
Abstract
Purpose
The Ehlers–Danlos syndromes (EDS) are a group of rare inherited connective tissue disorders. Vascular EDS (vEDS) is caused by pathogenic variants in COL3A1, most frequently glycine substitutions. We describe the phenotype of the largest series of vEDS patients with glutamic acid to lysine substitutions (Glu>Lys) in COL3A1, which were all previously considered to be variants of unknown significance.
Methods
Clinical and molecular data for seven families with three different Glu>Lys substitutions in COL3A1 were analyzed.
Results
These Glu>Lys variants were reclassified from variants of unknown significance to either pathogenic or likely pathogenic in accordance with American College of Medical Genetics and Genomics guidelines. All individuals with these atypical variants exhibited skin hyperextensibility as seen in individuals with classical EDS and classical-like EDS and evidence of tissue fragility as seen in individuals with vEDS.
Conclusion
The clinical data demonstrate the overlap between the different EDS subtypes and underline the importance of next-generation sequencing gene panel analysis. The three different Glu>Lys variants point toward a new variant type in COL3A1 causative of vEDS, which has consistent clinical features. This is important knowledge for COL3A1 variant interpretation. Further follow-up data are required to establish the severity of tissue fragility complications compared with patients with other recognized molecular causes of vEDS.
Similar content being viewed by others

INTRODUCTION
Ehlers–Danlos syndromes (EDS) are a group of clinically and genetically heterogeneous connective tissue disorders comprising 13 subtypes.1 Their overlapping features include joint hypermobility, skin and vascular fragility, and generalized connective tissue friability2 but the variability and clinical overlap can make the clinical diagnosis of a specific EDS subtype challenging. Establishing the correct clinical and molecular diagnosis has important implications for the management of the condition, including surveillance and treatment as well as accurate genetic counseling.
Vascular EDS (vEDS) is caused by pathogenic variants in COL3A1,3 which encodes the pro-α1 chain of type III collagen (OMIM 130050) and occasionally specific COL1A1 variants may lead to a vEDS phenotype. Estimated prevalence is around 1 in 90,000.2 The minimal criteria suggestive of vEDS according to the 2017 nosology are a family history of the disorder with a documented causative variant in COL3A1, a clinical history of arterial rupture, dissection or aneurysm under the age of 40, unexplained sigmoid colon rupture, uterine rupture, or a carotid-cavernous fistula. Minor criteria include spontaneous and/or easy bruising demonstrating tissue fragility, thin translucent skin, and early-onset varicose veins, as well as other nondermatological features.1,4 The most common causative variants are missense glycine substitutions that disrupt triple helical winding of the collagen III protein followed by shortening in-frame splice-site variants. These result in a dominant negative effect on the protein caused by the misfolding of procollagen III in the endoplasmic reticulum (ER) and retention of 7/8ths of the misfolded procollagen trimers. Pathogenic variants leading to haploinsufficiency are reported less frequently and lead to a 50% decrease of collagen III protein.5 There are genotype–phenotype correlations in that individuals with haploinsufficiency tend to have a milder phenotype6,7 and splice-site variants are associated with a more severe phenotype. Other data supporting a genotype–phenotype effect in vEDS have also been reported.8,9
The cardinal features of classical EDS (cEDS) (OMIM 130000 gravis [type I] and OMIM 130010 mitis [type II]) are hyperextensible skin with widened, atrophic scarring.10 Joint hypermobility is common and minor features include easy bruising, soft doughy skin, traumatic splitting of skin (skin fragility), molluscoid pseudotumors, and subcutaneous spheroids. Although hemosiderin deposition over shins and extensor surfaces is not in the major or minor nosology criteria, it is included under the footnote of atrophic scarring because these features are often seen together. Unlike vEDS, life-threatening features of tissue fragility such as vascular rupture are not a clinical hallmark and hollow organ rupture has not been reported. In over 90% of individuals with cEDS a dominant pathogenic variant in either COL5A1 or COL5A2 is identified.11 Specific variants in COL1A1 have also been described in both vEDS and cEDS. Three specific arginine to cysteine substitutions (Arg312Cys, Arg574Cys, and Arg1093Cys) of COL1A1 have been observed in cases described to have vEDS with a more “cEDS-like” presentation, “vascular-like EDS,” “classic vascular-like EDS,” and cEDS only (Arg312Cys).12,13,14 The description of these families in both the cEDS and vEDS literature has made the boundaries between these two phenotypes less distinct.
In this study, seven families with, on first clinical presentation, suspected cEDS, had DNA analysis for vEDS due to occurrence of tissue fragility-related complications. Individuals were identified to have glutamic acid to lysine substitutions (Glu>Lys) within the COL3A1 triple helix, which were initially classified as variants of unknown significance.
MATERIALS AND METHODS
Subjects selected for the study were evaluated in the EDS National Diagnostic Centre in London or within a Clinical Genetics department in the UK, France, or Canada. For the five families from the UK, according to the institutional review board (IRB) no formal research ethics approval or research and development approval was required as stipulated by the UK policy Framework for Health and Social Care Research and the Health Research Authority decision tool. The three families from France were recruited as part of a larger cohort study for which ethical approval was obtained from the Comité de Protection des Personnes. Details of the genetic analyses are available in the Supplementary Information.
RESULTS
Family 1
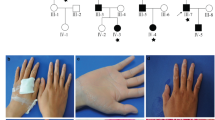
The family originated from Scotland. The proband (V.3) had “saggy skin exactly like his father’s” noted at birth. He bruised easily from the newborn period and developed “golf-ball sized” hematomas from minor injuries. At 43 years, he had an esophageal rupture secondary to vomiting. He was assessed in the UK in 2012 at age 44 and fulfilled the Villefranche criteria15 for both vEDS and cEDS with the presence of major criteria for both gastrointestinal (GI) rupture, marked skin hyperextensibility, a small number of widened atrophic scars, and joint hypermobility. Skin thinning was also noted at subsequent follow-up. Soon after initial assessment, it became apparent that a branch of the family was being investigated at the genetics service in Canada because of unexplained developmental delay (VI.6). On history and examination of the father (V.8), a connective tissue disorder was considered and genetic testing was carried out revealing a COL3A1 variant of uncertain significance: c.2044G>A p.Glu682Lys, which was also identified in the UK proband. Detailed segregation studies followed and according to American College of Medical Genetics and Genomics (ACMG) guidelines,16 the variant was deemed pathogenic (Fig. 1 and Table 2). Further family history elucidated that the UK proband’s paternal grandfather (III.1) with similar skin findings (Figs. 2–1, 2–2) had died from a perforation of the sigmoid colon at the age of 74 years. In addition, the maternal great-grandmother (II.6) of the Canadian proband V.8 died from a bowel perforation during routine colonoscopy age 89.

Clinical pedigrees of seven families with glutamic acid to lysine substitutions in COL3A1.

Clinical phenotype of patients with Glu>Lys variants in COL3A1. Consent for publication of patient photos was obtained for each patient. (1,2) F1/III.1 showing hyperextensible skin and abnormal feet. (3,4) F1/V.3 showing thin skin and scarring. (5,6) F1/VI.1 showing hyperextensible skin and large forehead hematoma with minimal trauma. (7–10) F2/II.6 showing hyperextensible skin, lack of hemosiderin deposition and scarring, temporary bruising following a fall, and florid cauliflower fibers on electron miscroscopy (EM). (11–15) F2/III.1 showing widened papyraceous scar, hyperextensible skin, striae rubrae, piezogenic papules, and occasional cauliflower fibers on EM. (16–21) F3/II.1 showing facial scarring age 8 and age 29, joint hypermobility, postsurgical scarring, and hyperextensible skin at knees. (Makeup does not enable full extent of scarring to be seen.) (22) F4/II.1 showing redundant skin at elbows. (23,24) F5/IV.1 showing scarring and varicose veins. (25–27) F5/V.1 showing lack of hemosiderin deposition and scarring, joint hypermobility, and piezogenic papules. (28) F5/V.3 showing flat feet with piezogenic papules.
Cardiac and vascular follow-up for the UK proband has identified a focal aneurysm of the celiac artery currently under 6-monthly surveillance, a mildly dilated aortic root, and mildly dilated left ventricle. Imaging also identified a large hiatus hernia. At age 50, he developed a rectal sheath hematoma secondary to a severe cough. It has been recommended that the proband commences celiprolol. Further details on individuals’ phenotypes are in Table 1.
Family 2
In family 2, the proband (II.6) was English. She presented with features of cEDS, and had not had a vascular event. However, at the time of a lumbar disc repair, the surgeon had commented on the “extreme tissue friability.” Genetic testing identified the COL3A1 variant c.3511G>A p.Glu1171Lys (unknown inheritance).17 Subsequent to this, her son with similar features on examination and also carrying the variant had a bowel rupture after a minor insult at the age of 24. Subsequent imaging for both the proband and her son has shown normal cardiac anatomy and normal angiographic appearances of the large arteries from neck to knees. They have been prescribed celiprolol.
Identification of the same variant in family 6, who present with a similar and consistent phenotype, and reanalysis using the ACMG guidelines has deemed this variant likely pathogenic (Table 2).
Family 3
The proband (II.1) was English. At 9 months, she was noted to have skin that tore easily following minimal trauma as well as a bruising tendency. A road traffic accident aged 3 years resulted in significant scarring to her face. She dislocated both her elbows repeatedly and fractured her left arm in three places in childhood following minimal trauma. An assessment aged 8 years showed scarring of the forehead and chin, a mild degree of scarring of the shins, and a Beighton score of 9 out of 9. Clinically she was thought to have cEDS—gravis type (type I) but genetic testing was not available at this time. At the age of 29, at 26 weeks gestation in her first pregnancy, she underwent blood gas sampling due to septicemia secondary to a urinary tract infection. This was complicated by a marked swelling of the left arm requiring urgent surgical repair of a left brachial artery rupture. COL3A1 gene testing identified a de novo COL3A1 variant c.721G>A (p.Glu241Lys). More recent evaluation of the variant has deemed this variant as likely pathogenic as the same variant has been identified in family 7. She is taking celiprolol and has some postural hypotension.
Family 4
The proband (II.1) was originally from Guyana. At the age of 29, he developed a spontaneous pneumothorax. The following year, age 30, he had a spontaneous small bowel rupture. Clinically he had hyperextensible skin with significant redundancy at the elbows. COL3A1 analysis identified the same pathogenic variant as in family 1: c.2044G>A p.Glu682Lys.17 He is being referred for further cardiovascular surveillance and offered celiprolol.
Family 5
Family 5 consists of a father and two daughters of Caucasian background. The father (IV.1) had been diagnosed clinically as a teenager with cEDS but was subsequently lost to follow-up. In later life he developed a left-sided subclavian vein aneurysm and a right-sided internal jugular venous aneurysm for which he had surgery age 48 but no genetic testing had been performed. His daughter presented when pregnant and was tested for classical and vascular EDS because of the above-stated family history and because she reported to have similar skin findings to her father. COL3A1 testing identified the Glu682Lys variant (identical pathogenic variant to family 1 and family 4), which was subsequently identified in the father (IV.1) and another daughter (V.3).
Family 6
The proband (II.1) is a 33-year-old male who was initially referred by a military physician after presenting at the age of 21 with a 43 cm by 20 cm muscular hematoma of the right leg sustained during military training. This patient was identified to have a presumed de novo variant of COL3A1 p.Glu1171Lys and was published in 2015 by Frank et al.8 (AN_002971), now classed as likely pathogenic (as per family 2). Work-up of the arterial tree revealed no arterial defects at the age of 22 years.
Family 7
The proband suffered from a rectal perforation during hemorrhoid surgery at the age of 54 and subsequently died. He had easy bruising, hyperextensible skin, scarring, and joint hypermobility. He has been found to have the same variant as the proband in family 3 (c.721G>A p.Glu241Lys).
Skin biopsy findings
Four probands out of seven and one affected relative had skin biopsies examined with electron microscopy (EM) in this cohort. EM of skin in patients with vEDS has identified a thinned dermis and a disorganization of collagen fibrils with a variable fibril diameter size compared with controls, although these features may not always be present.18,19 EM of skin in patients with cEDS often shows characteristic florid abnormally shaped collagen fibrils known as “collagen flowers’” although this is not specific to cEDS only.20 We identified collagen flowers only in one patient, variability of collagen fiber diameter in three patients, and both in one patient. One EM result was normal. No other EM abnormalities were reported. Interestingly, there was intrafamilial variation, with the proband of family 2 demonstrating florid collagen flowers and her son only having a small number of collagen flowers with some variability in fiber diameter. Collagen electrophoresis can be used to study the effect of the variant but the absence of demonstration of abnormal collagen III produced by fibroblasts does not exclude the diagnosis.21 In this cohort, collagen electrophoresis was normal in all cases tested (n = 5).
DISCUSSION
We identified 18 individuals (10 male and 8 female, age range 3–74 years) from seven different families, each harboring an atypical variant resulting in glutamic acid to lysine substitution in the COL3A1 gene. Seventeen individuals were confirmed with direct molecular testing and one (III.1 in family 1) was not alive when genetic confirmation became available but is an obligate carrier due to the segregation pattern within the family. Molecular analysis of COL5A1, COL5A2, and COL1A1 proved negative in all probands. Genetic testing for classical-like EDS (clEDS) was negative in the three families (families 4, 6, and 7) without a clear autosomal dominant history and not performed in the others. Molecular data for one individual (AN_002971) has previously been published in Frank et al. (2015) (p.Glu1171Lys family 6) and another two in Weerakkody et al. (2016) (p.Glu1171Lys family 2 and p.Glu682Lys family 4). Four families have not previously been reported and the three previously reported have not been reported as pathogenic or likely pathogenic according to ACMG guidelines. Molecular data is also available from a further two patients (father and son) with a glutamic acid to lysine substitution in COL3A1 (https://www.ncbi.nlm.nih.gov/clinvar/), which has been reported as a variant of uncertain significance (c.1351G>A p.Glu451Lys). The proband suffered from a perforation of the sigmoid colon at the age of 41. He has transparent, hyperextensible skin and a predisposition to hematoma formation as well as gingival fragility. He also has a pectus excavatum and surgery for an inguinal hernia. His father was also predisposed to hematoma formation and suffered a bowel perforation at the age of 48. Further reports of this variant in individuals with a similar phenotype may allow for this to be upgraded to a likely pathogenic variant according to ACMG guidelines.
At the time of first assessment, all probands described fulfilled clinical criteria for cEDS, which included three different classification criteria dependent on the year that they were assessed (Table 1) (Berlin,22 Villefranche, and 2017 New York criteria). Only two probands (families 1 and 4) fulfilled both clinical diagnostic criteria of cEDS and vEDS, as they both had experienced gastrointestinal ruptures. One other individual (family 6) was suspected to have vEDS on the basis of facial features, easy bruising, and thin translucent skin.
Skin features more specific to vEDS, for example, early-onset varicose veins (n = 3) and thin skin with visible veins (n = 6) are noted, as well as skin features found in both vEDS and cEDS such as easy bruising (n = 15). Skin features seen more exclusively in cEDS are also seen in this cohort: skin hyperextensibility (n = 18) and a variable degree of atrophic scarring (n = 7); skin hyperextensibility is universal in the cohort. Whilst several individuals in this cohort have a degree of atrophic scarring, the typical hemosiderin deposition seen, for example, over the shins in cEDS is not described. In addition, the degree of scarring is not a consistent feature in the patients described and perhaps this reflects the variation in age and activity levels of a small cohort. An additional striking feature is the propensity to hematoma formation (including nonsubcutaneous hematomas); however, this is not specifically mentioned and differentiated from bruising in either current or historical classification of cEDS or vEDS. Hematomas are deep bruises resulting in a collection of blood that lies within body tissues or cavities.23 Predisposition to hematomas was seen in 16 of 18 patients and were most frequently subcutaneous (see Fig. 2–6) but other more severe hematomas such as muscular hematomas requiring surgical intervention or hospitalization were also described. Only 3% of patients with cEDS caused by COL5A1 variants were noted to have hematomas in a recent review24 and there is no known literature of the incidence of hematomas in vEDS. Two of our cases had striae, a feature reported not to be present in patients with cEDS.10
Six of the seven families had at least one individual affected by a vascular or GI complication (7/18 individuals—[39%]). An additional GI perforation occurred in another family member known to be phenotypically affected but not included in Table 1 (F1-II.6—bowel perforation secondary to routine colonoscopy age 89 and subsequent death). Of these seven individuals with complications, three (F1-V.3, F3-II.1, and F5-IV.1) have a total of two vascular findings/complications each. Only one patient has had a vascular event (brachial artery rupture) and one has had elective surgery (jugular venous aneurysm). Five of 18 (28%) suffered from GI rupture. GI perforations included esophagus as well as small and large bowel. There is both intra- and interfamilial variability in the severity of the phenotype, and other genetic factors, lifestyle, and environment may potentially modify disease. However, despite these complications, several patients are older than the mean expected life expectancy for patients with vEDS25 and 11 patients have not had any GI or vascular complications, although 4 of these 11 are pediatric patients.
Patients with overlapping features of vascular and classical EDS have been reported in five possible cases as far back as the 1960s.26 In 1987, a case report describing a clinical diagnosis of EDS type I (cEDS) with reduced amount of collagen type III was published27 although molecular confirmation was not established as the gene for vEDS had not yet been published. Subsequently, there have been other cases described in the 1990s and 2000s and with availability of molecular analysis, COL1A1 variants have been identified in these cases.12,13,28,29 However, to our knowledge, this is the first description of a similar overlapping phenotype in individuals with COL3A1 variants, specifically Glu>Lys substitutions. One individual with hyperextensible skin has previously been reported in the literature30 with a COL3A1 variant that is not a Glu>Lys substitution (arginine to leucine in C-propeptide region) although the pathogenicity has not been confirmed.
Differential diagnoses
Adopting the current nosology, classical-like EDS (clEDS) (OMIM 606408) as well as cEDS ought to be considered in cases of skin hyperextensibility with minimal scarring. The presence of skin hyperextensibility with velvety skin texture and the absence of atrophic scarring, as well as joint hypermobility and easy bruising, should raise suspicion of clEDS, caused by biallelic TNXB variants.31 These features are all present in our cohort. Vascular complications are not reported in clEDS except for hematoma formation in 10/19 cases.24 GI rupture has also been described in individual case reports.32,33 However, an autosomal recessive mode of inheritance would not be compatible with several of the pedigrees in this cohort. In addition, musculocontractural EDS caused by variants in DSE and CHST14 and dermatoparaxis EDS caused by variants in ADAMSTS2 present with increased risk of hematomas and a novel recessive EDS subtype caused by variants in AEBP1 presents as classical-like EDS.34 These conditions would need to be considered in the differential diagnosis of these individuals but mostly present with additional features.
Skin biopsy analyses
Prior to molecular characterization, cEDS would have been diagnosed by clinical history, examination findings, and ultrastructural findings on electron microscopy (EM) of skin demonstrating collagen flowers when available. Collagen flowers are not usually seen in vEDS patients who are more likely to have variability in collagen diameter and loose packing of collagen fibers on EM. There is no consistent finding on EM in our cohort. With next-generation sequencing, molecular testing is now superceding EM analysis although it may have continued use in the event of unusual or complex findings such as a variant of uncertain significance or inconsistency of both clinical and molecular findings. Collagen profiling in those performed did not reveal any specific abnormal patterns.
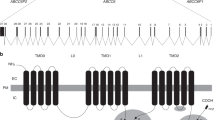
Common structural features of the pathogenic variants
There is no clear explanation for the resulting atypical phenotype for vascular EDS and further work is recommended. The common feature of all the variants is their occurrence in a local Gly-Glu-Arg sequence. The Gly-Glu-Arg sequence is fairly common in collagens, given the propensity for Glu residues to be found in the X position and Arg in the Y position of the Gly-X-Y repeat.35 As for Gly-Lys-Arg, there is only one natural occurrence of this sequence in the major fibrillar collagens (in COL1A1). Mapping of the variant sites on the collagen III interactome shows two of them (Glu241Lys and Glu682Lys) to be part of known integrin binding sites (Gly-Arg-Hyp-Gly-Glu-Arg and Gly-Ala-Hyp-Gly-Glu-Arg respectively) where the Glu residue plays an essential role in integrin binding.36 Glu1171Lys occurs in the sequence Gly-Asn-Arg-Gly-Glu-Arg, which does not appear to be an integrin binding site. Interestingly however, all three variants are closely aligned within the so-called overlap region of the collagen fibril, in which molecules are staggered by multiples of D37 (see Fig. 3).

Positions of the variants when molecules are in D-staggered array within the collagen fibril. All three variants (red dots) are aligned in the overlap region.
Implications for management and future directions
The reclassification from variants of uncertain significance to either pathogenic or likely pathogenic in accordance with ACMG guidelines, establishing a definitive diagnosis for an individual, is important for appropriate management, predictive testing for other family members, and the availability of reproductive options such as preimplantation genetic diagnosis (PGD). All individuals with these atypical variants exhibited skin hyperextensibility as seen in individuals with cEDS and clEDS and evidence of tissue fragility as seen in individuals with vEDS demonstrating the importance of gene panel testing.
At present, it is not clear if the risk of vascular complications in this subgroup of patients with Glu>Lys is different from typical pathogenic variants in COL3A1 involving glycine substitutions or other recognized molecular causes of vEDS. Only one patient in the cohort has had an arterial rupture/dissection. However, in total, there are three patients with vascular complications (not including varicose veins and hematomas) and each of them on further surveillance have been identified to have more than one vascular abnormality (see Table 1). Five molecularly proven cases from this cohort have had GI complications (1 small bowel perforation, 3× large bowel rupture, and 1 esophageal rupture). Therefore, at present, many of these individuals are being managed as vascular EDS. Although sudden arterial dissections may occur without a history of previous aneurysmal disease,38 surveillance is offered by vascular imaging. Based on a previously published clinical trial demonstrating the prophylactic use of cardioselective β- blocker celiprolol,39 some have been offered this β-blocker.
Given the clinical overlap, we propose that molecular confirmation of a suspected EDS subtype is performed where possible by gene panel testing. Patients with these Glu>Lys variants in COL3A1 should remain under the classification of vEDS because of the observed tissue fragility and managed per vEDS guidelines. With regard to the risk of vascular and other complications, a larger patient group and further long term follow-up studies are required to accurately assess risk and benefit of vascular surveillance and prophylaxis such as celiprolol. Further research to explain the pathological consequences of these variants leading to this specific and consistent phenotype is required.
References
Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26.
Malfait F Vascular aspects of the Ehlers-Danlos syndromes. Matrix Biol. 2018;71–72:380–395.
Supertifurga A, Steinmann B, Ramirze F, Byers PH. Molecular defects of type-3 collagen in Ehlers-Danlos syndrome type-4. Collagen Rel Res. 1988;8:509–509.
Byers PH, Belmont J, Black J, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175:40–47.
Chiarelli N, Carini G, Zoppi N, Ritelli M, Colombi M. Transcriptome analysis of skin fibroblasts with dominant negative COL3A1 mutations provides molecular insights into the etiopathology of vascular Ehlers-Danlos syndrome. PLoS One. 2018;13:e0191220.
Leistritz DF, Pepin MG, Schwarze U, Byers PH. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med. 2011;13:717–722.
Schwarze U, Schievink WI, Petty E, et al. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69:989–1001.
Frank M, Albuisson J, Ranque B, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet. 2015;23:1657–1664.
Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16:881–888.
Bowen JM, Sobey GJ, Burrows NP, et al. Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet. 2017;175:27–39.
Symoens S, Syx D, Malfait F, et al. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat. 2012;33:1485–1493.
Colombi M, Dordoni C, Venturini M, Zanca A, Calzavara-Pinton P, Ritelli M. Delineation of Ehlers-Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: report on a three-generation family without cardiovascular events, and literature review. Am J Med Genet A. 2017;173:524–530.
Malfait F, Symoens S, De Backer J, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28:387–395.
Gaines R, Tinkle BT, Halandras PM, Al-Nouri O, Crisostomo P, Cho JS. Spontaneous ruptured dissection of the right common iliac artery in a patient with classic Ehlers-Danlos syndrome phenotype. Ann Vasc Surg. 2015;29:595 e511–594.
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31–37.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Weerakkody RA, Vandrovcova J, Kanonidou C, et al. Targeted next-generation sequencing makes new molecular diagnoses and expands genotype-phenotype relationship in Ehlers-Danlos syndrome. Genet Med. 2016;18:1119–1127.
Pope FM, Burrows NP. Ehlers-Danlos syndrome has varied molecular mechanisms. J Med Genet. 1997;34:400–410.
Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G, Richards AJ. COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol. 1996;135:163–181.
Vogel A, Holbrook KA, Steinmann B, Gitzelmann R, Byers PH. Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Lab Invest. 1979;40:201–206.
Steinmann B, Superti-Furga A, Joller-Jemelka HI, Cetta G, Byers PH. Ehlers-Danlos syndrome type IV: a subset of patients distinguished by low serum levels of the amino-terminal propeptide of type III procollagen. Am J Med Genet. 1989;34:68–71.
Beighton P, de Paepe A, Danks D, et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am J Med Genet. 1988;29:581–594.
Dermnet New Zealand. Bleeding and bruising. https://www.dermnetnz.org/topics/bleeding-and-bruising/. Accessed 4 December 2018.
D'Hondt S, Van Damme T, Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers-Danlos syndrome: a systematic review. Genet Med. 2018;20:562–573.
Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis. 2007;2:32.
Beighton P. Lethal complications of the Ehlers-Danlos syndrome. Br Med J. 1968;3:656–659.
De Paepe A, Nicholls A, Narcisi P, et al. Ehlers-Danlos syndrome type I: a clinical and ultrastructural study of a family with reduced amounts of collagen type III. Br J Dermatol. 1987;117:89–97.
Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T, De Paepe A. Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am J Hum Genet. 2000;66:1398–1402.
Ritelli M, Dordoni C, Venturini M, et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J Rare Dis. 2013;8:58.
Stembridge NS, Vandersteen AM, Ghali N, et al. Clinical, structural, biochemical and X-ray crystallographic correlates of pathogenicity for variants in the C-propeptide region of the COL3A1 gene. Am J Med Genet A. 2015;167A:1763–1772.
Demirdas S, Dulfer E, Robert L, et al. Recognizing the tenascin-X deficient type of Ehlers-Danlos syndrome: a cross-sectional study in 17 patients. Clin Genet. 2017;91:411–425.
Besselink-Lobanova A, Maandag NJ, Voermans NC, van der Heijden HF, van der Hoeven JG, Heunks LM. Trachea rupture in tenascin-X-deficient type Ehlers-Danlos syndrome. Anesthesiology. 2010;113:746–749.
Sakiyama T, Kubo A, Sasaki T, et al. Recurrent gastrointestinal perforation in a patient with Ehlers-Danlos syndrome due to tenascin-X deficiency. J Dermatol. 2015;42:511–514.
Blackburn PR, Xu Z, Tumelty KE, et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers-Danlos syndrome. Am J Hum Genet. 2018;102:696–705.
Persikov AV, Ramshaw JA, Brodsky B. Prediction of collagen stability from amino acid sequence. J Biol Chem. 2005;280:19343–19349.
Raynal N, Hamaia SW, Siljander PR, et al. Use of synthetic peptides to locate novel integrin alpha2beta1-binding motifs in human collagen III. J Biol Chem. 2006;281:3821–3831.
Parkin JD, San Antonio JD, Persikov AV, et al. The collalphagen III fibril has a “flexi-rod” structure of flexible sequences interspersed with rigid bioactive domains including two with hemostatic roles. PLoS One. 2017;12:e0175582.
Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680.
Ong KT, Perdu J, De Backer J, et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet. 2010;376:1476–1484.
Acknowledgements
We express our gratitude to all the families who participated in the study. We thank Tim Aitman and Ruwan Weerakkody for the sequencing and research data for family 2 and family 4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ghali, N., Baker, D., Brady, A.F. et al. Atypical COL3A1 variants (glutamic acid to lysine) cause vascular Ehlers–Danlos syndrome with a consistent phenotype of tissue fragility and skin hyperextensibility. Genet Med 21, 2081–2091 (2019). https://doi.org/10.1038/s41436-019-0470-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0470-9
Keywords
This article is cited by
-
The Ehlers–Danlos syndromes
Nature Reviews Disease Primers (2020)
