
Abstract
Depression is a common mental disorder affecting more than 264 million people worldwide. The first-line treatment for most cases of depression are selective serotonin reuptake inhibitors (SSRIs), such as sertraline, reboxetine and fluoxetine. Recently, it has been found that one-quarter of depressed patients have excessive activation of the immune system. This potentially warrants sub-categorisation of depressed patients into inflammatory and non-inflammatory subtypes. Such a sub-category of depression already exists for those not responding to various traditional antidepressants and is known as treatment-resistant depression. Those with treatment-resistant depression are far more likely to have raised inflammatory markers relative to those whose depression is treatment-responsive. Chronic, low-level inflammation seems to trigger depression via a multitude of mechanisms. These include kynurenine pathway and microglial cell activation, resulting in a reduction in hippocampal volume. Raised inflammatory cytokines also cause perturbations in monoaminergic signalling, which perhaps explains the preponderance of treatment resistance in those patients with inflammatory depression. Therefore, if treatment-resistant depression and inflammatory depression are semi-synonymous then it should follow that anti-inflammatory drugs will display high efficacy in both sub-types. Ketamine is a drug recently approved for use in depression in the USA and displays a particularly good response rate in those patients with treatment resistance. It has been suggested that the antidepressant efficacy of ketamine results from its anti-inflammatory effects. Ketamine seems to produce anti-inflammatory effects via polarisation of monocytes to M2 macrophages. Furthermore, another anti-inflammatory drug with potential use in treatment-resistant depression is Celecoxib. Celecoxib is a long-acting, selective COX-2 inhibitor. Early clinical trials show that Celecoxib has an adjuvant effect with traditional antidepressants in treatment-resistant patients. This paper highlights the importance of classifying depressed patients into inflammatory and non-inflammatory subtypes; and how this may lead to the development of more targeted treatments for treatment-resistant depression.
Similar content being viewed by others
Introduction
Depression is a psychiatric disorder that affects mood, behaviour and overall health. The treatment for depression is a combination of counselling and pharmacotherapy. Depression treatment can be challenging due to drug side effects. Importantly, there is a group of depressed patients who do not respond adequately to multiple courses of appropriate pharmacotherapy, patients with treatment-resistant depression. Lack of response to antidepressants increases the risk of suicide, prolongs unnecessary suffering and is a large healthcare burden [1]. This makes finding alternative treatments for treatment-resistant patients a healthcare imperative. In this perspective, we will discuss the latest discoveries in treatment-resistant depression and how immunomodulatory drugs could be used to improve treatment responses.
Why would immunomodulators be useful in depression?
A comprehensive literature shows that depression is linked to an activation of the immune system. The primary evidence for this comes from measures of immune cytokines. Cytokines associated with Th1 activation, including tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6, are raised in the cerebrospinal fluid and plasma of depressed patients. Furthermore, activated microglial cells are found in patients during a depressive episode, relative to healthy control [2], and other biomarkers of inflammatory states, such as low serum iron and raised body temperature, are seen in major depression [3].
Worthy of inquiry is in which direction the relationship between inflammation and depression is causal. It is commonly known that depression-like behaviour can be induced in animals by the injection of lipopolysaccharide (LPS), which is highly pro-inflammatory [4]. In addition to this, clinical use of interferons in multiple sclerosis, among other disorders, can cause depression as a side effect. This implies that the inflammatory response can be causal in depression [5].
Further evidence for an immune system-depression link is provided by genomics data. Many genetic polymorphisms have been associated with depression but the existence of genetic roots for depression is somewhat perplexing. Through evolutionary history, depression would have increased one’s risk of death from suicide [6, 7]. This begs the question, therefore, as to why these genes would continue to persist in the gene pool? One answer is that depression-associated genes could provide a dual function. Single nucleotide polymorphisms identified by candidate genes and genome-wide association studies to be correlated with depression risk are regularly found in immune-related genes. Indeed, 8 of the 10 gene variants that most strongly increase depression risk also have immune or inflammatory function [3]. Such evolutionary trade-offs are common, as is the case with the malaria-protective effects of the haemoglobin mutation that causes sickle cell disease for example [8]. The fact that inflammation appears to trigger depression is likely more than just an evolutionary trade-off, however, as it has been proposed that depression is a prolonged form of sickness behaviour. Indeed, the symptoms of major depression are remarkably akin to sickness behaviour. Both are characterised by sleep pattern change (insomnia, hypersomnia), appetite changes, reduced sociability (social withdrawal), irritability, low mood and reduced motivation and interest in daily activities (anhedonia) [3].
The primary mechanism proposed to mediate Th1 immune response-triggered depression is the activation of indoleamine 2,3-dioxygenase (IDO). IDO is an enzyme involved in tryptophan metabolism and is up-regulated and activated by Th1-associated cytokines, particularly IFNγ [9, 10]. It generates kynurenine from tryptophan, diverting tryptophan away from serotonin synthesis. Kynurenine and its downstream metabolites are neurotoxic. This is due to the direct agonism of N-methyl-d-aspartate (NMDA) receptors and activation of glutamate and reactive oxygen species release from microglial cells. The resultant excitotoxicity and oxidative stress cause tissue damage, particularly in the hippocampus. Reduced hippocampal volume is a well-characterised marker seen in the brains of depressed people [11,12,13,14]. Further support for this hypothesis is found in the fact that LPS was unable to engender depression-like behaviour in IDO1 knockout mice, suggesting inflammation-induced depression is IDO-dependent [15].
In addition to activation of the kynurenine pathway causing hippocampal neurotoxicity, inflammation also seems to attenuate hippocampal neurogenesis. IL-6, IL-1 and TNFα, for example, all suppress the neurotrophin, brain-derived neurotrophic factor (BDNF). BDNF is known to ameliorate depressive symptoms, in part by increasing hippocampal neurogenesis, which is consistently decreased in depressed individuals [16]. Also, TNF-α increases the activity of nuclear factor kappa B, which further suppresses neurogenesis [17]. Furthermore, the inflammation affects monoaminergic signalling in the brain via mechanisms beyond those associated with IDO-induced tryptophan depletion. IL-1 and TNF-α increase phosphorylation of the serotonin transporter (SERT). This leads to increased translocation of SERT into the neuronal membrane, resulting in increased serotonin reuptake and reduced response to SERT-blocking antidepressants [18]. Furthermore, chronic, low-grade inflammation seems to reduce dopamine synthesis, packaging and release. Reduced dopamine neurotransmission increases depression risk, particularly symptoms of anhedonia, fatigue and psychomotor retardation [19].
Despite the link between inflammation and depression being incontrovertible, there is a large degree of heterogeneity between patients regarding inflammatory status. Around one-quarter of patients with depression have raised low-level inflammation [20]. This suggest that depressed patient can be categorised into non-inflammatory and inflammatory subtypes [21]. Furthermore, akin to the heterogeneity observed regarding the presence of inflammation in depressed populations, traditional antidepressants display a large degree of heterogeneity in their efficacy. On average, traditional antidepressants seem to show around a 25% remission rate, 75% response rate and 25% non-response rate, though this differs substantially from study to study [1, 22, 23]. Interestingly, treatment-resistant depression is far more often accompanied by increased Th1 cytokines relative to that which is treatment-responsive [24, 25]. This data suggests that the inflammatory depression subtype is also characterised by resistance to traditional antidepressants and, therefore, investigation of the efficacy of immunomodulators in treatment-resistant depression would be prudent.
The salience of an inflammatory depression subtype, appropriately targeted by immunosuppressives, is further supported by a number of other noteworthy facts from the literature. Firstly, depression that is co-morbid with previous childhood ill-treatment is associated with an increase in inflammation and is more often treatment-resistant [26]. That is, relative to depressed patients with a more normative parental environment. Similarly, acute stress seems to increase immune function, in direct contrast to chronic stress [27]. This is important in social stress, which increases pro-inflammatory cytokines in both humans and rodents [28, 29] (Fig. 1B). Furthermore, trials of cytokine inhibitor use in depression, such as infliximab (anti-TNFα) and sirukumab (anti-IL-6), selectively display efficacy in patients with low- inflammation levels prior to treatment [30, 31]. This selective efficacy will be a theme when discussing other immunomodulators (ketamine and celecoxib) during the rest of the paper.

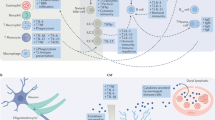
A A diagram comparing M1 and M2 type macrophages. M1 macrophages are activated by pro-inflammatory signals, including LPS, Th1 cytokines (IFNγ, TNFα) and Th1 cell surface proteins (CD40L). M2 macrophages are activated by Th2 cytokines (IL-4, IL-10, IL-13, IL-21) and by ketamine. M1 macrophages produce pro-inflammatory cytokines, as well as nitric oxide (NO) and neurotoxic kynurenine and reactive oxygen species and thus mediate the Th1 response. Ketamine causes the polarisation of monocytes to M2 macrophages. This acts to promote the release of anti-inflammatory TGFβ, IL-10 and ornithine. It also reduces monocyte differentiation to M1 macrophages, hampering the effects of Th1 dominance. B A schematic showing the possible causes of Th1 dominance in patients with treatment-resistant depression and the downstream consequences of this excessive Th1 immune activation. Indoleamine 2,3-dioxygenase (IDO) activation is central to inflammation-induced depression via its effects on tryptophan metabolism and generation of neurotoxicity. IDO-mediated changes in tryptophan metabolism reduce serotonin, as does phosphorylation of serotonin transporters (SERT). Brain-derived neurotrophic factor (BDNF) exacerbates the negative effects of IDO activation on hippocampal volume by reducing hippocampal neurogenesis, as does nuclear factor kappa B (NFkB) induction.
Ketamine
Ketamine is a drug that has been used for many years as a fast-acting non-barbiturate general anaesthetic. It also has well-established use in hypotensive shock, reactive airway disease, analgesia and procedural sedation. It is primarily an NMDA receptor antagonist, but its pharmacodynamics are complex. It also shows interactions with opioid, cholinergic, purinergic and adrenergic receptors, as well as with ion channels not gated by endogenous ligands [32]. More recently, ketamine has found its purpose in depression, being approved by the US food and drug administration in 2019 [33].
Ketamine has a remarkably high response rate in treatment-resistant depression (~65%) but the exact mechanisms underlying its antidepressant effects are unknown, particularly as other NMDA receptor antagonists possess non-comparable effects [34, 35]. While a multiplicity of mechanisms have been proposed, such as effects on opioid and AMPA receptors [36,37,38], ketamine seems to produce a number of its effects via immunomodulatory mechanisms. A systematic review of 9 human studies and 22 animal studies [39] found that ketamine consistently produced reductions in IL-1β, IL-6 and TNFα. Furthermore, in all but one study in which it was measured ketamine was found to decrease IDO activity and reduce the prevalence of downstream neurotoxic metabolites [39]. Moreover, the magnitude of reduction in IL-6 and IL-1β is associated with the magnitude of ketamine’s antidepressant effect [40].
Little is known about ketamine’s immunomodulatory mechanism. It is likely to be the result of some direct effect on leucocytes [41]. This is evidenced by the fact that ketamine can reduce pro-inflammatory cytokine production in isolated human blood. One current hypothesis as to how ketamine does this, is that it causes macrophage polarisation to an M2 phenotype [42]. Macrophages can be categorised into M1 and M2 type macrophages. Th1 cells induce differentiation to M1 macrophages via the expression of IFN-γ and CD40 ligand. These macrophages are highly pro-inflammatory. Meanwhile, Th2 cells induced differentiation to anti-inflammatory M2 macrophages via the expression of IL-4 and IL-13. M1 macrophages preferentially produce many of the pro-inflammatory immune cytokines seen in depression, such as IL-6 and TNF-α, as well as pro-inflammatory nitric oxide (NO) and reactive oxygen species (Fig. 1A). Therefore, by ketamine polarising macrophages to the M2 type, it may be opposing the downstream effects of the Th1 dominance observed in depression [43, 44]. Furthermore, IDO is more highly expressed in M1 macrophages, which may explain how ketamine reduces its activity [45]. The exact mechanism via which ketamine causes monocytes to differentiate into M2 macrophages is unclear but it seems to occur via an mTOR-dependent mechanism and involved increased expression of CD163 and MERTK [42].
Microglia also possess an M1 and M2 bifurcation in phenotype and LPS induces differentiation of microglia to an M1 type via activation of toll-like receptors, hence its neuroinflammatory effect [46]. Ketamine blocks LPS-induced M1 differentiation in both peripheral and central nervous system macrophages [47, 48]. Furthermore, M2 microglia preferentially produce transforming growth factor (TGF)-1β, which produces anti-inflammatory effects in the brain. Ketamine can prevent reductions in TGF-1β levels induced by chronic social defeat stress in mice (Fig. 1B). Meanwhile, the use of an anti-TGF-1β antibody in mice experiencing chronic social defeat stress blocks the antidepressant effects of ketamine [49].
Celecoxib
Celecoxib is a long-acting, selective cyclooxygenase-2 inhibitor. It is of similar potency to ibuprofen but is used in patients with mild to moderate pain and/or with arthritis, who cannot tolerate the gastrointestinal side effects of traditional non-steroidal anti-inflammatory drugs (NSAIDs). Celecoxib works by inhibiting pro-inflammatory protein synthesis [50]. Downstream effects of this protein synthesis inhibition are facilitated by alterations in cell-cell interactions, vascular tone and permeability, cytokine production and receptor expression and leucocyte maturation, migration and survival [51].
Several studies had shown that Celecoxib is useful in treatment-resistant and inflammatory depression. Greater over-the-counter NSAID use is associated with reduced depression rates in the Danish population, suggesting NSAIDs may be antidepressant per se [52]. Furthermore, depression is associated with raised body temperature, perhaps suggesting a role of prostaglandin E2 in its pathophysiology [43, 53]. The effects of NSAIDs on immune cytokines also indicate potential efficacy. As mentioned previously, IL-6 is the most commonly raised immune cytokine in depression. The synthesis of IL-6, downstream of CD40 activation in B cells, is dependent upon COX-2 [54]. This, among other mechanisms, means that NSAIDs reliably reduce levels of IL-1 and IL-6, leading to a reduction in the Th1 response [55, 56]. Lastly, one of the ten gene polymorphisms most strongly associated with depression is found is adcy3, which codes for adenylate cyclase 3. Adenylate cyclase 3 plays an integral role in the signal transduction downstream of prostaglandin receptors, further providing evidence of a role for COX-2 inhibition in depression [3].
Multiple clinical trials have been conducted using celecoxib in depressed individuals. In 2014, a meta-analysis of celecoxib trials was conducted [57]. Monotherapy and therapy adjunctive to traditional antidepressants in depressed individuals with and without co-morbid inflammatory disease were found to be superior to placebo and to traditional antidepressants alone [57]. It is worth noting that it is difficult to draw conclusions from trials in patients that had a co-morbid inflammatory disease (6 out of 11 of those included in the meta-analysis). This is because there is no mechanism to control for the potential that the antidepressant effect resulted indirectly via amelioration of discomfort (swelling, pain, etc.) rather than via some direct action. Saying this, more trials have been conducted since 2014 with some promising results. The effect of sertraline, a common drug to treat depression, with celecoxib versus sertraline alone in drug-naive depressed women was assessed [58]. Although no statistically significant difference in the mean reduction of Hamilton Depression Rating Scale scores was observed at eight weeks, the response rate and remission rate were much higher in the celecoxib group relative to the sertraline-only group. The response rate was 100% in the group taking both sertraline and celecoxib compared with 78% in the group taking sertraline alone. Furthermore, and perhaps most worthy of attention, the remission rate in the celecoxib group was 57% relative to 11% in the placebo group. This trial suggests that Celecoxib directly targets individuals who would normally be treatment-resistant (Fig. 2) Also, Celecoxib adjunct therapy was found to reduce serum IL-6 in depressed patients and the magnitude of the IL-6 reduction predicted the magnitude of the antidepressant effect [59]. Moreover, Celecoxib seems to target the IDO pathway. Higher kynurenine serum concentrations prior to treatment were found to predict remission in patients given Celecoxib adjunct therapy [60]. Lastly, Celecoxib monotherapy is better than placebo in rat models of depression and its use is associated with reduced levels of immune cytokines [61]. It also potentiates the effect of reboxetine and fluoxetine, common drugs to treat depression, on cortical noradrenaline and serotonin output in rats [62]. Considering this data, Celecoxib seems to be targeting treatment-resistant depression via the Th1-associated immune pathways.

A bar chart showing the percentage response rate, remission rate and non-response rate to antidepressant treatment. Responses to three drug treatments are compared: the average first-line selective serotonin reuptake inhibitor (SSRI) (numbers derived from analysis of the wider literature [1, 22, 23], shown on the left, sertraline in drug-naive women (data taken from [58]), shown in the centre, and sertraline in combination with celecoxib in drug-naive women [58], shown on the right. The addition of celecoxib to SSRI treatment dramatically increases the percentage rate of response (100% taking celecoxib vs 75–78% taking placebo/nothing) and rate of remission (57% taking celecoxib vs 11–25% taking placebo/nothing). This provides evidence as to the efficacy of celecoxib as an adjunctive treatment and suggests it may actively target the treatment-resistant depression subtype.
Unfortunately, there are contradictory results within the literature. For example, the only long-term trial of Celecoxib monotherapy in depression conducted to date, found no treatment effect [57, 63]. This could be due to the lack of selection for individuals with inflammatory depression, rather than a lack of drug effect. On the other hand, the dearth of long-term proven efficacy and tolerability of Celecoxib in depression is concerning. While the absence of COX1 inhibition obviates the risk of gastrointestinal side effects to Celecoxib use [50], its selectivity could increase the risk of blood clotting due to preferential inhibition of prostacyclin vs thromboxane A2 synthesis [64]. Saying this, in the 2014 meta-analysis [57], the authors found no significant increase in cardiovascular incidence when using Celecoxib in depression, but the lack of long-term trials included in the study makes this finding unconvincing at this point. Also noteworthy is that while NSAID use is correlated with antidepressant effects, this is not a black and white finding. Chronic use of over-the-counter NSAIDs or the use of high doses of aspirin was associated with increased depression incidence [52]. While this is only a correlative finding, it could indicate that Celecoxib would not be applicable to long-term depression treatment.
Conclusions and perspectives
Depression is a complex condition. It is well accepted that inflammation can be a core feature of depression. Treatment-resistant depression is a subtype of depression, frequently characterised by enhancement of the Th1 cell-mediated and inflammatory immune responses. This subtype of depression could be amenable to improvement with the use of immunomodulatory drugs. Th1 dominance seems to cause treatment-resistant depression by causing reductions in hippocampal volume, mediated by IDO activation, microglial activation and reduction in neurogenesis, and perturbations in monoaminergic neurotransmission. Clinical trials of immunomodulators in depression thus far have been thwarted by the lack of an inflammatory depression subtype classification but have still shown promising results nonetheless.
Ketamine is an exemplar of how effective a drug with immunomodulatory mechanisms can be in treatment-resistant depression. It seems to act via M2 polarisation of macrophages, which results in attenuation of the Th1 immune response. Celecoxib is a pharmacotherapy in earlier stages of development for treatment-resistant depression, which has shown exciting results. This is especially true when used as an ancillary to traditional antidepressants, notably increasing the response and remission rate. Further research is needed to elucidate the exact mechanism by which ketamine causes monocytes to differentiate into M2 macrophages and to what extent its efficacy in treatment-resistant depression is the result of immunomodulation. Moreover, future research should focus on confirming the efficacy, safety and tolerability of Celecoxib in long-term clinical trials in individuals with inflammatory depression.
Data availability
All the data used to support the arguments in this study are included within the article.
References
Jaffe DH, Rive B, Denee TR. The humanistic and economic burden of treatment-resistant depression in Europe: a cross-sectional study. BMC Psychiatry. 2019;19:247.
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 2015;72:268–75.
Raison CL, Miller AH. The evolutionary significance of depression in Pathogen Host Defense (PATHOS-D). Mol Psychiatry. 2013;18:15–37.
O’Connor JC, Lawson MA, Andro C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2009;14:511–22.
Pinto EF, Andrade C. Interferon-related depression: a primer on mechanisms, treatment, and prevention of a common clinical problem. Curr Neuropharmacol. 2016;14:743–8.
Humphrey N. The lure of death: suicide and human evolution. Philos Trans R Soc B. 2018;373:20170269.
Gourevitch D. Suicide among the sick in classical antiquity. Bull Hist Med. 1969;43:501–18.
Elguero E, D guerost Med t LM, Rougeron V, Arnathau C, Roche B, Becquart P, et al. Malaria continues to select for sickle cell trait in Central Africa. Proc Natl Acad Sci USA. 2015;112:7051–4.
Moreau M, Lestage J, Verrier D, Mormede C, Kelley KW, Dantzer R, et al. Bacille Calmette-GuU S Aect for sickle cell trait in Central Africad Preeral and brain indoleamine 2,3-dioxygenase in mice. J Infect Dis. 2005;192:537–44.
Müller N, Myint AM, Schwarz MJ. The impact of neuroimmune dysregulation on neuroprotection and neurotoxicity in psychiatric disorders-relation to drug treatment. Dialogues Clin Neurosci. 2009;11:319–32.
Colín-González AL, Maldonado PD, Santamaro A. 3-Hydroxykynurenine: an intriguing molecule exerting dual actions in the central nervous system. Neurotoxicology. 2013;34:189–204.
Dantzer R. Role of the kynurenine metabolism pathway in inflammation-induced depression: preclinical approaches. Curr Top Behav Neurosci. 2017;31:117–38.
Schlittler M, Goiny M, Agudelo LZ, Venckunas T, Brazaitis M, Skurvydas A, et al. Endurance exercise increases skeletal muscle kynurenine aminotransferases and plasma kynurenic acid in humans. Am J Physiol-Cell Physiol. 2016;310:C836–40.
Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85:2059–70.
Lawson MA, Parrott JM, McCusker RH, Dantzer R, Kelley KW, O lleyrr JC. Intracerebroventricular administration of lipopolysaccharide induces indoleamine-2,3-dioxygenase-dependent depression-like behaviors. J Neuroinflammation. 2013;10:87.
Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–41.
Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci USA. 2010;107:2669–74.
Zhu CB, Blakely RD, Hewlett WA. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology. 2006;31:2121–31.
Felger JC. The role of dopamine in inflammation-associated depression: mechanisms and therapeutic implications. Curr Top Behav Neurosci. 2017;31:199–219.
Osimo EF, Baxter LJ, Lewis G, Jones PB, Khandaker GM. Prevalence of low-grade inflammation in depression: a systematic review and meta-analysis of CRP levels. Psychol Med. 2019;49:1958–70.
Raison CL. The promise and limitations of anti-inflammatory agents for the treatment of major depressive disorder. Curr Top Behav Neurosci. 2017;31:287–302.
Gueorguieva R, Mallinckrodt C, Krystal JH. Trajectories of depression severity in clinical trials of duloxetine: insights into antidepressant and placebo responses. Arch Gen Psychiatry. 2011;68:1227–37.
Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry. 2001;62:869–77.
Yang C, Wardenaar KJ, Bosker FJ, Li J, Schoevers RA. Inflammatory markers and treatment outcome in treatment resistant depression: a systematic review. J Affect Disord. 2019;257:640–9.
Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ. Inflammation and clinical response to treatment in depression: a meta-analysis. Eur Neuropsychopharmacol. 2015;25:1532–43.
Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry. 2008;65:409–15.
Liu YZ, Wang YX, Jiang CL. Inflammation: the common pathway of stress-related diseases. Front Hum Neurosci. 2017;11:316.
Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology. 2010;35:2617–23.
Barrientos RM, Sprunger DB, Campeau S, Higgins EA, Watkins LR, Rudy JW, et al. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience. 2003;121:847–53.
MRC ImmunoPsychiatry Consortium, Wittenberg GM, Stylianou A, Zhang Y, Sun Y, Gupta A, et al. Effects of immunomodulatory drugs on depressive symptoms: a mega-analysis of randomized, placebo-controlled clinical trials in inflammatory disorders. Mol Psychiatry. 2020;25:1275–85.
Miller AH, Pariante CM. Trial failures of anti-inflammatory drugs in depression. Lancet Psychiatry. 2020;7:837.
Kurdi MS, Theerth KA, Deva RS. Ketamine: current applications in anesthesia, pain, and critical care. Anesth Essays Res. 2014;8:283–90.
Shin C, Kim YK. Ketamine in major depressive disorder: mechanisms and future perspectives. Psychiatry Investig. 2020;17:181–92.
Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–42.
Newport DJ, Carpenter LL, McDonald WM, Potash JB, Tohen M, Nemeroff CB, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172:950–66.
Deyama S, Bang E, Wohleb ES, Li XY, Kato T, Gerhard DM, et al. Role of neuronal VEGF signaling in the prefrontal cortex in the rapid antidepressant effects of ketamine. Am J Psychiatry. 2019;176:388–400.
Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533:481–6.
Williams NR, Heifets BD, Blasey C, Sudheimer K, Pannu J, Pankow H, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018;175:1205–15.
Kopra E, Mondelli V, Pariante C, Nikkheslat N. Ketamine's effect on inflammation and kynurenine pathway in depression: a systematic review. J Psychopharmacol. 2021;35:934–45.
Yang JJ, Wang N, Yang C, Shi JY, Yu HY, Hashimoto K. Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biol Psychiatry. 2015;77:e19–20.
Kawasaki T, Ogata M, Kawasaki C, Ogata J, Inoue Y, Shigematsu A. Ketamine suppresses proinflammatory cytokine production in human whole blood in vitro. Anesth Analg. 1999;89:665.
Nowak W, Grendas LN, Sanmarco LM, Estecho IG, Arena Ár, Eberhardt N, et al. Pro-inflammatory monocyte profile in patients with major depressive disorder and suicide behaviour and how ketamine induces anti-inflammatory M2 macrophages by NMDAR and mTOR. EBioMedicine. 2019;50:290–305.
Murphy K, Weaver C. Janeway’s immunology. 9th ed. New York, NY: Garland Science/Taylor & Francis Group, LLC; 2016. 904 p.
Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci. 2017;18:E1545.
Wang XF, Wang HS, Wang H, Zhang F, Wang KF, Guo Q, et al. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: focus on macrophage polarization of THP-1 cells. Cell Immunol. 2014;289:42–8.
Guo S, Wang H, Yin Y. Microglia polarization from M1 to M2 in neurodegenerative diseases. Front Aging Neurosci. 2022;14:815347.
Liu FL, Chen TL, Chen RM. Mechanisms of ketamine-induced immunosuppression. Acta Anaesthesiol Taiwan. 2012;50:172–7.
Chang Y, Lee JJ, Hsieh CY, Hsiao G, Chou DS, Sheu JR. Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediators Inflamm. 2009;2009:705379.
Zhang K, Yang C, Chang L, Sakamoto A, Suzuki T, Fujita Y, et al. Essential role of microglial transforming growth factor-uced microglial activationhage polarization of THP-1 cellse induces anti-inflamm. Transl Psychiatry. 2020;10:32.
Gong L, Thorn CF, Bertagnolli MM, Grosser T, Altman RB, Klein TE. Celecoxib pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22:310–8.
Harizi H. The immunobiology of prostanoid receptor signaling in connecting innate and adaptive immunity. Biomed Res Int. 2013;2013:683405.
Kessing LV, Rytgaard HC, Gerds TA, Berk M, Ekstrar CT, Andersen PK. New drug candidates for depression - a nationwide population-based study. Acta Psychiatr Scand. 2019;139:68–77.
Rausch JL, Johnson ME, Corley KM, Hobby HM, Shendarkar N, Fei Y, et al. Depressed patients have higher body temperature: 5-HT transporter long promoter region effects. Neuropsychobiology. 2003;47:120–7.
Dongari-Bagtzoglou AI, Thienel U, Yellin MJ. CD40 ligation triggers COX-2 expression in endothelial cells: evidence that CD40-mediated IL-6 synthesis is COX-2-dependent. Inflamm Res. 2003;52:18–25.
Kang BS, Chung EY, Yun YP, Lee MK, Lee YR, Lee KS, et al. Inhibitory effects of anti-inflammatory drugs on interleukin-6 bioactivity. Biol Pharm Bull. 2001;24:701–3.
Kusuhara H, Matsuyuki H, Okumoto T. Effects of nonsteroidal anti-inflammatory drugs on interleukin-1 receptor antagonist production in cultured human peripheral blood mononuclear cells. Prostaglandins. 1997;54:795–804.
Köhler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O. et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry. 2014;71:1381–91.
Majd M, Hashemian F, Hosseini SM, Vahdat Shariatpanahi M, Sharifi A. A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in treatment of drug-naive depressed women: a pilot study. Iran J Pharm Res. 2015;14:891–9.
Abbasi SH, Hosseini F, Modabbernia A, Ashrafi M, Akhondzadeh S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J Affect Disord. 2012;141:308–14.
Krause D, Myint AM, Schuett C, Musil R, Dehning S, Cerovecki A, et al. High kynurenine (a tryptophan metabolite) predicts remission in patients with major depression to add-on treatment with celecoxib. Front Psychiatry. 2017;8:16.
Müller N. COX-2 inhibitors, aspirin, and other potential anti-inflammatory treatments for psychiatric disorders. Front Psychiatry. 2019;10:375.
Johansson D, Falk A, Marcus MM, Svensson TH. Celecoxib enhances the effect of reboxetine and fluoxetine on cortical noradrenaline and serotonin output in the rat. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39:143–8.
Fields C, Drye L, Vaidya V, Lyketsos C, ADAPT Research Group. Celecoxib or naproxen treatment does not benefit depressive symptoms in persons age 70 and older: findings from a randomized controlled trial. Am J Geriatr Psychiatry. 2012;20:505–13.
Steffel J, L effel TF, Ruschitzka F, Tanner FC. Cyclooxygenase-2 inhibition and coagulation. J Cardiovasc Pharmacol. 2006;47:S15–20.
Acknowledgements
The authors thank Dr. Christine Edmead, University of Bath for advice during the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
CWB wrote the paper and prepared the figures; MVN-C corrected the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beckett, C.W., Niklison-Chirou, M.V. The role of immunomodulators in treatment-resistant depression: case studies. Cell Death Discov. 8, 367 (2022). https://doi.org/10.1038/s41420-022-01147-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-022-01147-6
This article is cited by
-
The antidepressive mechanism of Longya Lilium combined with Fluoxetine in mice with depression-like behaviors
npj Systems Biology and Applications (2024)
-
Distinguishing features of depression in dementia from primary psychiatric disease
Discover Mental Health (2024)
-
Effects of physical activity and depressive symptoms on cognitive function in older adults: National Health and Nutrition Examination Survey
Neurological Sciences (2024)
-
C-reactive protein as a biomarker for unipolar versus bipolar depression: a cross-sectional study
Middle East Current Psychiatry (2023)
-
Effect modification of tumor necrosis factor-α on the kynurenine and serotonin pathways in major depressive disorder on type 2 diabetes mellitus
European Archives of Psychiatry and Clinical Neuroscience (2023)
