
Abstract
Purpose
Both monogenic pathogenic variant cataloging and clinical patient diagnosis start with variant-level evidence retrieval followed by expert evidence integration in search of diagnostic variants and genes. Here, we try to accelerate pathogenic variant evidence retrieval by an automatic approach.
Methods
Automatic VAriant evidence DAtabase (AVADA) is a novel machine learning tool that uses natural language processing to automatically identify pathogenic genetic variant evidence in full-text primary literature about monogenic disease and convert it to genomic coordinates.
Results
AVADA automatically retrieved almost 60% of likely disease-causing variants deposited in the Human Gene Mutation Database (HGMD), a 4.4-fold improvement over the current best open source automated variant extractor. AVADA contains over 60,000 likely disease-causing variants that are in HGMD but not in ClinVar. AVADA also highlights the challenges of automated variant mapping and pathogenicity curation. However, when combined with manual validation, on 245 diagnosed patients, AVADA provides valuable evidence for an additional 18 diagnostic variants, on top of ClinVar’s 21, versus only 2 using the best current automated approach.
Conclusion
AVADA advances automated retrieval of pathogenic monogenic variant evidence from full-text literature. Far from perfect, but much faster than PubMed/Google Scholar search, careful curation of AVADA-retrieved evidence can aid both database curation and patient diagnosis.
Similar content being viewed by others

INTRODUCTION
Exome and genome sequencing are entering clinical practice to aid the identification of molecular causes of highly penetrant genetic diseases, particularly Mendelian disorders, where typically one or two of the patient’s genetic variants in a single gene are causative (diagnostic) of the patient’s disease. After variant filtering, a typical singleton patient exome contains 200–500 rare variants.1,2 Identifying causative variants is therefore time-consuming, as investigating each variant can take up to an hour.3 Various approaches strive to accelerate this process.4,5,6 Identifying diagnostic variants can be greatly accelerated if the patient’s genome contains a previously reported disease-causing variant that partly or fully explains their phenotype. The American College of Medical Genetics and Genomics (ACMG) guidelines for the interpretation of sequence variants recommend variant annotation using databases of reported pathogenic variants.7
Variant curation from the literature includes (1) retrieval of variant evidence from the literature and (2) variant evaluation, which assesses the validity and power of every piece of evidence, and combines all (possibly conflicting) evidence together to make variant- and gene-level diagnostic assertions. The rapidly growing literature on human genetic diseases,8 the costly process of manual variant curation,9 and improved computational access to the full text of primary literature10 serve to incentivize automating parts of the variant curation pipeline. Here, we focus on automating the process of variant evidence retrieval from the primary literature about Mendelian diseases without attempting to automate variant evaluation. Automatic pathogenic variant evidence retrieval from the primary literature involves finding papers about Mendelian diseases that include variant descriptions (such as “c.123A>G”), linking them to a transcript of the correct gene, and converting these to genomic coordinates for ease of downstream use. Previous work on automatic variant evidence retrieval from the literature has largely focused on finding variant descriptions in article titles and abstracts without converting the discovered variants to genomic coordinates11,12,13,14 or only to dbSNP15 variant identifiers (rsIDs). Mapping textual variant descriptions directly to reference genome coordinates requires significant effort, and has thus far largely been left to manually curated databases such as the Human Gene Mutation Database (HGMD)16 and ClinVar,17 which devote many worker-hours to the task.
The ClinGen9 project has proposed to “develop machine-learning algorithms to improve the throughput of variant interpretation” and notes that a rate-limiting factor for clinical use of variant information is the lack of openly accessible knowledgebases capturing known genetic variants. The Automatic VAriant evidence DAtabase (AVADA) is trained on a sample of manually curated variants (from ClinVar and HGMD), and then applied to the entire body of PubMed indexed literature for automatic retrieval of published variants in papers about Mendelian disease. We show that AVADA improves on the state of the art in automated variant retrieval by comparing it with tmVar 2.0, a best-in-class tool18 used to harvest variants from PubMed abstracts.
To show the amount of valuable evidence retrieved by AVADA, we also compare variants collected by our approach to the fully curated HGMD and ClinVar databases. We also show, on 245 real patients, that when AVADA is coupled with manual curation, it can aid diagnosis.
We note that the creators of HGMD (P.D.S. and D.N.C.) are coauthors of this study. They provided HGMD data and advised on validating and curating AVADA literature-extracted variant evidence and the comparison of AVADA to HGMD.
MATERIALS AND METHODS
Identification of relevant literature
PubMed is a database containing titles and abstracts of biomedical articles, only a subset of which contain descriptions of variants that cause human genetic disease. A document classifier is a machine learning classifier that takes as input arbitrary text and classifies it as “positive” (here, an article about genetic disease) or “negative” (otherwise). We trained a scikit-learn19 LogisticRegression20 classifier to identify relevant documents using positive input texts (titles and abstracts of articles cited in the “Allelic Variants” sections of OMIM,21 and all of HGMD,16 which curates germline disease-causing and disease-associated variants) and negative input texts (random PubMed titles and abstracts). Input texts were converted into a feature vector based on the frequency of words in input documents by means of a scikit-learn CountVectorizer followed by a term frequency–inverse document frequency20 (TF-IDF) transformer. After training the title/abstract document classifier, we applied it to all 25,793,020 titles and abstracts in PubMed to identify articles that might be relevant to the diagnosis of genetic diseases (if the title/abstract classifier returned a score of at least 0.5). Full-text PDFs of potentially relevant articles were downloaded, converted to text using pdftotext22 version 0.26.5, and evaluated for full-text relevance using a TF-IDF transformer, a full-text scikit-learn LogisticRegression classifier, and a threshold of 0.5 on the full-text relevance scores.
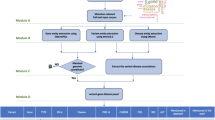
A total of 133,410 articles were downloaded and subsequently classified as relevant to the diagnosis of human genetic diseases based on the articles’ full text (Supplementary Methods). We refer to this set of articles as the “AVADA full-text articles” (Fig. 1).

Construction of the Automated Variant Evidence Database (AVADA). Identification of relevant literature: AVADA discovers potentially relevant articles (about the genetic causes of Mendelian diseases) from PubMed, downloads their full text, and again filters potentially relevant articles based on the articles’ full text. Variant mapping: Variant descriptions are detected in articles using 47 manually built regular expressions. Variant descriptions are then linked to mentioned genes to form gene–variant candidate mappings. Gene–variant candidate mappings are filtered using a gene–variant candidate classifier and converted to genomic coordinates. AVADA ultimately retrieves (unvalidated) evidence about 203,536 distinct genetic variants in 5827 genes from 61,116 articles.
To estimate recall (the fraction of relevant articles that are contained in the AVADA full-text article set) of our pipeline, we took the set of 27,361 articles associated with “likely/pathogenic” variants in ClinVar. Of these, 24,724 (90%) were classified relevant by the title/abstract classifier, 23,978 (88%) were downloaded in full text, and 20,014 (73%) were finally classified relevant by the full-text classifier. To determine the precision (fraction of relevant articles among all AVADA full-text articles) of our pipeline, we randomly selected 200 articles from the set of AVADA full-text articles and manually assessed their relevance. An article was considered relevant if it discussed a Mendelian disease and its causative gene. Ninety-nine (49.5%) articles were determined to be relevant (Supplementary Table S1).
Variant and gene mention detection
To retrieve genetic variants from full-text articles about human genetic disease and convert them to genomic coordinates, it is necessary to detect both mentions of genes and variant descriptions in articles about genetic disease. This is because variant descriptions in Human Genome Variation Society (HGVS)-like notation, such as “c.123A>G”, often do not contain the identity of the specific transcript or gene they reside in (Table 1).
AVADA extracts gene mentions from articles’ full text using a custom-built database of gene and protein name entries from the HUGO Gene Nomenclature Committee (HGNC) and UniProt databases. These were matched case-insensitive to word groups of length 1–8 in the document. To identify variant mentions, we manually developed 47 regular expressions based on commonly observed HGVS-like variant notations in articles about human genetic disease (Supplementary Methods, Supplementary Table S2 and Fig. 2a). At this step, we refer to every string that matches one of the 47 regular expressions as a “variant description.” In the AVADA full-text articles, variant descriptions were identified in 92,436 articles, with a mean of 11.1 variant descriptions per article (Fig. 1).

Automatic conversion of variant mentions to genomic coordinates from full-text literature. (a) AVADA uses a regular expression to detect a variant mention (e.g., p.M34T) in the full text of an article. The position of the variant in the transcript (34), reference (M) and alternative alleles (T) are parsed using the regular expression. (b) AVADA detects mentioned genes in the article using a list of gene names and synonyms, and the help of a classifier that decides if recognized words are indeed a gene mention. The variant description detected in step A forms gene–variant candidate mappings with those genes that have the reference “M” at amino acid number 34. (c) Gene–variant candidate mappings (variant = p.M34T and gene = GJB2 in this example, highlighted in green) are associated with 125 numerical features based on the relative positions of the closest mention of the candidate gene to the variant mention, information about the candidate gene’s importance in the article, and words and characters surrounding the gene and variant mentions and nearby gene mentions (the latter highlighted in red; see Supplementary Methods). (d) A machine learning classifier (implemented as a gradient boosting classifier) takes these 125 features as input and returns a score between 0 and 1 indicating the classifier’s assessment of whether the variant actually refers to the given candidate gene. If the classifier returns a score greater than 0.9, the gene–variant candidate mapping is transformed to Variant Call Format (chromosome, position, reference and alternative allele) and entered into the AVADA database. In the present example, AVADA correctly decides that p.M34T only maps to GJB2 and not connexin 30 (encoded by the gene GJB6). Example taken from PubMed ID 23808595.
Mentioned genes form gene–variant candidate mappings with all plausible mentioned variant descriptions
Variant descriptions and their host gene mentions do not necessarily occur in the same sentence or even the same paragraph or page. To identify which variant description maps to which mentioned gene in the article, AVADA first forms so-called gene–variant candidate mappings between each variant description and each mentioned gene if the variant matches at least one RefSeq23 transcript of the gene. For example, the variant description “c.123A>G” forms gene–variant candidate mappings with all mentioned genes that have an “A” at coding position 123 in at least one transcript (Supplementary Methods and Fig. 2b). A variant description can form gene–variant candidate mappings with multiple genes, which are filtered in the next step. Gene–variant candidate mappings are converted to genomic coordinates in the GRCh37/hg19 reference assembly and initially result in a mean of 4.6 different genomic coordinates per extracted variant description (Fig. 1).
Machine learning classifier selects the correct gene–variant mapping candidate
AVADA uses a scikit-learn GradientBoostingClassifier24 to decide which gene–variant candidate mappings are likely to be correct. The training set comprised positive gene–variant mappings extracted from the literature that were referenced in all ClinVar entries, and a set of negative gene–variant mappings created by assigning variants from the positive training set to other genes mentioned in the article. For classification, each gene–variant mapping was converted to a feature vector, including the Euclidean distance between the 2D coordinates (consisting of page number, x- and y-coordinates of a mention) of the closest mentions of the variant and the gene in the PDF, the number of words between variant and gene mentions, and a number of other textual features containing information about the relationship between gene and variant mentions (Supplementary Methods and Fig. 2c). Using a threshold of 0.9 (justified in the section “Picking the gene–variant candidate classifier threshold”), the gene–variant candidate classifier successfully reduced 4.6 candidate gene–variant mappings per variant description to a mean of 1.2 genomic coordinates (chromosome, position, reference, and alternative allele) in the final set of AVADA full-text articles (Supplementary Methods and Figs. 1, 2d).
RESULTS
AVADA retrieved 203,536 variants in 5827 genes from 61,116 articles
A total of 61,116 articles made it into the final AVADA database, with a mean of 8.8 identified variant descriptions per article. From these articles, 203,536 distinct variants (GRCh37/hg19 chromosome, position, reference allele, and alternative allele) in 5827 genes were automatically retrieved (Fig. 1). The distribution of types of rare variants (≤3% variant frequency in the healthy population2) in AVADA is strikingly similar to that of manually curated HGMD and ClinVar: for each of six variant categories (stoploss, nonframeshift indel, splicing, stopgain, frameshift, missense), the fraction of rare variants in AVADA are between the fraction of the respective category of rare variants in all of HGMD and ClinVar ±1% (Table 2). The articles used to construct AVADA are from a variety of journals, which are similar to the journals targeted by all of HGMD to curate its variants (9 of the top 10 journals being the same between AVADA and all of HGMD; Fig. 3a, b).

Automatic variant retrieval results. (a) Top ten journals in AVADA. AVADA retrieved variants from 3159 articles in Human Mutation, 2330 articles in American Journal of Human Genetics, 2042 articles in Human Molecular Genetics, etc. (b) Top ten journals in all of HGMD. Similar to AVADA, the top three journals are Human Mutation, American Journal of Human Genetics, and Human Molecular Genetics. Reassuringly, the two lists share nine of the top ten journals even though HGMD is manually curated whereas AVADA automatically retrieves variant evidence, but does not validate it. (c) (Unvalidated) AVADA variants intersected with all curated disease-causing variants in HGMD (“DM” variants only) and ClinVar (“likely/pathogenic” variants only). AVADA retrieves 85,888 variants also in the HGMD set (subset to disease-causing variants) and 26,033 variants also in the ClinVar set (subset to pathogenic and likely pathogenic variants). (d) AVADA’s potential value in patient diagnosis. We enumerate the number of patient diagnostic variants found in each of four databases, for 245 Deciphering Developmental Disorders (DDD) diagnosed patients. Curated HGMD and ClinVar (predating the DDD publication) are subset to disease-causing (“DM”), and “likely/pathogenic,” respectively. For tmVar and AVADA, we manually validated all diagnostic evidence shown. AVADA completely subsumes and almost triples abstract-based tmVar. And while ClinVar alone implicates 21 diagnostic variants, AVADA offers unvalidated evidence for an additional 27 variants, of which 18 are valid, virtually doubling ClinVar’s reach.
Each variant in AVADA is annotated with the PubMed ID(s) of publications where this variant was retrieved from, a HGNC25 gene symbol, an Ensembl ID26 and Entrez ID,27 the transcript RefSeq ID (e.g., NM_005101.3), and the exact variant description from the original article (e.g., “c.163C.T”). The latter allows AVADA users to later rapidly locate mentions of the variant within the body of the article.
AVADA is 61% precise at mapping gene–variant pairs to genomic coordinates
To estimate the precision of AVADA at extracting gene–variant candidate mappings in articles into genomic coordinates, 200 distinct random variants in AVADA were manually examined. For each of these variants, we selected the article associated with the (alphanumerically sorted) first PubMed ID in AVADA, and let two reviewers determine if the gene–variant candidate mapping from the article was correctly extracted to genomic coordinates using all lines of evidence in the article such as Sanger sequencing reads, University of California–Santa Cruz (UCSC) Genome Browser shots, etc. One hundred twenty-two (61%) random variants were correctly extracted by AVADA (Supplementary Table S3).
AVADA recovers nearly 60% of disease-causing HGMD variants directly from the primary literature
We compared AVADA to HGMD and ClinVar versions with synchronized timestamps (Supplementary Methods). In this section, we subset HGMD to “DM” (disease-causing) variants and ClinVar to variants marked as “likely/pathogenic”; 85,888 variants in AVADA coincided with variants marked as disease-causing (“DM”) in HGMD, corresponding to 61% of all disease-causing variants in HGMD. We selected 200 distinct random variants from this set for verification by two reviewers. A variant was counted as correct and likely disease-causing if the reviewers came to a consensus that the original gene–variant candidate mapping was converted to the variant’s genomic coordinates correctly in at least one of its associated articles, and the variant was described as likely disease-causing in the article. This was the case in 96.5% of the 200 variants (Supplementary Table S4). Thus, we infer that AVADA contains 58% of all disease-causing variants identified by HGMD.
We compared AVADA’s performance to the best previously published automatic variant retrieval tool, tmVar 2.0,18 which attempts to map variant mentions in all PubMed abstracts to dbSNP identifiers (rsIDs). We converted rsIDs in tmVar 2.0 to genomic coordinates using mappings provided by dbSNP. tmVar retrieved only 19,481 (14%) disease-causing (DM) HGMD variants (Supplementary Fig. 1 and Fig. 3c).
Considering only single-nucleotide variants (SNVs), the largest class of known disease-causing variants, AVADA contains 70% of all DM SNVs in HGMD. Similarly, AVADA contains 26,033 (55%) of all “likely/pathogenic” variants in ClinVar and 62% of all “likely/pathogenic” SNVs in ClinVar. tmVar 2.0 retrieved only 14,841, or 31%, of pathogenic or likely pathogenic variants in ClinVar. Strikingly, AVADA contains 62,180 variants noted to be disease-causing in HGMD (“DM”) but not in ClinVar (“likely/pathogenic”).
Two reviewers evaluated a subset containing 200 distinct random variants of the remaining 115,323 variants that were retrieved by AVADA, but not reported as disease-causing in either HGMD (“DM”) or ClinVar (“likely/pathogenic”). Sixty-eight (34%) of the 200 variants were correctly converted to genomic coordinates. Eight variants (4%) were further reported to be likely disease-causing (Supplementary Table S5). Seven of these 8 are contained in later versions of HGMD (“DM”), suggesting AVADA could both help curators accelerate variant retrieval as well as unearth a modest amount of undocumented likely disease-causing variants still hidden in the literature.
Diagnosis of patients with Mendelian diseases using AVADA
We analyzed the utility of known variant databases using 260 diagnostic (i.e., causative) variants from 245 patients with developmental disorders, diagnosed in Supplementary Table 4 of the Deciphering Developmental Disorders (DDD) study,28 obtained from European Genome–phenome Archive (EGA)29 study number EGAS00001000775 (Supplementary Methods).
Accuracy of variant annotation using AVADA, tmVar, HGMD, and ClinVar
The more complete a variant database is, the higher its sensitivity when annotating patient genomes and the higher the likelihood of finding a diagnostic variant in the patient’s genome. We determined how many of the 260 reported diagnostic DDD variants were found in AVADA, tmVar, HGMD (“DM” variants only), and ClinVar (“likely/pathogenic” variants only). The more disease-causing variants are contained in a database, the more rapidly some patients can be diagnosed. For this comparison, we subset AVADA and tmVar 2.0 to articles published until 2014 (before DDD publication), used only disease-causing DM variants entered until 2014 in HGMD, and used only likely/pathogenic variants from ClinVar version 20141202.
Of 260 different diagnostic variants reported by the DDD study, a total of 45 had evidence in AVADA from the scientific literature. Because AVADA retrieves variant evidence without validating it, all AVADA evidence needs to be manually assessed. Each patient variant found in AVADA was counted as correct if our two reviewers agreed that AVADA cited at least one article from which the variant was correctly mapped to genomic coordinates and the variant was reported as likely disease-causing in this article. Thirty-five of the 45 variants found by AVADA fulfilled these criteria (Supplementary Table S6). Only 21 DDD diagnostic variants were listed in ClinVar and ascribed a pathogenicity level of “likely/pathogenic.” Combining the free variant databases yielded 39 variants, almost as many as the 43 variants listed in HGMD (“DM”). Combining all three databases yielded 48 variants (Fig. 3d). tmVar 2.0 contained only 13 diagnostic variants (Supplementary Table S7), all of which were in AVADA as well (Fig. 3d).
We defined patient variants to be “candidate causative variants” if they were nonsilent exonic or core splice-site variants and occurred at an allele frequency of at most 0.5% in large databases of healthy controls2,30,31 (Supplementary Methods). The 245 patients’ data contained a mean of 435 nondiagnostic candidate variants each. To determine the variant annotation precision of AVADA, HGMD, ClinVar, and tmVar 2.0, we divided the number of distinct annotated diagnostic variants by the number of distinct annotated candidate variants across the 245 patients. A mean of 6.7 variants per patient was found in AVADA (2.8% precision), 3.5 in HGMD (“DM”) (6.2% precision), 1.6 in ClinVar (clinical significance 4 or 5) (7.2% precision) in accordance with previous observations,2 and 3.4 in tmVar 2.0 (2.2% precision). Therefore, AVADA was less precise than the manually curated databases, but more precise than the previously best-in-class automatic variant retrieval tool tmVar 2.0.
Gene–variant pairing prediction classifier is robust to changes in training data
To examine how robust the gene–variant candidate pairing prediction classifier is to changes in training data, we first trained it on HGMD (“DM”) instead of ClinVar data. The resulting variant set is highly similar to the original, having almost (−0.6%) the same size and 97% of it identical to the original (Supplementary Fig. 2).
Next, we retrained the gene–variant candidate classifier on three different subsets of the original ClinVar-based training data, each containing a random half of the articles in the original training data (Supplementary Methods). Again, after running variant extraction on the original set of AVADA articles, the variants returned from each retraining was highly similar to the original AVADA variants, (+2.7–0.6% bigger in size and containing 98% of the original set; see Supplementary Fig. 3).
Picking the gene–variant candidate classifier threshold
To set the gene–variant classifier threshold at 0.9, we evaluated potential thresholds between 0.5 and 0.99 (Supplementary Fig. 4). Including all gene–variant candidate mapping with a gene–variant candidate classifier score of at least 0.5 increased the number of distinct genetic variants to 291,281 (+43% compared with the original AVADA database). Since we previously selected 200 distinct variants to estimate AVADA precision (Supplementary Table S3), we now selected a proportional 87 distinct random variants between confidence levels 0.5 and 0.9 that were not already in the original AVADA variant set to manually determine database precision at lower gene–variant candidate classifier score levels. Again, two reviewers independently evaluated each variant.
Overall, these 87 variants were less than 21% correctly mapped to genomic coordinates (Supplementary Table S8), compared with 61% correct mapping in the original set. Inversely, if we subset the previously verified 200 AVADA variants (Supplementary Table S3) to variants arising from gene–variant mappings scored only above 0.95 and 0.99, respectively, variant extraction precision would modestly increase to 63.5–68.5% (from 61%), but the fraction of recovered HGMD variants would decrease by 2.4–15.1% (Supplementary Fig. 4). Based on this search, we chose a gene–variant candidate classifier score threshold of 0.9 that balances precision and recall (Supplementary Fig. 4).
DISCUSSION
We studied the potential and challenges of creating an end-to-end machine learning tool for the automatic retrieval of variant evidence directly from full-text literature about Mendelian disease. AVADA automatically retrieved nearly a hundred thousand disease-causing variants from tens of thousands of downloaded and parsed full-text articles. All AVADA variants are stored in a Variant Call Format32 (VCF) file that includes the chromosome, position, reference and alternative alleles, variant descriptions as reported in the original article, and PubMed IDs of the original articles mentioning the variants.
AVADA makes a special effort not to curate just any variant mention, but rather to process only abstracts, and later full-text papers that appear to our classifiers to describe pathogenic variants in the context of Mendelian diseases. While this undoubtedly removes a great number of false positive human variant mentions, AVADA’s estimated 73% recall and 49.5% precision over relevant papers suggests that more can be done to optimize this process. AVADA also takes full advantage of recent success in allowing computerized access to the scientific literature. Its large gains over abstract-based tmVar 2.0 justify the engineering feat. However, the length and complexity of biomedical texts also result in AVADA’s 61% precision in mapping mentioned variants to their correct genomic coordinates (currently not including mitochondrial variants). Clearly computational effort should continue to bring precision up. It is also worthwhile noting that while we wrote 47 regular expressions to capture most common variant mentions, HGVS formalism does request that variant mentions be preceded with an accepted reference sequence (e.g., NP_003995.2 for the GJB2 variant in Fig. 2c). The more journals enforce and the more papers are written in strict HGVS notation, the easier the computerized coordinate conversion task becomes.
AVADA currently only tackles the first step in variant curation, that of evidence collection. Assessing the validity and power of individual papers, combining evidence from multiple, potentially conflicting papers, to arrive at variant and gene-level assertions are even more challenging tasks. In this sense, AVADA can be thought of as a much quicker (and more hit-or-miss) means of evidence hunting than PubMed or Google Scholar. To assess the validity of AVADA’s evidence we used the HGMD and ClinVar human curated databases of pathogenic variants. Neither of these is entirely accurate. For example, the ExAC paper2 highlights 68 HGMD “DM” variants (55 of which are also in AVADA) that appear in >1% of at least one of its populations, requiring re-evaluation of HGMD’s linked papers–based conclusions. With these caveats in mind, we estimate roughly 44% of AVADA-collected variants as pathogenic Mendelian ones.
Despite these limitations, AVADA does recover nearly 60% of all disease-causing (“DM”) variants deposited in HGMD at a fraction of the cost of constructing a manually curated database,9 and over four times as many as the abstract–rsID based tmVar 2.0. AVADA offers an approximate 64,617 (=96.5% * 61,180 + 4% * 115,323; Fig. 3c) disease-causing variants not present in ClinVar (136% increase over ClinVar alone), at the cost of nearly twice as many (112,886; complement of the above) additional benign or incorrectly extracted variants. In patient context if one is willing to manually validate AVADA evidence (as one is obliged to do with any compelling HGMD or ClinVar evidence as well), over the DDD example, ClinVar had evidence for 21 pathogenic variants. AVADA offers unvalidated evidence for an additional 27 variants, of which 18 were manually validated to be correctly mapped to genomic coordinates and correctly reported as disease-causing, essentially doubling ClinVar’s reach.
AVADA shows the potential to (1) improve the state of the art in machine learning–based evidence collection of literature-mentioned pathogenic variants and their mapping to reference genome coordinates, (2) enable first attempts to automate aspects of variant curation, and (3) motivate curation of benign variants as well as variants in other domains (such as cancer, mouse models, and other research fields where manually curated data may be scarce33). Combining AVADA-based rapid variant retrieval with validation will enable the creation and upkeep of cheaper, better, faster updating variant databases, which will ultimately empower both rapid diagnosis9 and reanalysis.8
Web resources
AVADA code for automatic variant evidence retrieval, and the automatically retrieved (but not validated) variant evidence database, will be available for noncommercial use at http://bejerano.stanford.edu/AVADA.
Change history
31 October 2019
The Supplementary Tables were missing in the original online version of this Article. This has now been corrected.
References
Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47:717–726.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Dewey FE, Grove ME, Pan C, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311:1035.
Smedley D, Jacobsen JOB, Jäger M, et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc. 2015;10:2004–2015.
Jagadeesh KA, Birgmeier J, Guturu H, et al. Phrank measures phenotype sets similarity to greatly improve Mendelian diagnostic disease prioritization. Genet Med. 2019;21:464–470.
Deisseroth CA, Birgmeier J, Bodle EE, et al. ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genet Med. 2019;21:1585–1593.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2016;19:209–214.
National Human Genome Research Institute (NHGRI). Clinical Genome (ClinGen) Resource. https://www.genome.gov/27558993/clinical-genome-clingen-resource/. Accessed 27 September 2018.
Westergaard D, Stærfeldt H-H, Tønsberg C, Jensen LJ, Brunak S. A comprehensive and quantitative comparison of text-mining in 15 million full-text articles versus their corresponding abstracts. PLoS Comput Biol. 2018;14:e1005962.
Doughty E, Kertesz-Farkas A, Bodenreider O, et al. Toward an automatic method for extracting cancer- and other disease-related point mutations from the biomedical literature. Bioinformatics. 2011;27:408–415.
Wei C-H, Harris BR, Kao H-Y, Lu Z. tmVar: a text mining approach for extracting sequence variants in biomedical literature. Bioinformatics. 2013;29:1433–1439.
Jimeno Yepes A, Verspoor K. Mutation extraction tools can be combined for robust recognition of genetic variants in the literature. F1000Res. 2014;3:18.
Thomas P, Rocktäschel T, Hakenberg J, Lichtblau Y, Leser U. SETH detects and normalizes genetic variants in text. Bioinformatics. 2016;32:2883–2885.
Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311.
Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–677.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868.
Wei C-H, Phan L, Feltz J, Maiti R, Hefferon T, Lu Z. tmVar 2.0: integrating genomic variant information from literature with dbSNP and ClinVar for precision medicine. Bioinformatics. 2018;34:80–87.
Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–2830.
Jurafsky D, Martin JH. Speech and language processing. 2nd ed. Upper Saddle River, NJ: Prentice Hall; 2008.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798.
Poppler. https://poppler.freedesktop.org/. Accessed 24 September 2018.
O’Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745.
Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–1586. https://doi.org/10.1038/ng.3703.
Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 2015;43:D1079–D1085.
Yates A, Akanni W, Amode MR, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–D716.
Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52–D57.
Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228.
Lappalainen I, Almeida-King J, Kumanduri V, et al. The European Genome-phenome Archive of human data consented for biomedical research. Nat Genet. 2015;47:692–695.
1000 Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073.
The UK10K Consortium. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82.
Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158.
McMurry JA, Köhler S, Washington NL, et al. Navigating the phenotype frontier: the Monarch Initiative. Genetics. 2016;203:1491–1495.
Acknowledgements
We thank Mark A. Lemley and Henry T. Greely for advice. This work was funded in part by a Bio-X Stanford Interdisciplinary Graduate Fellowship to J.B.; by grants EMBO ALTF292-2011 and NIH/NHGRI 5U41HG002371-15 to M.H.; and by DARPA, the Stanford Pediatrics Department, a Packard Foundation Fellowship, a Microsoft Faculty Fellowship and the Stanford Data Science Initiative to G.B. We are obliged to thank the European Genome–phenome Archive29 (EGA) and the Deciphering Developmental Diseases28 (DDD) project. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). De-identified DDD data were obtained through EGA. This research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
P.D.S. and D.N.C are the creators of HGMD. They receive financial support for it from Qiagen Ltd through a License Agreement with Cardiff University. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Birgmeier, J., Deisseroth, C.A., Hayward, L.E. et al. AVADA: toward automated pathogenic variant evidence retrieval directly from the full-text literature. Genet Med 22, 362–370 (2020). https://doi.org/10.1038/s41436-019-0643-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0643-6
Keywords
This article is cited by
-
X-CAP improves pathogenicity prediction of stopgain variants
Genome Medicine (2022)
-
Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases
Genome Medicine (2021)
-
The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting
Human Genetics (2020)

{kind=link}