
Abstract
Spinal and bulbar muscular atrophy (SBMA) is an adult-onset hereditary neurodegenerative disease caused by the expansions of CAG repeats in the androgen receptor (AR) gene. Androgen-dependent nuclear accumulation of pathogenic AR protein causes degeneration of lower motor neurons, leading to progressive muscle weakness and atrophy. While the successful induction of SBMA-like pathology has been achieved in mouse models, mechanisms underlying motor neuron vulnerability remain unclear. In the present study, we performed a transcriptome-based screening for genes expressed exclusively in motor neurons and dysregulated in the spinal cord of SBMA mice. We found upregulation of Mid1 encoding a microtubule-associated RNA binding protein which facilitates the translation of CAG-expanded mRNAs. Based on the finding that lower motor neurons begin expressing Mid1 during embryonic stages, we developed an organotypic slice culture system of the spinal cord obtained from SBMA mouse fetuses to study the pathogenic role of Mid1 in SBMA motor neurons. Impairment of axonal regeneration arose in the spinal cord culture in SBMA mice in an androgen-dependent manner, but not in mice with non-CAG-expanded AR, and was either exacerbated or ameliorated by Mid1 overexpression or knockdown, respectively. Hence, an early Mid1 expression confers vulnerability to motor neurons, at least by inducing axonogenesis defects, in SBMA.
Similar content being viewed by others


Introduction
Spinal and bulbar muscular atrophy (SBMA) or Kennedy’s disease is an adult-onset X-linked hereditary neurodegenerative disease. SBMA is accompanied by the degeneration of lower motor neurons in the spinal cord and specific brainstem motor nuclei and atrophy of skeletal muscle resulting in progressive motor impairment [1]. Patients with SBMA carry trinucleotide CAG-repeat expansion which encodes the polyglutamine (polyQ) tract in the first exon of the androgen receptor (AR) gene [2].
Androgen-dependent nuclear accumulation of mutant AR protein causes neurological symptoms [3], typically affecting males. The nuclear accumulation of pathogenic AR has been shown to induce transcriptional dysregulation and subsequent defects in numerous cellular functions resulting in cell death of motor neurons [4,5,6,7,8,9,10]. Motor neurons in the mouse model of SBMA have been shown to exhibit axonal dysfunction and atrophy without noticeable cell death [3, 7], suggesting that axonal dysfunctions can induce symptoms. The early, presymptomatic dysregulation of axonal transport by pathogenic AR has been proposed as a causative factor for axonal defects [9, 11, 12]. Hence, further exploration of the mechanisms of early axonal vulnerability is essential for elucidating SBMA pathogenesis.
Androgen-dependent regulation of axonogenesis in motor neurons has been extensively studied so far [13]. Androgen has been shown to promote axonogenesis by upregulating brain-derived neurotrophic factor (BDNF) in motor neurons [14] and inducing the expression of cpg15/neuritin [15]. Inversely, pathogenic AR impairs axonal transport and the BDNF-dependent growth of neurites in cell culture [11, 12].
Several mechanisms have been proposed for how pathogenic AR disrupts axonal transport. The aggregates of pathogenic nuclear AR cause toxicity, which downregulates the gene expression of dynactin1 and impairs retrograde axonal transport driven by dynein [9]. Non-aggregated and oligomeric pathogenic AR induces phosphorylation-mediated inhibition of kinesin via c-Jun N-terminal kinase activation, leading to impairment of anterograde axonal transport [12]. Hence, the previous studies suggest that transcriptional dysregulation and post-transcriptional regulation by pathogenic AR might be involved in the disruption of axonal transport and axonogenesis in SBMA. Translation of dysregulated cytoplasmic mRNA has been increasingly considered to be crucial for the pathogenesis of neurodegenerative diseases, including polyQ diseases [16]. Pathogenic proteins with a polyQ expansion have been shown to induce endoplasmic reticulum stress, which inhibits eukaryotic translation initiation factor 2 (eIF2), leading to cell death of vulnerable neurons in cell culture [17, 18]. Midline 1 (human MID1 and its mouse orthologue Mid1), an ubiquitin E3 ligase, was shown to form a microtubule-associated ribonucleotide complex with purine-rich sequence motif in mRNAs [19], which promotes translation of mRNAs harboring a CAG-repeat via mechanistic target of rapamycin (mTOR) signaling [20, 21]. These CAG-repeat mRNAs include AR with a non-expanded CAG repeat [22]. However, the cellular processes through which pathogenic AR causes translational dysregulation in SBMA remains elusive.
Here, we performed a transcriptome-based screening of genes and identified Mid1, a development-related gene, specifically expressed in the spinal motor neurons and upregulated in the spinal cord of SBMA mice. Using spinal cord slice cultures from the embryos of SBMA mice, we found that overexpression of Mid1 increased the pathogenic AR protein levels, thereby inducing the androgen-dependent impairment of axonogenesis. Our findings indicate a mechanism for the Mid1-associated axonal vulnerability in SBMA motor neurons.
Materials and methods
DNA constructs
The plasmids pCR3.1-AR-24Q and pCR3.1-AR-97Q, which drive expression of full-length human AR containing 24 and 97-expanded CAGs, respectively, under the control of a cytomegalovirus enhancer and a chicken β-actin promoter as previously described [23] were used in the current study. Mouse Mid1 cDNA was amplified from the cDNA obtained from mouse spinal cord on embryonic day 14.5 (E14.5) and was cloned into the NotI/EcoRI site of pcDNA3.1-mycHisC to create pcDNA3.1-Mid1-mycHisC. Infusion system (Invitrogen) was used for cloning Mid1 cDNA together with the sequence encoding the self-cleaving peptide 2 A into pLenti-GFP(C), resulting in pLenti-GFP-2A-Mid1. The plasmids for shRNA expression were obtained by inserting the target sequences into the AgeI/EcoRI site of pLKO.1 puro (Addgene). The target sequences of shRNA for mouse Mid1 and human AR were GACTTGCGTTACTTGTGAA [24] and GAAAGCACTGCTACTCTTCAGCATTATTCCA [25], respectively.
Cell culture and transfection
Mouse NSC34 motor neuron-like cells (kindly provided by N.R. Cashman, University of British Columbia, Vancouver, Canada) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). In addition, cells were maintained in a 37 °C incubator with a humidified atmosphere of 95% air/5% CO2. The plasmids were transfected using OPTI-MEM (Gibco) and Lipofectamine 2000 (Invitrogen). Ten hours after lipofection, the culture medium was changed to DMEM containing 2% FBS and 50 nM dihydrotestosterone (DHT). In the case of cycloheximide (CHX) treatment experiments, the culture medium was changed to DMEM containing 2% FBS and 80 μg/mL CHX, 24 h after lipofection.
Immunoblotting
Protein isolation was performed using Cellytics MT Cell Lysis Reagent (Sigma-Aldrich). We separated equal amounts of protein on 5–20% SDS-PAGE gels (FUJIFILM Corp.) and transferred them to Hybond-P membranes (GE Healthcare). The following primary antibodies were used with indicated dilutions: rabbit anti-AR pAb (H-280, Santa Cruz Biotechnology., 1:2000), mouse anti-Gapdh mAb (Millipore, 1:5000), rabbit anti-MID1 pAb (Abcam, 1:1000), rabbit anti-PP2Ac pAb (Cell Signaling Technology, 1:1000), rabbit anti-GFP pAb (MBL, 1:1000). The density of each band was quantified using ImageJ software (NIH, Bethesda, MD, USA). For RNA pull-down assay, cell lysate containing ~300 μg of total protein derived from HEK293T cells overexpressed with Mid1 was incubated for 1 h with 3 μg of in vitro-transcribed RNA biotinylated with Biotin-16-UTP (Sigma-Aldrich). It was followed by incubation with Dynabeads M-280 streptavidin (Invitrogen) for 1 h at 4 °C and washed with wash buffer three times.
Transcriptome data
Previously published microarray data on Gene Expression Omnibus, GSE39865 [4], was normalized using Robust Multichip Analysis of Bioconductor based on R (https://www.r-project.org). The resultant data was subjected to the differential gene expression analysis using an integrated web application for Differential Expression and Pathway analysis (iDEP) [26]. Cell type specificity of candidate genes was examined online using single-cell transcriptional atlas [27] (http://spinalcordatlas.org).
RT-PCR
Total RNA was extracted from NSC34 cells or spinal cord slice culture using TRIzol (Invitrogen) and RNeasy Mini Kit (QIAGEN). The extracted RNA was then reverse-transcribed into first-strand cDNA using ImProm-II Reverse Transcription system (Promega). Quantitative PCR was performed using THUNDERBIRD SYBR qPCR Mix (TOYOBO), and the amplified products were detected with the iCycler system (Bio-Rad Laboratories). The following primers were used for PCR: Mid1 forward/reverse, 5'-AGCCCTTTACAGGCCATCG-3'/5'-AGATTAACTGGTCATCGGTCACA-3'; AR forward/reverse, 5'-CGGAAGCTGAAGAAACTTGG-3'/5'-ATGGCTTCCAGGACATTCAG-3'; β2-microglobulin forward/reverse, 5'-CTGACCGGCCTGTATGCTAT-3'/5'-CCGTTCTTCAGCATTTGGAT-3'. The gene expression of beta2-microglobulin was used for normalization.
RNA immunoprecipitation
RNA immunoprecipitation (RIP) was performed using the total cell lysates from HEK293T cells co-expressing His-tagged Mid1 and AR-24Q or AR-97Q and anti-His-tag mAb-Magnetic beads (MBL) (or nonspecific Mouse IgG2a (isotype control)-Magnetic beads (MBL) antibody) for 1 h at 4 °C and washed with Cellytics MT for three times, followed by RNA purification using TRIzol (Invitrogen) and RNeasy Mini Kit (QIAGEN) and examined by RT-PCR using rTaq DNA polymerase (TaKaRa) and the AR forward/reverse primers mentioned above.
Immunofluorescence
Cultured spinal cord slices were fixed with 4% PFA, immersed in 20% sucrose, embedded in OCT compound, and cryo-sectioned (16 μm thickness). Sections were incubated overnight at 4 °C with the following primary antibodies with the indicated dilution: rabbit anti-MID1 pAb (ab70770, Abcam, 1:400); mouse anti-Islet1&2 mAb (39.4D5, DSHB, 1:100); rabbit anti-AR (D6F11, Cell Signaling Technology, 1:400) mAb; mouse anti-non-phosphorylated NF-H mAb (SMI32, BioLegend, 1:2000); rabbit anti-Pnpt1 (Invitrogen, 1:200). After PBS washes, the sections were incubated for 1 h at room temperature with the following secondary antibodies: Donkey anti-Rabbit IgG-Alexa488 and Donkey anti-Mouse IgG-Alexa546 (Invitrogen, 1:1000). After incubation, the sections were counterstained with DAPI and mounted with a mounting solution. Images were acquired using CoolSNAP CCD camera (Photometrics) assembled on BX60 upright microscope (Olympus).
Virus production
Lentiviral particles were produced in HEK293T cells by transfection of pLenti-GFP(C), pLenti-GFP-2A-Mid1, pLKO.1-contol shRNA (SHC016, Sigma), pLKO.1-Mid1 shRNA, or pLKO.1-human AR shRNA together with pLP1, pLP2, and pVSV-G (Invitrogen) using Lipofectamine 2000 (Invitrogen). The lentiviral-containing supernatants were collected 48 h after transfection and were concentrated by ultracentrifugation using Lenti-X Concentrator (Takara Bio USA). The viral titers were measured using Lenti-X qRT-PCR Titration Kit (Applied Biological Materials). The final concentration of lentiviral particles in the culture medium was ~5.0 × 107 IU/mL.
Maintenance of transgenic animals
AR-97Q transgenic mice expressing full-length AR containing 97-expanded CAGs under the control of a cytomegalovirus enhancer and a chicken β-actin promoter were generated in our previous study [3]. The mice were bred, maintained on a C57BL/6 J mouse background in the animal facility and were genotyped by PCR amplification using DNA extracted from the tail. The following primers were used for PCR: AR forward/reverse, 5'-CTTCTGGCGTGTGACCGGCG-3'/ 5'-TGAGCTTGGCTGAATCTTCC-3'; Sry forward/reverse, 5'-TGGGACTGGTGACAATTGTC-3'/5'-GAGTACAGGTGTGCAGCTCT-3'. Mice were fed with a standard diet and maintained on a 12 h light/dark cycle. Pregnant mice were obtained by crossing female AR-97Q transgenic mice with wild-type C57BL/6 J male mice.
Spinal cord slice culture
Cross-sectional slices of the spinal cord (250–300 μm thickness) were prepared as previously described for embryonic cerebral wall [28, 29] and cerebellar primordium [30]. As a prerequisite for the experiment, silicon rubber was solidified (5–10 mm thick and transparent) in a Petri dish by mixing KE-103 and CAT-103 (Shin-Etsu Chemical). Embryos were dissected in a Petri dish containing DMEM/F12, and vertebral bones and meninges were removed by tweezers. The naked spinal cords were cooled on ice for 2–3 h during the genotyping, transferred to the previously solidified silicon rubber, and sliced manually using micro-knives purchased from Alcon (V-Lance Ophthalamic corneal/scleral knife, 20 Gage). Using a 200 μL Pipetman (Gibson) with a large orifice (1–2 mm) tip, slices were transferred with ~200 μL enriched medium [DMEM/F12 supplemented with 5% horse serum, 5% FBS, N2 (1:100, Thermo Fisher Scientific)] to 35 mm dish (IWAKI). Slices were mixed with ~200 μL of 2 × type I collagen solution (1.5–1.6 mg/ml in distilled water, 5 × DMEM/F12 and neutralizing buffer; Cell matrix IA; Nitta Gelatin), prepared according to the manufacturer’s instructions, and was kept on ice until use. The gel (final concentration 0.7–0.8 mg/ml) was immediately spread on the dish base using a pipette tip. The transverse plane of the spinal cord slices was arranged horizontally to the dish base using a tungsten needle. The dishes were placed in an incubator (37 °C; 5% CO2, 40% O2) to allow for further gel solidification (5–10 min). An enriched medium (1 ml) was added to the gel and spread over the entire dish surface. Lentivirus (5.0 × 107 IU/mL in a final concentration) or DHT (50 nM in a final concentration) was directly added to the dish at the start of culture (day 0). Images of cultured spinal cord slices were acquired using ORCA-flash 4.0 v3 camera (Hamamatsu Photonics) assembled on an IX71 inverted microscope (Olympus).
Statistical analysis
We analyzed the data with unpaired two-sided t-test to compare two groups and analysis of variance (ANOVA) with Tukey’s test for multiple comparisons using Prism 8 Software (GraphPad). We considered p < 0.05 to indicate statistical significance. The data were checked for normality using Shapiro-Wilk test and for equality of variances using F-test. In the microarray data analysis, the false discovery rate (FDR) < 0.05 was considered to be statistically significant.
Results
Mid1 stands as a candidate gene for motor neuron vulnerability in SBMA
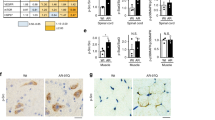
To identify genes involved in the degeneration of motor neurons, we reanalyzed the transcriptomes from the spinal cord of AR-97Q mice at different ages: 7–9, 10–12, and 13–15 weeks [4] and combined them with the single-cell transcriptional atlas [27] to extract genes specifically dysregulated in the spinal motor neurons of AR-97Q mice. Among the genes whose expression was significantly altered by more than 2-fold in AR-97Q mice compared to the control AR-24Q mice, we identified four upregulated genes (two genes during 7–9 weeks and two genes during 10–12 weeks) and three downregulated genes (all genes during 7–9 weeks) expressed in cholinergic neurons which express choline acetyltransferase, a motor neuron marker (Fig. 1a; Supplementary Table 1).

a The spinal cord transcriptome data of SBMA mice [4] contained seven upregulated and 27 downregulated genes. Analyses of these genes in single-cell transcriptional atlas [27] identified four upregulated and three downregulated genes expressed in the cholinergic neurons in the spinal cord of adult mice. b A binary matrix summarizing the expression of the genes in the other cell types in the spinal cord using the single-cell transcriptional atlas [27]. The expression was indicated by the filled matrices. c Relative quantification of Mid1 expression in the spinal cord of AR-97Q mice at 12 weeks of age (*p < 0.05, unpaired two-tailed two-sample t-test). d Immunostaining of Mid1 in the spinal cord of the mouse at 10 weeks of age. Isl1 is a motor neuron marker. Scale bar 50 μm.
Selected vulnerability of lower motor neurons within the central nervous system in SBMA suggests that genes exclusively expressed in motor neurons are involved in the pathogenesis of SBMA. Therefore, we examined the expression of the candidate genes in the other cell types in the spinal cord using the single-cell transcriptional atlas [27] to determine their properties for motor neuron specificity. Among the differentially expressed genes we identified, Mid1, which encodes a microtubule-associated RNA binding protein [19], had the largest fold change (Supplementary Table 1). In addition, the expression of Mid1 was restricted to the spinal motor neurons according to the single-cell transcriptional atlas (Fig. 1b). We used quantitative RT-PCR to confirm the upregulation of Mid1 in the spinal cord at the early symptomatic stages of AR-97Q mice (Fig. 1c). Furthermore, immunostaining confirmed that Mid1 expression is specific to the spinal motor neurons in AR-97Q mice (Fig. 1d), suggesting that the upregulation of Mid1 associated with pathogenic AR selectively occurs in the motor neurons. Moreover, both spinal cord samples from SBMA patients and previously published RNA-seq data using induced pluripotent stem cell (iPSC)-derived motor neurons from SBMA patients [31] displayed an increased level of Mid1 (Supplementary Fig. 1), confirming the similar trends in human. We also examined the expression of Pnpt1, which had the second-largest fold change, using immunofluorescence, but the expression pattern of it was not motor neuron-specific in the spinal cord (Supplementary Fig. 2). Therefore, we focused on Mid1 henceforth.
Mid1 increases pathogenic AR protein in a cellular model of SBMA
Previous studies on cell lines have shown that Mid1 binds to several mRNAs containing a CAG-repeat in a length-dependent manner [20, 21] to increase their translation. Although Mid1 protein is reported to bind to the CAG repeat of AR mRNA to regulate its translation [22], whether the translation of pathogenic AR with an expanded CAG repeat can similarly be increased was not known. Therefore, we tested the effects of overexpressing Mid1 on the level of pathogenic AR protein in the mouse Neuroblastoma-Spinal Cord hybrid (NSC) 34 cell line [32] (Fig. 2a, b). As we performed a short-term culture within 24 h, administration of DHT resulted in only moderate increases in AR protein levels. The protein levels of Mid1 was decreased by treatment with DHT (Fig. 2a), possibly due to a negative regulation of Mid1 by activated AR regardless of expanded polyglutamine repeats [22]. In this condition, overexpression of Mid1 increased the protein level of AR-97Q, but not AR-24Q, in the presence of DHT. Hence, the results suggested that the increase of AR protein levels by Mid1 overexpression depended on both the length of a CAG repeat and DHT levels.

a Immunoblots showing levels of human AR protein in NSC34 cells expressing AR-24Q or AR-97Q, which were cultured with or without DHT treatment and transfected with Mid1-expressing or empty vector. b Quantitative densitometry analyses of human AR levels in a (n = 3 samples per group). The data were normalized to the AR levels without DHT treatment and Mid1 transfection. Significant difference was detected by Tukey’s multiple comparisons test (*p < 0.005, **p < 0.05) after one-way ANOVA (*p < 0.005 for AR-97Q samples). c Immunoblots showing the time course of human AR protein levels in NSC34 cells expressing AR-97Q and transfected either with Mid1 or empty vector. Hours after the administration of CHX are shown. d Quantitative densitometry analyses of the time course of human AR protein levels in e (n = 3 samples per group). The data were normalized to the AR/Gapdh level at 0 h. No significant difference was detected by Two-way ANOVA. E Quantification of human AR expression levels in NSC34 cells transfected with Mid1-expressing vector or empty vector (n = 3 samples per group; Wilcoxon rank-sum test). n.s. not significant. f Immunoblots showing Mid1 in the total cell lysate (loading control), RNA-protein pull-down of Mid1 with no RNA, RNA containing 24 or 97 CAG repeats (AR-24Q, AR-97Q). Gapdh was used as a negative control. g An agarose gel showing RT-PCR products of AR from total cell lysate (loading control) and RIP products of HEK293T cells overexpressing AR-24Q/AR-97Q and Mid1.
The increase of AR protein levels could result from an increase in translation, a decrease in protein degradation, and/or an increase in transcription. CHX pulse-chase experiment ruled out the second possibility: Mid1 overexpression did not change the time course of pathogenic AR protein levels under CHX treatment (Fig. 2c, d), suggesting that the degradation of pathogenic AR protein was not affected by Mid1. To exclude the third possibility, the transcript level of AR transgene, which was driven by constitutive CAG promoter [3], was measured with quantitative RT-PCR. The result showed no change in AR transcript levels after Mid1 overexpression, confirming that AR was not upregulated at the transcript level (Fig. 2e). Therefore, an increase in translation appears to be a plausible mechanism for the rise of AR protein levels induced by Mid1 overexpression. As Mid1 is known to facilitate translation of mRNAs harboring a CAG-repeat [20, 21], we tested whether this protein binds to AR mRNA in a CAG-repeat length-dependent manner. The results of an RNA pull-down assay demonstrated that Mid1 had a higher affinity for the mRNA encoding AR-97Q than AR-24Q (Fig. 2f). Furthermore, immunoprecipitation of His-tagged Mid1 followed by RT-PCR showed greater amount of AR-97Q mRNA bound by Mid1 as compared to AR-24Q mRNA (Fig. 2g). Hence, Mid1 binding to AR depends on the length of CAG-repeat expansion, leading to an increase in the protein levels of pathogenic AR potentially through translational upregulation.
Mid1 is expressed in developing motor neurons
Mid1 expression has been reported to be ubiquitous during early mouse development but restricted to specific organs during later development [33, 34]. We found that motor neurons showed Mid1 expression as early as the prenatal stage (Fig. 3a). Notably, the spinal cord from the prenatal AR-97Q mouse showed nuclear localization of AR (Fig. 3b), suggesting that the motor neurons were already exposed to circulating androgen during the embryonic stage. The finding of co-occurrence of Mid1 expression and AR accumulation in the nuclei of motor neurons raises the possibility that the translational upregulation of pathogenic AR by Mid1 might affect the development of motor neurons.

a Immunostaining of Mid1 and Isl1 in the fetal mouse spinal cord. The anterior (ventral) side is toward the bottom. Boxed regions in the upper panels were magnified in the lower panels. Arrowheads and arrows indicate the overlap of strong and weak Mid1 expression with Isl1 expression, respectively. b Immunostaining of human AR and Isl1 in the fetal mouse spinal cord. Arrowheads indicate the overlap of nuclear AR and Isl1 expression. Scale bars 50 μm.
Spinal cord slice cultures from the SBMA mouse model show the androgen-dependent impairment of axonogenesis
To examine the effects of pathogenic AR on the development of spinal motor neurons, we utilized the organotypic spinal cord slice culture obtained from AR-97Q mice, which allows the assessment of axon and motor-neuron phenotypes associated with neurodegeneration in an anatomically preserved environment [35, 36]. The spinal cord slices were embedded in the collagen gel and cultured up to 2 days (Fig. 4a). In this system, administration of DHT from the beginning of the culture resulted in greater levels of AR protein compared with the culture without DHT (Fig. 4b). Moreover, the spinal cord slices cultured with DHT showed nuclear localization of AR in the motor neurons similar to that observed in the naive embryonic spinal cord (Fig. 3b). This was also observed in the spinal cord slices cultured without DHT, albeit to a lesser extent (Fig. 4c).

a A schematic procedure for the spinal cord slice culture from AR-97Q mouse fetus at the embryonic day 14.5. Cultured spinal cord slices were collected on Day 2 and used for subsequent analyses. Phase-contrast images were acquired, focusing on the anterior side with the axons extended into the collagen gel. b Increased pathogenic AR protein levels. The left and right panels show the immunoblots and quantitative densitometry analyses for human AR protein levels in the spinal cord slice culture from AR-97Q mice, respectively (n = 5 pairs of slice cultures per group). The data were normalized to the AR protein levels without DHT treatment. Statistically, significant difference was detected by unpaired two-tailed one-sample t-test (*p < 0.05). c Co-immunostaining for nuclear human AR and Isl1, a motor neuronal marker, under DHT treatment but not without DHT in the spinal cord slice cultures from AR-97Q mice. d Phase-contrast images showing the effects of DHT treatment on the axonogenesis of the spinal cord slice culture from AR-97Q and wild-type male mice. e Quantification of the axonal distribution using Neurite-J [37]. The number of intersections was shown as a function of the distance from the center of the culture bodies. Mean ± standard error of the mean (s.e.m.) are shown (n = 3–7 samples per group). f Comparisons of the maximal number of axons detected in an image (Nmax) calculated from the plots in e. Mean ± standard deviation (s.d.) are shown. Statistically, significant difference (*p < 0.01) was detected by Tukey’s multiple comparisons test.
We then investigated how androgen affects axonogenesis in this system. The spinal cord slices from AR-97Q mouse fetus showed substantial impairment of axonogenesis when cultured with DHT (Fig. 4d). We applied the previously reported method of image analysis to quantify axonogenesis in the spinal cord slice cultures (Fig. 4e) [37]. We found the reduction in the index for the number of axons (Nmax) in the cultured AR-97Q spinal cord slices treated with DHT (Fig. 4f). In the spinal cord slices from the wild-type littermates, the axonogenesis was not perturbed by the administration of DHT (Fig. 4d–f). These results showed that the androgen-dependent impairment of axonogenesis in the spinal cord slices from AR-97Q mice occurred mainly due to the toxicity of pathogenic AR.
Axons but not cell bodies are primarily affected in the spinal cord slice culture from a mouse model of SBMA
To further elucidate the androgen-dependent impairment of axonogenesis in the spinal cord slice cultures from AR-97Q mice, we performed immunohistochemical staining of the spinal cord slices using an antibody against non-phosphorylated neurofilament heavy chain (NF-H), which strongly stains the cell bodies and weakly stains the axons of the motor neurons [38]. We found that the axons stained for non-phosphorylated NF-H were originated from the cell bodies stained for non-phosphorylated NF-H (Fig. 5a). Again, the genesis of the stained axons was abrogated by the administration of DHT (Fig. 5a, b). A previous study reported that caspase-3 activation is involved in cell death induced by pathogenic AR [8]. Immunostaining revealed no significant increase in the number of caspase-3-positive motor neurons (Fig. 5c, d). These results suggest that axons but not cell bodies of the motor neurons were primarily affected in the spinal cord slice culture from AR-97Q mice.

a Intensity-coded images of non-phosphorylated NF-H. The double-headed arrows indicate the region of collagen gel where axons are embedded. b Motor neuronal cell bodies positive for non-phosphorylated NF-H were classified either with axon or no axon. Statistically, significant difference was detected in the percentage of motor neuronal cell bodies with axons (n > 30 per group) by Fisher’s exact test (*p < 0.001). c Immunostaining of cleaved caspase 3 (cCasp3) in the spinal cord slice cultures of AR-97Q mice with or without DHT treatment. Immunostaining of cCasp3 is nearly absent in Isl1-positive motor neurons under DHT treatment. d Quantification of motor neuronal nuclei (n > 50 per group) positive for cCasp3 with or without DHT treatment. No statistically significant difference was detected by Fisher’s exact test.
Mid1 exacerbates the androgen-dependent impairment of axonogenesis in the spinal cord slice culture from the mouse model of SBMA
If Mid1 could exacerbate the androgen-dependent impairment of axonogenesis in the spinal cord slices from AR-97Q mice, Mid1 overexpression would further impair the axonogenesis defects. To test this possibility, we introduced Mid1 to the spinal cord slice culture by lentivirus-mediated transduction (Fig. 6a, Supplementary Fig. 3). Overexpression of Mid1 resulted in increased pathogenic AR protein levels in a similar manner to that observed in the cellular model (Figs. 6b, 2a). Since the spinal cord slice culture from AR-97Q male mice showed a nearly complete absence of axonogenesis (Fig. 4c), we used the culture from AR-97Q female mice that showed a milder impairment of axonogenesis (Supplementary Fig. 4a, b) and thus allowed morphological analysis of axons for this purpose. Mid1 overexpression alone without DHT treatment resulted in only moderate impairment of axonogenesis, while Mid1 overexpression showed severe impairment of axonogenesis compared with that caused by DHT alone (Fig. 6c), which was confirmed by the quantification of axons (Fig. 6d, e). These results indicate that Mid1 exacerbates the androgen-dependent impairment of axonogenesis in the spinal cord slice culture from AR-97Q mice, especially under androgen exposure.

a Immunoblots showing levels of Mid1, human AR, and Gapdh. b Quantitative densitometry analyses of Mid1 and human AR levels in a (n = 4 samples per group). c Phase-contrast images showing the effects of Mid1 overexpression on the axonogenesis of the spinal cord slice culture from AR-97Q mouse fetus with or without DHT treatment. d For the spinal cord slice cultures from female AR-97Q female mouse fetus treated with DHT and overexpressed with GFP (control) or Mid1, plots showing the quantification of the intersection profile was obtained by Neurite-J. e Comparisons of the maximal number of axons detected in an image (Nmax). Statistically significant difference (*p < 0.05, **p < 0.001) was detected by Tukey’s multiple comparisons test.
Mid1 knockdown ameliorates the androgen-dependent impairment of axonogenesis in the spinal cord slice culture from a mouse model of SBMA
We further examined whether Mid1 knockdown rescues the axonogenesis defects. We first confirmed that knockdown of Mid1 did not affect axonogenesis in the spinal cord slice culture from wild-type mice (Supplementary Fig. 5). Next, in the spinal cord slice culture from AR-97Q male mice that had severe impairment of axonogenesis than female mice (Fig. 4d, Supplementary Fig. 4), we found that Mid1 knockdown decreased the pathogenic AR protein levels (Fig. 7a–c). Furthermore, Mid1 knockdown increased the protein levels of PP2Ac, a ubiquitination target of Mid1 involved in the translational regulatory complex of mTOR [39] (Supplementary Fig. 6). As expected, Mid1 knockdown ameliorated the axonogenesis defects (Fig. 7d–f), suggesting that endogenous Mid1 is responsible for the androgen-dependent impairment of axonogenesis in the spinal cord slice culture from AR-97Q mice. In addition, knockdown of the human AR transgene rescued the impairment of axonogenesis regardless of co-silencing of Mid1 (Fig. 7d–f, Supplementary Figs. 7, 8). Overall, Mid1 expressed in motor neurons played a crucial role in the androgen-dependent impairment of axonogenesis in the spinal cord slice culture from AR-97Q mice.

a Quantification of Mid1 expression levels in the spinal cord slice cultures from wild-type mouse fetus transfected with lentivirus expressing control shRNA or Mid1 shRNA. Statistically, significant difference (*p < 0.05) was detected by an unpaired two-tailed one-sample t-test. b Immunoblots showing levels of Mid1, AR, and Gapdh in the spinal cord slice culture from AR-97Q male mouse fetus transfected with control shRNA or Mid1 shRNA. c Quantitative densitometry analyses of Mid1 and human AR levels in a (n = 3 samples per group). d Phase-contrast images showing the effects of Mid1 and/or human AR knockdowns on the axonogenesis of the spinal cord slice culture from AR-97Q mouse fetus treated with DHT. e For the spinal cord slice cultures from AR-97Q male mouse fetus treated with DHT and shRNA (control, Mid1, human AR, or both Mid1 and human AR), plots showing the quantification of the intersection profile was obtained by Neurite-J. f Comparisons of the maximal number of axons detected in an image (Nmax). Statistically, significant differences (*p < 0.05) were detected by Tukey’s multiple comparisons test.
Discussion
The present study demonstrates that Mid1, a microtubule-associated RNA binding protein exclusively expressed in the motor neurons of the spinal cord, interacts with CAG-repeat expanded AR mRNA and increases the protein levels of pathogenic AR in both cellular and organotypic slice culture models of SBMA. The transcriptional upregulation of Mid1 by pathogenic AR induced the translational upregulation of pathogenic AR in the spinal cord of early symptomatic SBMA mice and accelerated the motor neuron pathology in SBMA. Interestingly, AR has been shown to repress the transcription of Mid1, forming a negative feedback loop leading to suppression of AR protein levels [22]. Hence, a loss-of-function property of pathogenic AR might affect this negative feedback regulation, resulting in increased Mid1 transcription. The bi-directional feedbacks between pathogenic AR and Mid1 could contribute to the vulnerability of motor neurons in SBMA.
Although SBMA is an adult-onset disease, studies in the primary culture of embryonic motor neurons from the mouse model of SBMA have suggested that both transcriptional and translational dysregulation by pathogenic AR impair motor neuron homeostasis as an early dysregulation during development [17, 40]. Our finding that pathogenic AR accumulated in the nuclei of embryonic motor neurons in AR-97Q mouse fetus, probably due to prenatal exposure to androgen [41], further suggests that pathogenic AR begins affecting motor neurons during embryonic stage itself. Substantial impairment of axonogenesis in the spinal cord slice cultures from AR-97Q mice in the presence of androgen may be attributed to, at least in part, the degree of pathogenic AR accumulation during embryogenesis. Regarding slice cultures, the greater impairment of axonogenesis in males compared to the females indicates the role of prenatal exposure to androgens in SBMA pathogenesis, especially in males. Moreover, the spinal cord slices from AR-97Q mice cultured without androgen showed axonogenesis levels similar to wildtype (Fig. 4f), suggesting that the effect of embryonically accumulated pathogenic AR could be offset by androgen deprivation. In our previous study [9], this reversibility was consistent with the rescue of axonal dysfunctions in AR-97Q mice by androgen depletion. Considering the role of Mid1 in the translation of pathogenic AR, targeting Mid1 may be effective as therapy against motor axonal vulnerability in SBMA even at an adult stage.
Our study indicates that the impairment of axonal growth by pathogenic AR partly depends on the translational upregulation of pathogenic AR by Mid1. It has also been reported that axonal growth could be directly regulated by Mid1-PP2A system [24, 42]. Still, this mechanism is unlikely to explain the axonogenesis defects in the spinal cord slice cultures from AR-97Q mice for three reasons. First, the impairment of axonogenesis in the spinal cord slice culture from AR-97Q mice is dependent on androgen levels but not on Mid1 alone (Figs. 4, 6). Second, axonogenesis in spinal cord slices from wild-type mice was not affected by the knockdown of Mid1 (Supplementary Fig. 3). Third, double knockdown experiments of Mid1 and AR suggest that the impairment of axonogenesis by Mid1 depends on the pathogenic AR (Fig. 7). Another interesting feature of Mid1 is its association with microtubules. Mid1 itself is transported along the axonal microtubule in a manner dependent on the dephosphorylation by PP2A [43]. Thus, the association of Mid1 with microtubule might also contribute to the effect of pathogenic AR on axonal transport via the increase in its local translation of pathogenic AR. In light of previous reports, cytoplasmic oligomers of pathogenic AR disrupt axonal transport, thereby impairing the neurite growth [8, 9]. Accordingly, increased translation of pathogenic AR by Mid1 in the vicinity of the microtubule may disrupt axonal transport and be involved in the substantial impairment of axonogenesis observed in the present study. If this mechanism involves the axonal vulnerability of motor neurons observed in the in vivo settings [6, 7, 9] needs to be explored in future studies.
Our study demonstrated that the embryonic spinal cord slice culture is a useful model for elucidating the axonal vulnerability in SBMA, which might reflect the early dysregulation of motor neurons in vivo. In the organotypic slice culture, axonogenesis is supported by the surrounding extracellular matrix and glial cells [35], in addition to rich trophic support by the culture medium. These conditions allowed axonogenesis of wild-type or androgen-untreated SBMA motor neurons but could not support axonogenesis of androgen-treated SBMA motor neurons that may have experienced higher cytotoxic stresses. In this sense, the analysis of axonogenesis in the spinal cord slice culture may serve as stress load testing for SBMA motor neurons. Also, spinal cord slice cultures were easily transduced with a lentivirus vector without the requirement of its in vivo administration, especially for embryos. This advantageous property further enabled us to elucidate the mechanism of androgen-dependent neuronal and axonal vulnerabilities of SBMA motor neurons. Studies of the developmental origin of functional deficits in motor axons using spinal cord slice culture models will advance our understanding of neuronal pathogenesis and lead to the development of targeted therapeutics in SBMA.
Ethics
All animal experiments were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and with the approval of the Nagoya University Animal Experiment Committee.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Hashizume A, Fischbeck KH, Pennuto M, Fratta P, Katsuno M. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (SBMA). J Neurol Neurosurg Psychiatry. 2020;91:1085–91.
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79.
Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–54.
Minamiyama M, Katsuno M, Adachi H, Doi H, Kondo N, Iida M, et al. Naratriptan mitigates CGRP1-associated motor neuron degeneration caused by an expanded polyglutamine repeat tract. Nat Med. 2012;18:1531–8.
Katsuno M, Adachi H, Minamiyama M, Waza M, Doi H, Kondo N, et al. Disrupted transforming growth factor-beta signaling in spinal and bulbar muscular atrophy. J Neurosci. 2010;30:5702–12.
Kemp MQ, Poort JL, Baqri RM, Lieberman AP, Breedlove SM, Miller KE, et al. Impaired motoneuronal retrograde transport in two models of SBMA implicates two sites of androgen action. Hum Mol Genet. 2011;20:4475–90.
Poort JE, Rheuben MB, Breedlove SM, Jordan CL. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum Mol Genet. 2016;25:3768–83.
Young JE, Garden GA, Martinez RA, Tanaka F, Sandoval CM, Smith AC, et al. Polyglutamine-expanded androgen receptor truncation fragments activate a Bax-dependent apoptotic cascade mediated by DP5/Hrk. J Neurosci. 2009;29:1987–97.
Katsuno M, Adachi H, Minamiyama M, Waza M, Tokui K, Banno H, et al. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J Neurosci. 2006;26:12106–17.
Iida M, Sahashi K, Kondo N, Nakatsuji H, Tohnai G, Tsutsumi Y, et al. Src inhibition attenuates polyglutamine-mediated neuromuscular degeneration in spinal and bulbar muscular atrophy. Nat Commun. 2019;10:4262.
Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52.
Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–16.
Fargo KN, Galbiati M, Foecking EM, Poletti A, Jones KJ. Androgen regulation of axon growth and neurite extension in motoneurons. Horm Behav. 2008;53:716–28.
Ottem EN, Beck LA, Jordan CL, Breedlove SM. Androgen-dependent regulation of brain-derived neurotrophic factor and tyrosine kinase B in the sexually dimorphic spinal nucleus of the bulbocavernosus. Endocrinology. 2007;148:3655–65.
Marron TU, Guerini V, Rusmini P, Sau D, Brevini TAL, Martini L, et al. Androgen-induced neurite outgrowth is mediated by neuritin in motor neurones. J Neurochem. 2005;92:10–20.
Kapur M, Monaghan CE, Ackerman SL. Regulation of mRNA translation in neurons—a matter life death neuron. Neuron. 2017;96:616–37.
Montague K, Malik B, Gray AL, La Spada AR, Hanna MG, Szabadkai G, et al. Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy. Brain. 2014;137:1894–906.
Leitman J, Barak B, Benyair R, Shenkman M, Ashery U, Hartl FU, et al. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS One. 2014;9:e90803.
Aranda-Orgillés B, Rutschow D, Zeller R, Karagiannidis AI, Köhler A, Chen C, et al. Protein phosphatase 2A (PP2A)-specific ubiquitin ligase MID1 is a sequence-dependent regulator of translation efficiency controlling 3-phosphoinositide-dependent protein kinase-1 (PDPK-1). J Biol Chem. 2011;286:39945–57.
Krauss S, Griesche N, Jastrzebska E, Chen C, Rutschow D, Achmüller C, et al. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat Commun. 2013;4:1511.
Griesche N, Schilling J, Weber S, Rohm M, Pesch V, Matthes F, et al. Regulation of mRNA translation by MID1: a common mechanism of expanded CAG repeat RNAs. Front Cell Neurosci. 2016;10:226.
Köhler A, Demir U, Kickstein E, Krauss S, Aigner J, Aranda-Orgillés B, et al. A hormone-dependent feedback-loop controls androgen receptor levels by limiting MID1, a novel translation enhancer and promoter of oncogenic signaling. Mol Cancer. 2014;13:146.
Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, et al. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–95.
Lu T, Chen R, Cox TC, Moldrich RX, Kurniawan N, Tan G, et al. X-linked microtubule-associated protein, Mid1, regulates axon development. Proc Natl Acad Sci USA. 2013;110:19131–6.
Cheng H, Snoek R, Ghaidi F, Cox ME, Rennie PS. Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer Res. 2006;66:10613–20.
Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinforma. 2018;19:534.
Blum JA, Klemm S, Shadrach JL, Guttenplan KA, Nakayama L, Kathiria A, et al. Single-cell transcriptomic analysis of the adult mouse spinal cord reveals molecular diversity of autonomic and skeletal motor neurons. Nat Neurosci. 2021;24:572–83.
Miyata T, Kawaguchi A, Okano H, Ogawa M. Asymmetric inheritance of radial glial fibers by cortical. Neurons Neuron. 2001;31:727–41.
Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M. Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development. 2004;131:3133–45.
Miyata T, Ono Y, Okamoto M, Masaoka M, Sakakibara A, Kawaguchi A, et al. Migration, early axonogenesis, and Reelin-dependent layer-forming behavior of early/posterior-born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev. 2010;5:23.
Lim WF, Forouhan M, Roberts TC, Dabney J, Ellerington R, Speciale AA. et al. Gene therapy with AR isoform 2 rescues spinal and bulbar muscular atrophy phenotype by modulating AR transcriptional activity. Sci Adv. 2021;7:eabi6896.
Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, et al. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–21.
Dal Zotto L, Quaderi NA, Elliott R, Lingerfelter PA, Carrel L, Valsecchi V, et al. The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region. Hum Mol Genet. 1998;7:489–99.
Pinson L, Augé J, Audollent S, Mattéi G, Etchevers H, Gigarel N, et al. Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome. J Med Genet. 2004;41:381–6.
Bucchia M, Merwin SJ, Re DB, Kariya S. Limitations and challenges in modeling diseases involving spinal motor neuron degeneration in vitro. Front Cell Neurosci. 2018;12:61.
Croft CL, Futch HS, Moore BD, Golde TE. Organotypic brain slice cultures to model neurodegenerative proteinopathies. Mol Neurodegener. 2019;14:45.
Torres-Espín A, Santos D, González-Pérez F, del Valle J, Navarro X. Neurite-J: an Image-J plug-in for axonal growth analysis in organotypic cultures. J Neurosci Methods. 2014;236:26–39.
Avossa D, Grandolfo M, Mazzarol F, Zatta M, Ballerini L. Early signs of motoneuron vulnerability in a disease model system: characterization of transverse slice cultures of spinal cord isolated from embryonic ALS mice. Neuroscience. 2006;138:1179–94.
Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, et al. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat Genet. 2001;29:287–94.
Malik B, Devine H, Patani R, La Spada AR, Hanna MG, Greensmith L. Gene expression analysis reveals early dysregulation of disease pathways and links Chmp7 to pathogenesis of spinal and bulbar muscular atrophy. Sci Rep. 2019;9:3539.
Clarkson J, Herbison AE. Hypothalamic control of the male neonatal testosterone surge. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150115.
Zhu L-Q, Zheng H-Y, Peng C-X, Liu D, Li H-L, Wang Q, et al. Protein phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP2. J Neurosci. 2010;30:3839–48.
Aranda-Orgillés B, Aigner J, Kunath M, Lurz R, Schneider R, Schweiger S. Active transport of the ubiquitin ligase MID1 along the microtubules is regulated by protein phosphatase 2A. PLoS One. 2008;3:e3507.
Acknowledgements
This work was funded by Grant-in-Aid (KAKENHI) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Nos. 20H00527 to M.K. and 19H03545 to K.S.). No other agencies provided fundings, and the investigators had sole discretion over the study design; collection, analysis, and interpretation of data; writing of the report; and the decision to submit it for publication. The authors thank Dr. Kanako Saito (Fujita Medical University) for technical assistance on the slice culture experiments.
Author information
Authors and Affiliations
Contributions
Project planning was performed by Y.O., K.S., and M.K.; The project was supervised by M.K. and T.M.; experiments were performed by Y.O., K.S., T.H. M.I., and M.K.; and data were analyzed by Y.O., K.S., and T.H. and M.K. The first draft of the manuscript was prepared by Y.O., K.S., and M.K.; the manuscript layout was designed by Y.O., K.S., and M.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Alexei Verkhratsky
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ogura, Y., Sahashi, K., Hirunagi, T. et al. Mid1 is associated with androgen-dependent axonal vulnerability of motor neurons in spinal and bulbar muscular atrophy. Cell Death Dis 13, 601 (2022). https://doi.org/10.1038/s41419-022-05001-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05001-6
This article is cited by
-
Critical Roles of Protein Arginine Methylation in the Central Nervous System
Molecular Neurobiology (2023)
