
Abstract
Pathogenic variants in the SRCAP (SNF2-related CREBBP activator protein) gene, which encodes a chromatin-remodeling ATPase, cause neurodevelopmental disorders including Floating Harbor syndrome (FLHS). Here, we report the discovery of a de novo transposon insertion in SRCAP exon 13 from trio genome sequencing in a 28-year-old female with failure to thrive, developmental delay, mood disorder and seizure disorder. The insertion was a full-length (~2.8 kb), antisense-oriented SVA insertion relative to the SRCAP transcript, bearing a 5’ transduction and hallmarks of target-primed reverse transcription. The 20-bp 5’ transduction allowed us to trace the source SVA element to an intron of a long non-coding RNA on chromosome 12, which is highly expressed in testis. RNA sequencing and qRT-PCR confirmed significant depletion of SRCAP expression and low-level exon skipping in the proband. This case highlights a novel disease-causing structural variant and the importance of transposon analysis in a clinical diagnostic setting.
Similar content being viewed by others


Introduction
Floating-Harbor syndrome (FLHS [OMIM #136140]) is an ultra-rare, autosomal dominant, neurodevelopmental disorder caused by truncating variants in exons 33–34 of the SRCAP gene [1,2,3,4,5]. It is characterized by short stature, delayed bone age, characteristic facial appearance, and expressive and receptive language delays [5, 6]. Recently, a novel SRCAP-related neurodevelopmental disorder (NDD) was described, which is caused by SRCAP truncating variants outside of the FLHS locus (exons 33–34) [7]. Despite some phenotypic similarity, these individuals with a NDD lacked key features of FLHS and were more likely to have autism, psychiatric and behavioral problems, and hypotonia.
SRCAP encodes a SNF2-related CREBBP activator protein, which increases gene transcription by incorporating histone variant H2A.Z into nucleosomes [8, 9]. SRCAP contains a N-terminal Helicase/SANT-associated domain, a SNF2-like ATPase, a CPB binding domain, and three AT-hook domains at C-terminal [1]. Interestingly, all reported pathogenic mutations are truncating mutations, and most are located within the last two exons, upstream of the AT-hook motifs [1,2,3,4,5]. Beyond the recent report on DNA methylation signature [7], the consequences of variants located outside of the FLHS locus are still poorly understood.
Advances in sequencing technologies have accelerated clinical discovery of causal variants in various Mendelian disorders. However, a large fraction of such cases remain undiagnosed, in part because conventional analytic methods fail to capture the full spectrum of genomic variations such as transposable elements (TEs). TEs are large DNA elements (several hundreds to kilobases in length) that mobilize in the human genome. TE polymorphisms account for ~30% of structural variation discovered from short-read whole-genome sequencing (WGS) in the human population [10]. Currently, three TE families (LINE-1, Alu, and SVA) can retrotranspose in a copy-and-paste manner via RNA intermediates, with estimated rates in 1/20~200 births [11, 12]. While TE insertions could be disease-causing by altering RNA expression and splicing [13,14,15], they are mostly not evaluated in clinical sequencing analysis, leading to missed diagnoses in some patients.
Here, we identified a de novo exonic SVA insertion in the SRCAP exon 13 in a proband with a NDD. RNA sequencing and qRT-PCR demonstrated significantly decreased abundance of SRCAP in the proband, confirming the pathogenicity of this insertion.
Results
Clinical history and presentation
Following an uncomplicated pregnancy, the proband was born via spontaneous vaginal delivery at 40 weeks gestation with normal Apgar scores. She weighed 2.86 kg and measured 49.5 cm long with normal head circumference. In the first year of life, she was diagnosed with failure to thrive, hypotonia, and developmental delay. From age five to seven, she experienced several afebrile seizures, which were accompanied by a normal brain MRI and EEGs suggestive of a focal seizure disorder. Her seizures recurred at 16 years of age, became intractable, and included myoclonic and generalized tonic-clonic type. The proband was also diagnosed with autism, anxiety, tic, and mood disorders. Additional investigation revealed uterine agenesis and single fused pelvic kidney. The proband had myopia and unique facial features including bitemporal narrowing, large mouth, thick lips, prognathia, high-arched palate with crowded teeth and hypoplastic enamel. Most fingers had fetal fat pads and feet were flat with bilateral 2–3 toe cutaneous syndactyly.
Early metabolic studies were suspicious for short-chain acyl-coA dehydrogenase deficiency due to elevated ethylmalonate; however, no pathogenic variants were found by ACADS gene sequencing beyond a homozygous missense polymorphism (c.625 G > A; p.G209S) classified as benign/likely benign (ClinVar ID: 3831) according to ACMG-AMP guidelines [16, 17]. Additional genetic testing included chromosomal microarray, fragile X, FISH testing for 7q11.23, mitochondrial genome, and 70-gene epilepsy panel; all were considered non-diagnostic. The proband and her family were enrolled, but research trio exome sequencing was unrevealing. Given these negative findings, WGS was then completed and analyzed as described below.
A de novo exonic SVA insertion identified by WGS
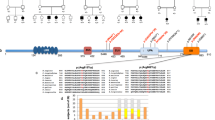
Evaluation of single nucleotide variants (SNVs) and indels with the genome analysis toolkit (GATK) failed to identify pathogenic mutations in any disease-causing genes, including SRCAP. However, application of GATK-SV to discover structural variants revealed a putative de novo SVA insertion in SRCAP exon 13 in the proband. Further in silico review at the predicted integration site showed multiple features of target-primed reverse transcription (TPRT)-mediated retrotransposition [18]: (1) soft-clipped reads or junction-spanning reads were fused either to unmapped repeat sequences on one end or unmapped poly-T sequences on the other end, which indicate the beginning and ending (poly-A tails) of the insertion, respectively. (2) discordant read-pairs, in which one of the paired-end reads was mapped to the flanking genomic sequence of the breakpoints, whereas the other was aligned to a TE at unexpected distance or orientation. (3) breakpoints offset by target site duplication (TSD) (Fig. 1A).

A The Integrative Genomics Viewer (IGV) plot showed a de novo insertion in exon 13 of the SRCAP gene in the proband and its absence in the parents. This non-reference insertion was supported by soft-clipped reads (colored bases) that span the insertion breakpoints (red dashed lines) with clear hallmarks of TPRT-mediated retrotransposition, including target site duplication (TSD) and poly-A tail. B, C Representative gel images of full-length PCR and 5’ junction PCR validations for the de novo exonic insertion. gDNA samples were extracted from blood samples from the trio and two unrelated individuals. D Confirming the presence/absence of the insertion in the proband and four unaffected siblings using 5’ junction PCR. gDNA from siblings were extracted from saliva samples. Black asterisks indicated bands with target size.
To experimentally validate this insertion, we performed full-length PCR, where primers annealed to the flanking sequences on both sides of the insertion. This long-range PCR supported a de novo ~3-kb insertion in the proband (Fig. 1B). We also confirmed the presence of the insertion using both 5′ and 3′ junction PCR assays (Fig. 1C and Fig. S1) and confirmed the absence of the insertion in four unaffected siblings (Fig. 1D).
Variant characterization
To characterize the insertional mechanism, we fully resolved the exonic insertion at single-base resolution using both Sanger sequencing on junction amplicons and next-generation sequencing on sonicated fragments of full-length amplicon (Fig. S2A and Supplementary File 1). This revealed a full-length SVA insertion landed in an antisense orientation in exon 13, accompanied by a 15-bp TSD, a polyA tail longer than 70-bp, and an L1-endonuclease cleavage site at 5′–GC/AAGA–3′ (Fig. 2A). These hallmarks confirmed a L1-mediated retrotransposition via TPRT. This variant was represented as NC_000016.10: g.30712369_30712370ins[SVA;30712355_30712369].

A The schematic diagram showed the fully resolved exonic insertion at single base resolution. It was an ~2.8 kb anti-sense SVA insertion with 5′ transduction. It carried a 15-bp TSD, > 70 bp poly-A tail, and L1 endonuclease cleavage site. B Multiple sequence alignment of the 5′ end of the de novo SVA insertion in the proband (the top row) and other SVA subfamilies (from Repbase consensus sequences). The de novo SVA insertion carried a 20-bp 5′ transduction sequence and an alternative hexamer variant ((CCCTCT)2 CCCGTCT)n. C The source locus traced by the 5′ transduction sequence of the insertion was identified in intron 1 of a lncRNA (RefSeq: NR_135014) region. The IGV plot showed the chimeric reads spanning the 5′ junction of SVA-genome where soft-clipped sequences aligned to exon 13 of the SRCAP gene. The additional SVA insertion led to increased depth of coverage at the source locus. D The exonic SVA insertion caused a significant decrease of SRCAP gene expression in the proband. SRCAP TPM (Transcripts Per Million) levels are shown for the blood RNA-seq of the proband compared to GTEx RNA-seq of fibroblasts, lymphoblastoid cell lines (LCLs), and whole blood from healthy donors. E SYBR green qRT-PCR assay demonstrated reduced SRCAP RNA abundance in the proband compared to two unrelated and unaffected controls. Experiments were performed in triplicate.
Unlike the canonical 5′ junction of SVA insertion beginning with hexameric repeat sequences (CCCTCT) [19], this insertion carried a 20-bp 5ʹ transduction, which allowed for tracing back to the source locus (Fig. 2B, C). This 20-bp sequence uniquely mapped to an uncharacterized lncRNA locus (RefSeq: NR_135014 — SNRPF divergent transcript, transcript variant 1) that was specifically expressed in testis. A recent study reported a similar insertion in the exon of MSH6 gene, which was derived from the same locus (chr12: 96233959–96236309 [hg19]) [20]. Both SVA insertions bear the same CCCTCT hexamer variant ((CCCTCT)2 CCCGTCT)n (Fig. S2B)), which suggests that this donor SVA is active in the human genome.
Confirmation of pathogenicity
To understand the consequence of the exonic SVA insertion, we first performed in silico analysis on the triplet reading frame. Although it was hard to precisely determine the length of polyA tail of the anti-sense insertion, all possible open reading frames contained early stop codon, truncating the SRCAP gene and rendering it nonfunctional (Supplementary File 2). We then investigated the effect of this insertion on RNA using blood RNA-seq data. The proband showed a significant depletion of SRCAP expression compared to controls in the Genotype-Tissue Expression (GTEx) dataset (Fig. 2D). The SYBR green qRT-PCR assay further confirmed the decreased SRCAP gene expression in the proband compared to unaffected controls (Fig. 2E).
Discussion
In this study, we identified a de novo full-length SVA insertion in exon 13 of the SRCAP gene in a proband with a NDD. RNA-seq and qPCR assay confirmed the pathogenicity of the insertion. As SRCAP variants typically cause FLHS, this proband had a partial phenotypic overlap (speech delay, genitourinary malformation, seizures, and dental issues) but lacked the short stature and delayed bone age historically observed in FLHS patients (Table 1). While most reported FLHS cases are caused by truncating variants in exons 33–34, a recent publication reported 33 cases with variants outside the FLHS locus causing a related NDD [7]. Like our proband, most patients with variants proximal to the FLHS locus had autism and mood/behavioral problems and also lacked the short stature and delayed bone age typical to FLHS (Table 1). Our case supports a broader phenotypic spectrum and further expands the type and location of SRCAP mutations causing NDDs. To our knowledge, this is the first NDD case caused by a transposon insertion in the SRCAP gene.
To test whether this antisense insertion bears a novel splicing site and leads to alternative RNA splicing, we reconstructed a patient customized genome and re-aligned RNA-seq reads to the revised reference. Although splicing analysis predicted several novel splicing signals, no splicing events were observed within this ~3-kb exonic insertion (Supplementary File 3). Notably, low-level exon 13 skipping was observed from RT-PCR (Fig. S3A). As a functional validation at protein level, two commercial SRCAP antibodies were tested for western blot, but neither produced a specific band with expected molecular weight (data not shown).
SVA retrotransposition produces 5′ transduced sequences via alternative transcription start sites at the rates of 9.17% (220 out of 2398) for reference insertions [21] and 8.33% (45 out of 540) for non-reference insertions [22]. Utilizing the unique 20-bp 5′ transduction, this SVA insertion was traced to a source locus in an intron of a lncRNA that is almost exclusively expressed in testis (Fig. S3B). This suggests the possible origin of the de novo insertion in the sperm where the SVA locus was highly active for transcription. Since there are no informative heterozygous SNVs or other types of variants near the insertion, we were not able to phase the SVA insertion onto parental haplotypes. Because both older and younger siblings do not carry the same insertion event, we can speculate that the retrotransposition likely occurred in a very small proportion of sperm cells or even a single sperm that eventually gave rise to the phenotype in the proband. Accurate mosaicism could not be measured due to a lack of the paternal sperm sample.
Genomic sequencing has revolutionized our ability to discover causal genetic variants, however, conventional analysis restricted to SNVs and small indels fail to capture the spectrum of mutational mechanisms accessible to clinical sequencing data. Our case highlights the importance of detailed computational and functional characterization of TE insertions, which represent an important and underexplored source of pathogenic variants in clinical genomic studies. To understand the prevalence and contribution of such pathogenic insertions to genetic diseases, systematic TE profiling needs to be performed on genomic sequencing data from large patient cohorts.
Data availability
Sequencing data supporting the findings of this study are available upon reasonable request.
References
Hood RL, Lines MA, Nikkel SM, Schwartzentruber J, Beaulieu C, Nowaczyk MJM, et al. Mutations in SRCAP, encoding SNF2-related CREBBP activator protein, cause Floating-Harbor syndrome. Am J Hum Genet. 2012;90:308–13. https://doi.org/10.1016/j.ajhg.2011.12.001
Kehrer M, Beckmann A, Wyduba J, Finckh U, Dufke A, Gaiser U, et al. Floating-Harbor syndrome: SRCAP mutations are not restricted to exon 34. Clin Genet. 2014;85:498–9. https://doi.org/10.1111/cge.12199
Seifert W, Meinecke P, Krüger G, Rossier E, Heinritz W, Wüsthof A, et al. Expanded spectrum of exon 33 and 34 mutations in SRCAP and follow-up in patients with Floating-Harbor syndrome. BMC Med Genet. 2014;15:127. https://doi.org/10.1186/s12881-014-0127-0
Goff CL, Mahaut C, Bottani A, Doray B, Goldenberg A, Moncla A, et al. Not all floating-harbor syndrome cases are due to mutations in exon 34 of SRCAP. Hum Mutat2013;34:88–92. https://doi.org/10.1002/humu.22216
Nikkel SM, Dauber A, de Munnik S, Connolly M, Hood RL, Caluseriu O, et al. The phenotype of Floating-Harbor syndrome: clinical characterization of 52 individuals with mutations in exon 34 of SRCAP. Orphanet J Rare Dis. 2013;8:63. https://doi.org/10.1186/1750-1172-8-63
Robinson PL, Shohat M, Winter RM, Conte WJ, Gordon-Nesbitt D, Feingold M, et al. A unique association of short stature, dysmorphic features, and speech impairment (Floating-harbor syndrome). J Pediatrics. 1988;113:703–6. https://doi.org/10.1016/s0022-3476(88)80384-6
Rots D, Chater-Diehl E, Dingemans AJM, Goodman SJ, Siu MT, Cytrynbaum C, et al. Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am J Hum Genet. 2021;108:1053–68. https://doi.org/10.1016/j.ajhg.2021.04.008
Ruhl DD, Jin J, Cai Y, Swanson S, Florens L, Washburn MP, et al. Purification of a human SRCAP complex that remodels chromatin by incorporating the histone variant H2A.Z into nucleosomes. Biochemistry. 2006;45:5671–7. https://doi.org/10.1021/bi060043d
Wong MM, Cox LK, Chrivia JC. The chromatin remodeling protein, SRCAP, is critical for deposition of the histone variant H2A.Z at promoters. J Biol Chem 2007;282:26132–9. https://doi.org/10.1074/jbc.M703418200
Collins RL, Brand H, Karczewski KJ, Zhao X, Alfoldi J, Francioli LC, et al. A structural variation reference for medical and population genetics. Nature. 2020;581:444–51. https://doi.org/10.1038/s41586-020-2287-8
Borges-Monroy R, Chu C, Dias C, Choi J, Lee S, Gao Y, et al. Whole-genome analysis reveals the contribution of non-coding de novo transposon insertions to autism spectrum disorder. Mob DNA. 2021;12:28. https://doi.org/10.1186/s13100-021-00256-w
Feusier J, Watkins WS, Thomas J, Farrell A, Witherspoon DJ, Baird L, et al. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019;29:1567–77. https://doi.org/10.1101/gr.247965.118
Hancks DC, Kazazian HH Jr. Roles for retrotransposon insertions in human disease. Mob DNA. 2016;7:9.https://doi.org/10.1186/s13100-016-0065-9
Aneichyk T, Hendriks WT, Yadav R, Shin D, Gao D, Vaine CA, et al. Dissecting the causal mechanism of X-Linked Dystonia-Parkinsonism by integrating genome and transcriptome assembly. Cell. 2018;172:897–909.e821. https://doi.org/10.1016/j.cell.2018.02.011
Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019. https://doi.org/10.1056/NEJMoa1813279
Juhl Corydon M, Vockley J, Rinaldo P, James Rhead W, Kjeldsen M, Winter V, et al. Role of common gene variations in the molecular pathogenesis of short-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 2001;49:18–23. https://doi.org/10.1203/00006450-200101000-00008
Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med2017;19:1105–17. https://doi.org/10.1038/gim.2017.37
Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–910.
Hancks DC, Kazazian HH Jr. SVA retrotransposons: evolution and genetic instability. Semin Cancer Biol. 2010;20:234–45. https://doi.org/10.1016/j.semcancer.2010.04.001
Yamamoto G, Miyabe I, Tanaka K, Kakuta M, Watanabe M, Kawakami S, et al. SVA retrotransposon insertion in exon of MMR genes results in aberrant RNA splicing and causes Lynch syndrome. Eur J Hum Genet. 2020. https://doi.org/10.1038/s41431-020-00779-5
Damert A, Raiz J, Horn AV, Lower J, Wang H, Xing J, et al. 5′-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res. 2009;19:1992–2008. https://doi.org/10.1101/gr.093435.109
Ebert P, et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021. https://doi.org/10.1126/science.abf7117
Acknowledgements
We thank the patient and family for their participation and support. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal.
Funding
EAL was supported by the NIH (DP2 AG0724), Suh Kyungbae Foundation, Charles H. Hood foundation, and the Allen Discovery Center program of the Paul G. Allen Family Foundation. BZ was supported by the Manton Center Pilot Project Award and Rare Disease Research Fellowship. MHW is supported by an Early Career Award from the Thrasher Research Fund. PBA’s research work is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, R01AR068429-01, National Human Genome Research Institute, 1R01HG011798-01A1 and “Because of Bella” foundation. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141, as well as the National Institutes of Health grants MH115957 and HD081256. Sanger sequencing was performed by the Boston Children’s Hospital IDDRC Molecular Genetics Core Facility supported by NIH award U54HD090255 from the National Institute of Child Health and Human Development.
Author information
Authors and Affiliations
Contributions
BZ characterized the insertion variant, designed and analyzed experimental validation assays, performed RNA-seq analysis, qRT-PCR, and splicing analysis for pathogenicity confirmation, made all figures and supplementary materials, and wrote the manuscript. JAM coordinated samples, clinical record, and sequencing data collections, made Table 1 for clinical presentation, and wrote the manuscript. JL performed all experimental validation assays, and edited the manuscript. GTB enrolled the patient. MHW helped analyze genome sequencing results that identified the potential variant, and edited the manuscript. XZ, HB, and MT performed SV data processing, identified the potential variant, and edited the manuscript. EAL and PBA directed the overall research and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
PBA is on the Scientific Advisory Board of Illumina, Inc. and GeneDx, Inc. Other authors declare no competing interests.
Ethics approval and consent to participate
The proband and her family were enrolled into the Gene Discovery Core at The Manton Center for Orphan Disease Research approved by the IRB (10-02-0053) at Boston Children’s Hospital. Written informed consents were obtained from the patient and family members.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhao, B., Madden, J.A., Lin, J. et al. A neurodevelopmental disorder caused by a novel de novo SVA insertion in exon 13 of the SRCAP gene. Eur J Hum Genet 30, 1083–1087 (2022). https://doi.org/10.1038/s41431-022-01137-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01137-3
This article is cited by
-
Combined exome and whole transcriptome sequencing identifies a de novo intronic SRCAP variant causing DEHMBA syndrome with severe sleep disorder
Journal of Human Genetics (2024)
-
PhenoSV: interpretable phenotype-aware model for the prioritization of genes affected by structural variants
Nature Communications (2023)
-
2022: the year that was in the European Journal of Human Genetics
European Journal of Human Genetics (2023)
-
Guidelines, guidelines everywhere—and still I’m not sure what to do
European Journal of Human Genetics (2022)
