
Abstract
The most frequent Y-chromosomal (chrY) haplogroups in northern and eastern Europe (NEE) are well-known and thoroughly characterised. Yet a considerable number of men in every population carry rare paternal lineages with estimated frequencies around 5%. So far, limited sample-sizes and insufficient resolution of genotyping have obstructed a truly comprehensive look into the variety of rare paternal lineages segregating within populations and potential signals of population history that such lineages might convey. Here we harness the power of massive re-sequencing of human Y chromosomes to identify previously unknown population-specific clusters among rare paternal lineages in NEE. We construct dated phylogenies for haplogroups E2-M215, J2-M172, G-M201 and Q-M242 on the basis of 421 (of them 282 novel) high-coverage chrY sequences collected from large-scale databases focusing on populations of NEE. Within these otherwise rare haplogroups we disclose lineages that began to radiate ~1–3 thousand years ago in Estonia and Sweden and reveal male phylogenetic patterns testifying of comparatively recent local demographic expansions. Conversely, haplogroup Q lineages bear evidence of ancient Siberian influence lingering in the modern paternal gene pool of northern Europe. We assess the possible direction of influx of ancestral carriers for some of these male lineages. In addition, we demonstrate the congruency of paternal haplogroup composition of our dataset with two independent population-based cohorts from Estonia and Sweden.
Similar content being viewed by others
Introduction
Genetic studies investigating uniparental and fine-scale autosomal variation in Estonia [1] and in its neighbouring populations in NEE [2,3,4,5,6] observed that the regional genetic structure correlates closely with geography. In addition, recent ancient DNA studies have begun to uncover the settlement history of NEE, which is distinct from that of central and southern parts of the continent [7,8,9].
The four most common chrY haplogroups (hgs) with incidence above 5% (R1a-M198, N3-TAT, I-M170, R1b-M343) constitute over 90% of the chrY pool in NEE [3, 10,11,12]. Several studies have analysed these hgs in a wide phylogeographic context.
Besides the four most common hgs, several paternal lineages belonging on the basic level to hgs E2, J2, G and Q with frequency up to 5%, complement the pool of Y-chromosomes in NEE [3, 4, 13,14,15]. In Europe, hgs E2a, J2 and G are common in the southern Mediterranean populations and form 20–30% of their chrY lineages. In NEE, the frequency of hg E2a’d is ~2–3%, hgs J2 and G respectively reach ~1–2% and ~1% of the total pool of chrY lineages [3, 5, 6, 15, 16]. Hg Q has a frequency of 1–3% in most European populations with the highest incidence in Sweden [3, 4]. Hg Q is otherwise widely spread in Siberian populations and is among the major founding male lineages in the peopling of the Americas [17, 18]. These rare hgs that make up less than 10% of NEE male lineages, are mostly left unexplained and are often regarded as recent scattered entries into populations. The small sample sizes and low phylogenetic resolution has not allowed separation of rare lineages beyond the major hg labels. The sequencing of complete Y-chromosomes provides a way to resolve the inner structure of lineages on the phylogenetic tree regardless of their prevalence in populations [13, 14, 19, 20]. Sequencing a considerable number of well dispersed samples from NEE reveals the distribution of rare lineages on the entire phylogenetic tree and provides sufficiently granular data to estimate their split times. This builds the necessary geographic and chronological context for surveying patterns of uncovered lineage clusters stemming from a single node and hallmarking local expansions. The coalescence ages of ancestral internal nodes and phylogenetically well-defined clusters nested within disclose the geography and timeframe of local expansions as well as possible gene flow involving ancestral carriers of rare male lineages in Estonia, Sweden and their neighbouring populations.
Here we aim to analyse the previously understudied rare chrY lineages with a focus on Estonia and Sweden together with their NEE neighbours and Germany to account for the historic influence of the Baltic Germans. Additional populations are included to widen the geographic context. We combined full sequences of Y-chromosomes from populations inhabiting Estonia, Sweden, Finland, Latvia, Lithuania, Poland, Germany, Ukraine and the Russian Federation to build updated phylogenetic trees for haplogroups rare in NEE. In order to mitigate sampling bias that might influence any conclusions drawn from such a rare substratum present among the populations, we tested the representativeness of our two largest cohorts sampled from the Estonian and Swedish populations by comparing their frequency compositions with sample sets independently obtained from the same two populations.
Materials and methods
Samples
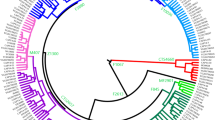
We screened the occurrence of rare hgs in a sample of 1160 chrY sequences from male donors (selected randomly by county of birth) from the population-based Estonian Biobank [21]. The Estonian chrY sequences are part of the whole genome sequencing (WGS) data set autosomally first described in Mitt et al. [22] for constructing a population-specifc imputation panel. Only chrY sequences of the haplogroups rare in NEE (N = 64) are included in the current study. Next, in scientific collaboration with the commercial genetic testing company Gene by Gene (Houston, Texas, USA), we screened the collection of customers who had provided informed consent for their data to be used in scientific inquiry. This resulted in a total of 2018 male donors with self-reported ancestry from Sweden, Finland, Latvia, Lithuania, Poland, Germany, Ukraine and the Russian Federation. If the database contained more than 500 samples from a respective country, individuals with identical self-reported paternal and maternal origin were preferably selected. In case of smaller available sample sets, all samples with self-reported paternal origin from the respective country were selected. From the resulting set of 2018 samples, we detected 222 Y-chromosomes belonging to the rare NEE haplogroups and these samples were included in the current study. We collected additional 139 chrY sequences from published sources resulting in the final set of 421 chrY sequences (Supplementary Table S1) used for reconstruction of phylogenetic trees for rare hgs E2 (129 samples), J2 (136 samples), Q (83 samples) and G (71 samples) (Figs. 1, 2 and Supplementary Figs. S1–S7).

The calibrated trees were constructed using BEAST v.1.7.5 software package. Internal nodes, sub-clade names and population names (numbers show the number of samples) are indicated. Internal nodes with posterior probabilities <0.73 are not shown. Samples from Estonia and Sweden are marked in blue and orange, respectively. a A schematic phylogenetic tree of hg E2a is based on 132 high-coverage chrY sequences. Neighbour-clade E2b and its sublineages are marked in grey. Detailed tree can be found in Supplementary Materials (Supplementary Fig. S5). Age estimates can be found in Supplementary Table S8. All the subclade (node) defining mutations and marker names are presented in Supplementary Table S4. b A schematic phylogenetic tree of hg J2b is based on 136 high-coverage chrY sequences. Neighbour-clade J2a and its sublineages are marked in grey. Detailed tree can be found in Supplementary Materials (Supplementary Fig. S6). Age estimates can be found in Supplementary Table S9. All the sub-clade (node) defining mutations and marker names are presented in Supplementary Table S5.

A detailed phylogenetic tree of hg Q-M242 is based on 84 high-coverage chrY sequences. Two hg R1a sequences were used for an outgroup. The detailed calibrated tree was constructed using BEAST v.1.7.5 software package. Internal nodes, sub-clade names and population names are indicated. Internal nodes with posterior probabilities <0.73 are not shown. Age estimates can be found in Supplementary Table S10. All the subclade (node) defining mutations and marker names are presented in Supplementary Table S6. Samples from Estonia and Sweden are marked in blue and orange, respectively.
To test for possible sampling bias in the two largest sequencing cohorts, we screened the haplogroup frequencies of two independent datasets – a total of 505 chrY sequences available from the SweGen project (samples specifically selected to be representative of the historic Swedish population [23]) and a randomly selected non-overlapping set of genotyped 7949 Estonian male donors from the Estonian Biobank.
Sequencing, mapping and genotyping
ChrY sequences from the Estonian Biobank and the SweGen project were generated with Illumina Inc. (Illumina, San Diego, CA, USA) using HiSeq instruments (PCR-free protocol) and targeted 30x genome-wide coverage. The personal genetic testing company dataset was generated using the proprietary BigY Illumina-based targeted chrY capture sequencing service (https://learn.familytreedna.com/wp-content/uploads/2014/08/BIG_Y_WhitePager.pdf).
We used the same processing pipeline for all Illumina data. Fastq files were mapped with BWA-MEM (v0.7.12) [24] on the human reference hs37d5 (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence). Read duplicates were removed with Picard (v2.12.0) (http://broadinstitute.github.io/picard/) and remaining unique reads realigned around known indels, followed by base quality score recalibration (BQSR) using GATK (v3.8) [25]. Variant calling was performed with GATK tool HaplotypeCaller in haploid mode. All-sites VCF files were filtered with bcftools (v1.9) [26]. The Illumina data and previously filtered data from Complete Genomics (Supplementary Table S1) were merged with CombineVariants from GATK (v3.8) [25]. We extracted the effective overlap between the two datasets by masking out all positions with 5% or higher proportion of missing genotypes in either Illumina or Complete Genomics datasets. We additionally excluded regions with poor mappability as described previously [13] resulting in a total of 9.7 Mb of analysed sequence. Within this sequence, the resulting numbers of variant positions used for phylogenetic reconstruction in each haplogroup are given in Supplementary Tables S4–S7.
Haplogroup assignment
We assigned chrY haplogroups using yHaplo [27] for the Illumina capture and WGS data. We used SNAPPY [28] for chrY haplogroup assignment of the genome-wide array genotyping data.
Comparisons with an independent Estonian cohort
To validate the representativeness of sequenced Estonian chrY samples (N = 1160), we compared the hg frequencies of this cohort against a ~7 times larger cohort of 7949 Estonian male samples genotyped with the Illumina Infinium Global Screening Array v2 (Illumina, San Diego, CA, USA) containing 6638 Y-specific single nucleotide variants (SNVs). To do this, we first assessed the accuracy of haplogroup assignments obtained from this particular set of SNVs. We sub-sampled the 6638 array-specific Y-SNVs from the Estonian WGS data and used SNAPPY software to determine the haplogroups from the extracted set of SNVs. We compared the results against those from the software yHaplo [27]. The latter utilises the full set of SNPs in the WGS samples. The results are identical on the highest level of the major branches and only differ slightly at the finest resolution due to the lower number of array-genotyped SNPs available to SNAPPY for detecting the haplogroups. However, this shows that hg assignments based on the 6638 array-specific Y-positions are accurate enough to be compared to hg assignments based on full sequencing. The comparison of the hg frequencies of the WGS-based and array-based Estonian datasets was performed using a Wilcoxon signed rank test with continuity correction. We only used array-based data for comparing haplogroup frequencies between two independent cohorts. For the phylogeny reconstruction and phylogeographic analysis full sequencing data were used.
Phylogeny reconstruction of rare paternal haplogroups
We reconstructed phylogenies and estimated the coalescent times with the software package BEAST v.1.7.5 [29]. We used a Bayesian skyline coalescent tree prior, the general time reversible (GTR) substitution model with gamma-distributed rates and a relaxed lognormal clock. The run was performed with the piecewise-constant coalescent model. The mutation rate used was 0.74 × 10−9 (95% CI: 0.63–0.95 × 10−9) per base pair per year [13]. The results were visualised and checked for effective sample size above 200 in Tracer v.1.4. Coalescence time estimates were computed with normally distributed age priors with 10% standard deviation from previously published phylogeny [13] and are in Supplementary Table S3. Lineages from hg R and I were used as outgroups for hgs Q and G, respectively. Each run had thirty million chains logged every 3000 steps and 10% discarded as burn-in. Two parallels with different random number seeds were combined with LogCombiner.
The manually annotated phylogenetic trees, mutation lists and coalescence age estimates are available in Supplementary Figs. S1–S4 and Supplementary Tables S4–S11. This study’s updated nomenclature follows the criteria set in Karmin et al. [13].
Bayesian phylogeographic analysis
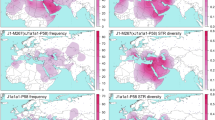
To illustrate the potential direction of influx of the primarily Estonian subclades in hgs E2a1-CTS1273 and J2b2-L283, we performed Bayesian phylogeographic analyses in continuous space. For this we used available geographic coordinates for 59 sequences belonging to hg E2a1-CTS1273 and for 41 sequences belonging to hg J2b2-L283. This method has been originally developed and successfully used to reveal the ancestral location and spatial dynamics of viruses in continuous space [30, 31]. We conducted the analysis according to the publication exploring the history of Y-chromosomal hg J1 [32] in BEAST v1.10.4 [33] using BEAGLE library v3.1.2 [34] for accelerated likelihood evaluation. This statistically robust and absolutely data-driven method uses molecular sequence data and geographic coordinates of the samples to infer phylogeography in a continuous landscape while simultaneously reconstructing the evolutionary history in time. It draws the confidence area of ancestral locations where the root and internal nodes originated together with the directions and the speed of the diffusion (Fig. 3). The uncertainties of the maximum clade credibility tree node locations were visualised with SpreaD3 v0.9.7.1rc software [35]. This inference approach accounts for the coalescent, phylogenetic, molecular clock, location, and other uncertainties within a single framework. Additional details are provided in Supplementary Note 1.

Maps indicate the phylogeographic spread of a J2b2-L283 around 6 kya, 4 kya, 2 kya and in the present, and b E2a1-CTS1273 around 5–6 kya, 4 kya, 2 kya and in the present. Shaded in pink are the 80% HPD areas of the node locations inferred by Bayesian continuous phylogeographic analysis with Beast v1.10.4 software. White circles indicate the median locations of the nodes, while black lines indicate the branches of the maximum clade credibility tree.
Results
Phylogeny of rare lineages in NEE
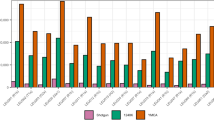
The studied 1160 high-coverage sequences of Y-chromosomes from Estonia disclose 64 samples carrying male lineages rare in NEE (frequency of each under 3%), amounting to ~6% of the total paternal lineage pool in Estonia. The most frequent minor lineage in Estonia belongs to hg E2 (2.5%), followed by hgs J2 (1.9%) and hg G (0.9%), whereas hg Q is the rarest (0.3%) (Supplementary Table S2). Our second largest sample set consists of a total of 746 males from Sweden and discloses 78 samples with rare NEE chrY lineages. The most common minor haplogroup in the Swedish cohort is hg Q (4.6%); followed by hgs G (3%), E2 (1.7%) and J2 (1.2%) (Supplementary Table S2).
To verify the robustness of our frequency estimates, we compared hg frequencies of our Swedish sample set and the SweGen cohort (N = 505) [23]. The Wilcoxon signed rank test showed no statistically significant differences between the two, either considering all hgs (p value = 0.4689) or minor hgs with major hgs collapsed (p value = 0.6602). Similarly, a comparison of hg frequencies between the Estonian sample set and an independent non-overlapping set of 7949 genotyped male samples from the Estonian Biobank yielded no statistically significant differences in their hg composition, either considering all haplogroups (Wilcoxon signed rank test p value = 0.4896) or rare hgs with major hgs collapsed (Wilcoxon signed rank test p value = 0.9219).
Hg E originated in Africa with its sublineage E1 distributed solely on the African continent, whereas the neighbour-lineage hg E2 displays a notably wider distribution. Subclade E2-V13 is common (~10–20%) among south-eastern European populations [4, 6, 14, 16], falling to 10% in Anatolia and the Middle East [36] and declining towards northern Europe to 1–2% in Scandinavia [4].
Here we reconstruct the phylogeny of hg E2a’b’c’d-M35. Its subclade E2a-M78 is largely confined to Europe with a coalescence time of ~14 kya (95% CI: 10,432–18,566) (Fig. 1a, Supplementary Table S8). Within this subclade, L618 marker unites almost all European samples that split ~13 kya (95% CI: 9,682–17,360) from the neighbouring clade E2a2-V22. The latter consists primarily of samples from the Middle East with deeper diversification times (Supplementary Fig. S5). The absolute majority (25/29) of hg E samples from Estonia belong to subclade E2a1-V13 (Supplementary Table S2). The bulk of Estonian samples form clearly distinguishable clusters: lineage E2a1-S7461 contains an Estonian founding cluster that splits from the neighbour lineages with Swedish and Middle Eastern origin ~4 kya (95% CI: 3,146–5,752, Supplementary Table S8) and a radiation time of ~2 kya (95% CI: 1,398–2,999; Supplementary Table S8). Similar pattern can be seen in the hg E2a1-B409 that has lineages from Germany and Sweden and an exclusively Estonian cluster defined by marker Z37869 with a radiation time of ~2 kya (95% CI: 1,150–2,428; Supplementary Table S8).
Hg J is one of the most common haplogroups in Western Asia and in regions surrounding the Mediterranean Sea and thus was initially connected to the dispersion of male farmers from the Fertile Crescent. Phylogenetic studies of hg J have shown surviving ancient sublineages with radiation signs in the Bronze Age [37, 38]. Additionally, hg J2a and an unresolved hg J have been discovered in ancient DNA from hunter-gatherer samples excavated in the Caucasus [39] and Karelia [40]. In southern Europe, the most common hg J subclade is J2-M172, which, however, becomes rare throughout the northern latitudes [4, 16].
Here we reconstruct the phylogenetic tree of hg J2-M172 (Fig. 1b and Supplementary Fig. S6) with 134 individuals. A substantial part of NEE individuals belong to sublineages within hg J2b2-L283 (Fig. 1b) which splits from its neighbouring clade at ~16 kya (95% CI: 11,860–20,018; Supplementary Table S9). Hg J2b2-L283 itself split ~7 kya (95% CI: 5,000–8,912) into two major sublineages J2b2-Z2505 and J2b2-YP29. The latter is an exclusively Estonian cluster encompassing over half of all hg J samples from Estonia (12 of 22) with an expansion time of ~2 kya (95% CI: 1,446–3,027) (Fig. 1b, Supplementary Fig. S6, Supplementary Table S9).
The other major hg J subbranch – J2a-M410 – contains samples from broad Eurasian background which are distributed in subclades mostly coalescing during the early post-Last Glacial Maximum – a much deeper time estimate than in the neighbouring hg J2b-M12 phylogeny (Supplementary Fig. S6). Lack of information on detailed geographic or ethnic origin hinders any further conclusions regarding the single-origin clusters from the Russian Federation (Supplementary Fig. S6). Based on published research, lineages of hg J2a-M67 are among the most common (~20%) paternal haplogroups of the North Caucasus region [41], whereas in ethnic Russians this haplogroup amounts to less than 2% [5, 6].
Hg Q is frequent in Siberian populations and is carried by over 85% of male Native Americans [16,17,18, 42]. In Europe, the occurrence of hg Q is uneven and the general frequency is low (~0.42%) [42], but hg Q is somewhat more frequent in the populations of Sweden and Norway [3, 4]. It is the most numerous minor haplogroup in both of our Swedish sample sets with frequencies of 2.6% and 4.6% (Supplementary Table S2). In the datasets of Karlsson et al. [4] and Lappalainen et al. [3] the frequency of hg Q fluctuates between 1% and 5% in different regions of Sweden. On the updated phylogenetic tree, Swedish samples fall into two main clusters that separated from each other around the peak of the Last Glacial Maximum ~20 kya (Fig. 2). About a third of the Swedish hg Q samples are defined by marker L804. Hg Q1a-L804 coalesces ~16 kya (95% CI: 12,456–19,874; Supplementary Table S10) with haplogroup Q1a-M3, which today describes the overwhelming majority of Native American Y-chromosomes [42]. The rapid diversification among Swedes in the L804-defined clade began ~3 kya (95% CI: 1,961–3,917; Supplementary Table S10).
Haplogroup G-M201 is common in the Caucasus and the Middle East. Hg G is one of the most prevalent male lineages in Sardinia and Corsica, but displays low frequencies elsewhere in Europe [4, 14, 15]. Hg G splits into two basal lineages – hgs G1 and G2, of which the former occurs infrequently in Western and Central Asia and is almost absent in Europe [15]. Almost all hg G samples from NEE belong to hg G2-P287 that ~22 kya (95% CI: 17,620–26,973) split into two main subclades – G2a-P15 and G2b-M377 (Supplementary Fig. S7, Supplementary Table S11). The bulk of sampled European individuals belong to subclade G2a2-P303 (Supplementary Table S2). Downstream, in hg G2a2-Z727, the absolute majority of Swedish hg G samples forms localised clusters with a variety of coalescence times (Supplementary Fig. S7, Supplementary Table S11).
Discussion
In case of Estonia, our sequenced samples were collected across the country avoiding large settlements with recorded extensive migration history. Considering a census size of roughly 1 million, rare lineages amount to roughly 30,000 men evenly sampled across the country and thus cannot be exclusively ascribed to any random influx of recent migrants.
From the screened sample of 506 Finnish males we did not detect any rare NEE lineages as almost all Finnish samples belong to hgs common among neighbouring populations – a probable reflection of either differing migration history or of demographic bottleneck(s) that have affected the Finnish population [43, 44].
Hg E sublineages have been associated with Neolithic demic diffusion into Europe [16], but current ancient DNA data has shown this haplogroup to be uncommon among the first agriculturalists in Europe [40]. In the resolved phylogenetic tree, the primarily Middle Eastern neighbouring clade with deeply diverged lineages supports a possible Levantine source of the European hg E2a1-V13. However, the split time predates the Neolithic transition in Europe and matches better with the age of the Villabruna hunter-gatherer cluster that displays earliest autosomal affinities to the Middle Eastern populations detected in ancient samples from Europe [45]. The coalescence age of the primarily European clades of hgs E3a1-V13 and J2b2-Z2505 underpins mid-Holocene as the starting point of chrY variation growth in Western Europe (Fig. 1) and indicates a possible influx of male lineages from the Levant or the Caucasus.
The coalescence ages of Estonia-specific clades J2b2-YP29, E2a1-Z37869 and E2a1-Y28220 broadly correspond to the Late Bronze Age and Iron Age period in Northern Europe (Supplementary Fig. S8). Additional sampling might certainly affect the coalescence age of these clusters. However, the geographical spread across all Estonian counties and current age estimates suggest that these expansions are not the result of any migratory events from the recent recorded (last ~800 years) history of this region. To infer the potential directions of influx of the clades J2b2-YP29, E2a1-Z37869, and E2a1-Y28220, we conducted continuous Bayesian phylogeographic analysis of parent hgs J2b2-L283 and E2a1-CTS1273. The estimated diffusion rate of hg J2b2-L283 equals 0.27 (95% HPDs: 0.1992–0.3478) and for hg E2a1-CTS1273 0.231 (95% HPDs: 0.175–0.295) kilometres/year. The 80% HPD of the putative geographic centre of diffusion for the hg J2b2 covers the area focused in present-day Poland, with a partial covering of central and southeastern Europe, spreading further north and south (Fig. 3a). The area for hg E2a1 ancestral location similarly covers central and eastern Europe with a focus on Poland (Fig. 3b), but the focal point appears to be more condensed.
From a conservative standpoint, all three subclades most probably arrived to present-day Estonia from the direction of central Europe. However, based on currently available data, it is not possible to say whether the evident local expansions initially began in Estonia or were the carriers already sufficiently diversified on arrival.
Within hg Q, clusters defined by L804 and Y4838 capture almost all of Swedish hg Q diversity, marking these lineages as an inherent, albeit scarce, part of the pool of male lineages in Sweden. The scarcity of internal nodes on the branches leading to the two now predominantly Swedish clusters hinders any discussion regarding a potential direction of influx or ancestral centre of diffusion. Due to the glacial coverage, the split between lineages Q1a-L804 and the Native American Q1a-M3 could not have happened in Scandinavia. Ancient DNA research confirms the presence of hg Q in the remains of hunter-gatherers (~8 kya) from Latvia and Lower Volga Region in Russia [46]. Today, European Q1 lineages are restricted to NEE with occasional findings in other populations (single L804 derived English chrY sample in Grugni et al. [47]). Precursors to current European hg Q1 sublineages could have been widely present in North Eurasia during the Last Glacial Maximum and followed a primarily northern (Siberian) route of dispersal into Europe. The presumptively common ancient gene pool is reflected in the autosomal European affinity of 24,000-old Mal’ta sample from the vicinity of Lake Baikal [48]. Alternatively, the prevalence of hg Q in Sweden could testify of a more recent Siberian influence deduced both from modern and ancient DNA analysis in northeastern Europe [2, 8]. Studies have demonstrated minor eastern affinities in the autosomes and in the maternal lineages of the modern Saami, but small sample sizes have not revealed Saami male lineages belonging to hg Q [2]. Further sampling across Northern Eurasia might provide additional insights about these peculiar North Eurasian hg Q lineages. A total of two out of the three Estonian hg Q samples form a subset of the Swedish Y4838-defined cluster. It is most parsimonious to assume that the paternal ancestors of the two Y4838-derived individuals arrived in Estonia around 1–2 kya from Scandinavia.
Hg G2a has become firmly associated with the early Neolithic farmers of Europe [40, 46, 49]. Most of European hg G2a inner lineages started to diverge around 5–7 kya (Supplementary Fig. S7, Supplementary Table S11) – within the timeframe of the European agricultural transition. In Sweden, it is the second most frequent minor chrY haplogroup. The majority of Swedish carriers demonstrate a strong expansion signal approximated to ~1 kya (nodes 52 and 60 in Supplementary Fig. S7), whereas Estonian samples are not part of the Swedish hg G2a diversity.
In conclusion, we demonstrate that in NEE, rare paternal lineages are not just single lineages scattered across different subclades in the phylogeny. We identified several population-specific clusters among less common haplogroups, which testify of radiation events that have occurred in various timeframes and can be used to tentatively suggest possible influx directions.
This study demonstrates the power of large-scale re-sequencing of Y-chromosomes to explore and compare the male demographic history of single populations. Current survey of rare lineages paves the way for future research involving large datasets of re-sequenced genomes with a focus on those maternal and paternal lineages that have left a major demographic impact on modern populations in NEE and elsewhere.
Data availability
The Estonian WGS data are available on demand through the Estonian Biobank: https://www.geenivaramu.ee/en/biobank.ee/data-access. In accordance to the consent form signed by the customers of Gene by Gene commercial genetic testing company, the sequencing data included in this study is used for the sole purpose of scientific inquiry and is reported here on an aggregate level in the form of phylogenetic trees. For both the Estonian Biobank and the Gene by Gene samples, summary-level data including variable positions and their frequency in the cohort population have been deposited to dbSNP with links to BioProject accession number PRJNA718714 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/). The Swedish data from the SweGen Project is available upon request from the original authors of the project [23].
References
Pankratov V, Montinaro F, Kushniarevich A, Hudjashov G, Jay F, Saag L et al. Differences in local population history at the finest level: the case of the Estonian population. Eur J Hum Genet. 2020; 28:1580–1591.
Tambets K, Yunusbayev B, Hudjashov G, Ilumäe AM, Rootsi S, Honkola T, et al. Genes reveal traces of common recent demographic history for most of the Uralic-speaking populations. Genome Biol. 2018;19:1–20.
Lappalainen T, Laitinen V, Salmela E, Andersen P, Huoponen K, Savontaus ML, et al. Migration waves to the baltic sea region. Ann Hum Genet. 2008;72:337–348.
Karlsson AO, Wallerström T, Götherström A, Holmlund G. Y-chromosome diversity in Sweden - a long-time perspective. Eur J Hum Genet. 2006;14:963–970.
Balanovsky O, Rootsi S, Pshenichnov A, Kivisild T, Churnosov M, Evseeva I, et al. Two sources of the Russian patrilineal heritage in their Eurasian context. Am J Hum Genet. 2008;82:236–250.
Kushniarevich A, Utevska O, Chuhryaeva M, Agdzhoyan A, Dibirova K, Uktveryte I et al. Genetic heritage of the balto-slavic speaking populations: A synthesis of autosomal, mitochondrial and Y-chromosomal data. PLoS One. 2015; 10. https://doi.org/10.1371/journal.pone.0135820.
Jones ER, Zarina G, Moiseyev V, Lightfoot E, Nigst PR, Manica A, et al. The Neolithic transition in the Baltic was not driven by admixture with early European farmers. Curr Biol. 2017;27:576–582.
Lamnidis TC, Majander K, Jeong C, Salmela E, Wessman A, Moiseyev V et al. Ancient Fennoscandian genomes reveal origin and spread of Siberian ancestry in Europe. Nat Commun. 2018; 9. https://doi.org/10.1038/s41467-018-07483-5.
Saag L, Laneman M, Varul L, Malve M, Valk H, Razzak MA, et al. The arrival of Siberian ancestry connecting the Eastern Baltic to Uralic speakers further East. Curr Biol. 2019;29:1701–1711.e16.
Myres NM, Rootsi S, Lin AA, Järve M, King RJ, Kutuev I, et al. A major Y-chromosome haplogroup R1b Holocene era founder effect in Central and Western Europe. Eur J Hum Genet. 2011;19:95–101.
Underhill PA, Poznik GD, Rootsi S, Järve M, Lin AA, Wang J, et al. The phylogenetic and geographic structure of Y-chromosome haplogroup R1a. Eur J Hum Genet. 2015;23:124–131.
Ilumäe AM, Reidla M, Chukhryaeva M, Järve M, Post H, Karmin M, et al. Human Y chromosome haplogroup N: a non-trivial time-resolved phylogeography that cuts across language families. Am J Hum Genet. 2016;99:163–173.
Karmin M, Saag L, Vicente M, Wilson Sayres MA, Järve M, Gerst Talas U, et al. A recent bottleneck of Y chromosome diversity coincides with a global change in culture. Genome Res. 2015;25:459–466.
Batini C, Hallast P, Zadik D, Delser PM, Benazzo A, Ghirotto S et al. Large-scale recent expansion of European patrilineages shown by population resequencing. Nat Commun. 2015; 6. https://doi.org/10.1038/ncomms8152.
Rootsi S, Myres NM, Lin AA, Järve M, King RJ, Kutuev I, et al. Distinguishing the co-ancestries of haplogroup G Y-chromosomes in the populations of Europe and the Caucasus. Eur J Hum Genet. 2012;20:1275–1282.
Cruciani F, La Fratta R, Trombetta B, Santolamazza P, Sellitto D, Colomb EB, et al. Tracing past human male movements in northern/eastern Africa and western Eurasia: New clues from Y-chromosomal haplogroups E-M78 and J-M12. Mol Biol Evol. 2007;24:1300–1311.
Karafet TM, Osipova LP, Gubina MA, Posukh OL, Zegura SL, Hammer MF. High levels of Y-chromosome differentiation among native Siberian populations and the genetic signature of a boreal Hunter-Gatherer way of life. Hum Biol. 2002;74:761–789.
Dulik MC, Zhadanov SI, Osipova LP, Askapuli A, Gau L, Gokcumen O, et al. Mitochondrial DNA and Y chromosome variation provides evidence for a recent common ancestry between Native Americans and indigenous Altaians. Am J Hum Genet. 2012;90:229–246.
Hallast P, Batini C, Zadik D, Delser PM, Wetton JH, Arroyo-Pardo E, et al. The Y-chromosome tree bursts into leaf: 13,000 high-confidence SNPs covering the majority of known clades. Mol Biol Evol. 2014;32:661–673.
Poznik GD, Xue Y, Mendez FL, Willems TF, Massaia A, Wilson Sayres MA, et al. Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences. Nat Genet. 2016;48:593–599.
Leitsalu L, Haller T, Esko T, Tammesoo ML, Alavere H, Snieder H, et al. Cohort profile: Estonian biobank of the Estonian genome center, university of Tartu. Int J Epidemiol. 2015;44:1137–1147.
Mitt M, Kals M, Pärn K, Gabriel SB, Lander ES, Palotie A, et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur J Hum Genet. 2017;25:869–876.
Ameur A, Dahlberg J, Olason P, Vezzi F, Karlsson R, Martin M, et al. SweGen: A whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur J Hum Genet. 2017;25:1253–1260.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013. https://arxiv.org/abs/1303.3997.
Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Auwera GA Van der et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2017. https://doi.org/10.1101/201178.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079.
Poznik GD. Identifying Y-chromosome haplogroups in arbitrarily large samples of sequenced or genotyped men. bioRxiv. 2016. https://doi.org/10.1101/088716.
Severson AL, Shortt JA, Mendez FL, Wojcik GL, Bustamante CD, Gignoux CR. SNAPPY: single nucleotide assignment of phylogenetic parameters on the Y chromosome. bioRxiv. 2018. https://doi.org/10.1101/454736.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973.
Lemey P, Rambaut A, Welch JJ, Suchard MA. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol. 2010;27:1877–1885.
Pybus OG, Suchard MA, Lemey P, Bernardin FJ, Rambaut A, Crawford FW, et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc Natl Acad Sci USA. 2012;109:15066–15071.
Sahakyan H, Margaryan A, Saag L, Karmin M, Bahmanimehr A, Parik J. et al. Origin and diffusion of human Y chromosome haplogroup J1‑M267. Sci Rep. 2021; https://doi.org/10.1038/s41598-021-85883-2.
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:1–5.
Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO, et al. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst Biol. 2012;61:170–173.
Bielejec F, Baele G, Vrancken B, Suchard MA, Rambaut A, Lemey P. SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol Biol Evol. 2016;33:2167–2169.
Trombetta B, D’Atanasio E, Massaia A, Myres NM, Scozzari R, Cruciani F, et al. Regional differences in the accumulation of SNPs on the male-specific portion of the human y chromosome replicate autosomal patterns: Implications for genetic dating. PLoS One. 2015;10:1–18.
Finocchio A, Trombetta B, Messina F, D’Atanasio E, Akar N, Loutradis A, et al. A finely resolved phylogeny of y chromosome Hg J illuminates the processes of Phoenician and Greek colonizations in the Mediterranean. Sci Rep. 2018;8:3–11.
Zalloua PA, Platt DE, El Sibai M, Khalife J, Makhoul N, Haber M, et al. Identifying genetic traces of historical expansions: phoenician footprints in the mediterranean. Am J Hum Genet. 2008;83:633–642.
Jones ER, Gonzalez-Fortes G, Connell S, Siska V, Eriksson A, Martiniano R et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat Commun. 2015; 6. https://doi.org/10.1038/ncomms9912.
Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503.
Yunusbayev B, Metspalu M, Ja M, Kutuev I, Rootsi S, Metspalu E, et al. The Caucasus as an asymmetric semipermeable barrier to ancient human migrations research article. Mol Biol Evol. 2012;29:359–365.
Zegura SL, Karafet TM, Zhivotovsky LA, Hammer MF. High-resolution SNPs and microsatellite haplotypes point to a single, recent entry of Native American Y chromosomes into the Americas. Mol Biol Evol. 2004;21:164–175.
Kittles RA, Bergen AW, Urbanek M, Virkkunen M, Linnoila M, Goldman D, et al. Autosomal, mitochondrial, and Y chromosome DNA variation in Finland: evidence for a male-specific bottleneck. Am J Phys Anthropol. 1999;108:381–399.
Martin AR, Karczewski KJ, Kerminen S, Kurki MI, Sarin AP, Artomov M, et al. Haplotype sharing provides insights into fine-scale population history and disease in Finland. Am J Hum Genet. 2018;102:760–775.
Fu Q, Posth C, Hajdinjak M, Petr M, Mallick S, Fernandes D, et al. The genetic history of Ice Age Europe. Nature. 2016;534:200–205.
Mathieson I, Alpaslan-Roodenberg S, Posth C, Szécsényi-Nagy A, Rohland N, Mallick S, et al. The genomic history of southeastern Europe. Nature. 2018;555:197–203.
Grugni V, Raveane A, Ongaro L, Battaglia V, Trombetta B, Colombo G, et al. Analysis of the human Y-chromosome haplogroup Q characterizes ancient population movements in Eurasia and the Americas. BMC Biol. 2019;17:1–14.
Raghavan M, Skoglund P, Graf KE, Metspalu M, Albrechtsen A, Moltke I et al. Upper palaeolithic Siberian genome reveals dual ancestry of native Americans. Nature. 2014; 505. https://doi.org/10.1038/nature12736.
Marchi N, Winkelbach L, Schulz I, Brami M, Hofmanová Z. The mixed genetic origin of the first farmers of Europe. bioRxiv. 2020. https://doi.org/10.1101/2020.11.23.394502.
Funding
This work was supported by institutional research funding IUT24-1 of the Estonian Ministry of Education and Research, Estonian Research Council grants PRG243, PRG1071 and project No. 2014-2020.4.01.16-0024 (MOBTT53) granted by the European Regional Development Fund, European Union Horizon 2020 research and innovation programme (grant No. 810645), European Regional Development Fund project no. MOBEC008. A-MI is supported by Finnish Academy (DIGIHUM project URKO, decision number 329257). High-coverage genome data for five 1000 Genomes samples were generated at the New York Genome Center with funds provided by NHGRI Grant 3UM1HG008901-03S1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
DMB and CB declare stock ownership at Gene by Gene, Ltd. LFS in an employee of Gene by Gene.
Ethics approval
All donors have provided informed consent and all experiments were performed in accordance with the relevant guidelines and regulations of collaborating institutions. Access to genetic data in Estonian Biobank was approved by the Research Ethics Committee of the University of Tartu (permission number 1.1.-12/659 granted by the Research Ethics Committee of the University of Tartu, Estonia). The chrY sequences included from customers of the commercial personal genetic testing service were only from individuals who had provided informed consent for the use of their data in scientific research and for publication in aggregated form. The list of IDs along with additional sample information is presented in Supplementary Table S1.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ilumäe, AM., Post, H., Flores, R. et al. Phylogenetic history of patrilineages rare in northern and eastern Europe from large-scale re-sequencing of human Y-chromosomes. Eur J Hum Genet 29, 1510–1519 (2021). https://doi.org/10.1038/s41431-021-00897-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00897-8
