zhang X, et al. Oocyte stage-specific effects of MTOR determine granulosa cell fate and oocyte quality in mice. Proc Natl Acad Sci USA. 2018;115:E5326–E5333.
Wang CX, Cui GS, Liu X, Xu K, Wang M, Zhang XX, et al. METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol. 2018;16:e2004880.
Ratnadiwakara M, Anko ML. mRNA stability assay using transcription inhibition by actinomycin D in mouse pluripotent stem cells. Bio Protoc. 2018;8:e3072.
Acknowledgements
We thank Prof. Youqiang Su from State Key Laboratory of Reproductive Medicine of Nanjing Medical University for providing protocols for the collection and culture of mouse oocytes. We thank Dr. Tuo Zhang and Dr. Yanli Dai for their help with the quantification of ovarian follicles and histological analysis. We thank Dr. Paula Stein from the National Institute of Environmental Health Sciences, National Institutes of Health and Dr. Bingying Liu for help with oocyte microinjection. We thank Prof. Sen Wu and Prof. Xuguang Du for lending us FemtoJet 4i Microinjector.
Funding
This work was supported by the National Natural Science Foundation of China (31970825, 31772601, 31601941, 31625016, and 31571497), China National Basic Research Program (2016YFA0100202), the National Key R&D Program of China (2019YFA0110901), Shanghai Municipal Science and Technology Major Project (2017SHZDZX01), the Youth Innovation Promotion Association of Chinese Academy of Sciences (CAS2018133), Plan 111 (B12008), Beijing Nova Program (Z201100006820104), and Research Programs from the State Key Laboratories of Agrobiotechnology and College of Biological Sciences (2020SKLAB1–3, 31051378, and 10052521-01).
Author information
Authors and Affiliations
Contributions
H.M. and D.Z. designed, executed, and analyzed most of the experiments with assistance from J.L., L.Y., B.S., Z.L. and Z.-B.W.; T.Z. performed bioinformatic analysis of RNA-seq and MeRIP-seq datasets with assistance from D.G. and L.Z.; Y.Y constructed librarys for MeRIP-seq using mouse oocytes with the assistance from Y.H.; J.G. performed western blot assay; X.C. drew the working model; Q.-Y.S. and M.-H.T. provided transgenetic mouse model and advice on this project; J.-Y.H. and Y.-G.Y. conceived this project, supervised the study and interpreted the data. J.H., Y.-G.Y., H.M., T.Z. and Y.Y. wrote the manuscript. All coauthors provided feedback on the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr. Kim McCall
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mu, H., Zhang, T., Yang, Y. et al. METTL3-mediated mRNA N6-methyladenosine is required for oocyte and follicle development in mice. Cell Death Dis 12, 989 (2021). https://doi.org/10.1038/s41419-021-04272-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04272-9
This article is cited by
-
RNA N6-methyladenosine modification in female reproductive biology and pathophysiology
Cell Communication and Signaling (2023)
-
scm6A-seq reveals single-cell landscapes of the dynamic m6A during oocyte maturation and early embryonic development
Nature Communications (2023)
-
The RNA m6A landscape of mouse oocytes and preimplantation embryos
Nature Structural & Molecular Biology (2023)
-
Single-cell multi-omics profiling reveals key regulatory mechanisms that poise germinal vesicle oocytes for maturation in pigs
Cellular and Molecular Life Sciences (2023)
-
Transcriptome-wide N6-methyladenine methylation in granulosa cells of women with decreased ovarian reserve
BMC Genomics (2022)
