
Abstract
Dystrophin deficiency due to genetic mutations causes cardiac abnormalities in Duchenne’s muscular dystrophy. Dystrophin is also shown to be downregulated in conventional failing hearts. Whether restoration of dystrophin expression possesses any therapeutic potential for conventional heart failure (HF) remains to be examined. HF mouse model was generated by transverse aortic constriction (TAC). In vivo activation of dystrophin transcription was achieved by tail-vein injection of adeno-associated virus 9 carrying CRISPR/dCas system for dystrophin. We found that activation of dystrophin expression in TAC mice significantly reduced the susceptibility to arrhythmia of TAC mice and the mortality rate. We further demonstrated that over-expression of dystrophin increased cardiac conduction of hearts in TAC mice by optical mapping evaluation. Activation of dystrophin expression also increased peak sodium current in isolated ventricular myocytes from hearts of TAC mice as recorded by the patch-clamp technique. Immunoblotting and immunofluorescence showed that increased dystrophin transcription restored the membrane distribution of Nav1.5 in the hearts of TAC mice. In summary, correction of dystrophin downregulation by the CRISPR-dCas9 system reduced the susceptibility to arrhythmia of conventional HF mice through restoring Nav1.5 membrane distribution. This study paved the way to develop a new therapeutic strategy for HF-related ventricular arrhythmia.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
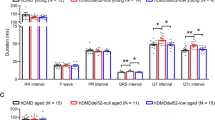


Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28.
Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–17.
Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000;150:1209–14.
Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–40.
Koenig X, Ebner J, Hilber K. Voltage-dependent sarcolemmal ion channel abnormalities in the dystrophin-deficient heart. Int J Mol Sci. 2018;19:3296.
Min YL, Bassel-Duby R, Olson EN. CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med. 2019;70:239–55.
Yoshida H, Takahashi M, Koshimizu M, Tanonaka K, Oikawa R, Toyo-oka T, et al. Decrease in sarcoglycans and dystrophin in failing heart following acute myocardial infarction. Cardiovasc Res. 2003;59:419–27.
Prado FP, Dos Santos DO, Blefari V, Silva CA, Machado J, Kettelhut IDC, et al. Early dystrophin loss is coincident with the transition of compensated cardiac hypertrophy to heart failure. PLoS One. 2017;12:e0189469.
Kawada T, Masui F, Tezuka A, Ebisawa T, Kumagai H, Nakazawa M, et al. A novel scheme of dystrophin disruption for the progression of advanced heart failure. Biochim Biophys Acta. 2005;1751:73–81.
Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, et al. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation. 2003;107:2920–5.
Schoger E, Carroll KJ, Iyer LM, McAnally JR, Tan W, Liu N, et al. CRISPR-mediated activation of endogenous gene expression in the postnatal heart. Circ Res. 2020;126:6–24.
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–51.
Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–63.
Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10:977–9.
Li DS, Xue GL, Yang JM, Li CZ, Zhang RX, Tian T, et al. Knockout of interleukin-17A diminishes ventricular arrhythmia susceptibility in diabetic mice via inhibiting NF-kappaB-mediated electrical remodeling. Acta Pharmacol Sin. 2022;43:307–15.
Zhang Y, Sun L, Xuan L, Pan Z, Hu X, Liu H, et al. Long non-coding RNA CCRR controls cardiac conduction via regulating intercellular coupling. Nat Commun. 2018;9:4176.
Zhan G, Wang F, Ding YQ, Li XH, Li YX, Zhao ZR, et al. Rutaecarpine targets hERG channels and participates in regulating electrophysiological properties leading to ventricular arrhythmia. J Cell Mol Med. 2021;25:4938–49.
Wang Y, Hill JA. Electrophysiological remodeling in heart failure. J Mol Cell Cardiol. 2010;48:619–32.
Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431–88.
Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, et al. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304.
Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244.
Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91.
Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017;8:14454.
Armoundas AA, Wu R, Juang G, Marban E, Tomaselli GF. Electrical and structural remodeling of the failing ventricle. Pharmacol Ther. 2001;92:213–30.
Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C, et al. Mechanisms contributing to cardiac remodelling. Clin Sci (Lond). 2017;131:2319–45.
Cutler MJ, Jeyaraj D, Rosenbaum DS. Cardiac electrical remodeling in health and disease. Trends Pharmacol Sci. 2011;32:174–80.
Han F, Lu YM, Hasegawa H, Kanai H, Hachimura E, Shirasaki Y. et al. Inhibition of dystrophin breakdown and endothelial nitric-oxide synthase uncoupling accounts for cytoprotection by 3-[2-[4-(3-chloro-2-methylphenyl)-1-piperazinyl]ethyl]-5,6-dimethoxy-1-(4-imidazo lylmethyl)-1H-indazole dihydrochloride 3.5 hydrate (DY-9760e) in left ventricular hypertrophied mice. J Pharmacol Exp Ther. 2010;332:421–8.
Moretti A, Fonteyne L, Giesert F, Hoppmann P, Meier AB, Bozoglu T, et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med. 2020;26:207–14.
Luo L, Ning F, Du Y, Song B, Yang D, Salvage SC, et al. Calcium-dependent Nedd4-2 upregulation mediates degradation of the cardiac sodium channel Nav1.5: implications for heart failure. Acta Physiol (Oxf). 2017;221:44–58.
Shy D, Gillet L, Ogrodnik J, Albesa M, Verkerk AO, Wolswinkel R, et al. PDZ domain-binding motif regulates cardiomyocyte compartment-specific NaV1.5 channel expression and function. Circulation. 2014;130:147–60.
Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, et al. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407–14.
Rougier JS, Gavillet B, Abriel H. Proteasome inhibitor (MG132) rescues Nav1.5 protein content and the cardiac sodium current in dystrophin-deficient mdx (5cv) mice. Front Physiol. 2013;4:51.
Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, et al. dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol Ther. 2020;28:235–53.
Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, et al. Rescue of Fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172:979–92.e6.
Funding
This work was supported by National Natural Science Foundation of China (82070344, 81870295 to ZP) and HMU Marshal Initiative Funding (HMUMIF-21017 to ZP).
Author information
Authors and Affiliations
Contributions
RZ, JL, and GX performed experiments, analyzed data, and prepared the manuscript. JY, DL, TT, XZ, and KG helped perform experiments and collect data. ZP designed the project, oversaw the experiments, and prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Compliance with ethical standards.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, R., Liu, J., Xue, G. et al. Forced activation of dystrophin transcription by CRISPR/dCas9 reduced arrhythmia susceptibility via restoring membrane Nav1.5 distribution. Gene Ther 30, 142–149 (2023). https://doi.org/10.1038/s41434-022-00348-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-022-00348-z
