
Abstract
Current therapeutic options for treating colorectal cancer have little clinical efficacy and acquired resistance during treatment is common, even following patient stratification. Understanding the mechanisms that promote therapy resistance may lead to the development of novel therapeutic options that complement existing treatments and improve patient outcome. Here, we identify RAC1B as an important mediator of colorectal tumourigenesis and a potential target for enhancing the efficacy of EGFR inhibitor treatment. We find that high RAC1B expression in human colorectal cancer is associated with aggressive disease and poor prognosis and deletion of Rac1b in a mouse colorectal cancer model reduces tumourigenesis. We demonstrate that RAC1B interacts with, and is required for efficient activation of the EGFR signalling pathway. Moreover, RAC1B inhibition sensitises cetuximab resistant human tumour organoids to the effects of EGFR inhibition, outlining a potential therapeutic target for improving the clinical efficacy of EGFR inhibitors in colorectal cancer.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the second commonest cause of cancer-related mortality with highly variable disease outcome and response to therapies. Part of these differences are believed to be due to extensive genomic and transcriptomic heterogeneity leading to the acquisition of rapid resistance to available treatments1. For example, the effectiveness of the anti-epidermal growth factor (EGF) antibodies cetuximab or panitumumab, which are utilised in a subgroup of KRAS wild-type (WT) CRCs is limited, with resistance rapidly emerging via multiple and diverse mechanisms2,3. Recent efforts to understand CRC complexity has led to the definition of a number of distinct consensus molecular subtypes (CMSs) based on gene expression patterns of tumour biopsies or purified tumour epithelial cells1,4. These different subtypes classify tumours with distinguishing features such as microsatellite instability (MSI), high/hypermutated/BRAF mutated (CMS1), WNT activated (CMS2), metabolic/KRAS mutated (CMS3) and EMT/TGF-β activated (CMS4). Classification into different subtypes provides useful predictive information such as patient prognosis and predicted response to therapies. For example, patients with CMS2 tumours preferentially benefit from anti-epidermal growth factor receptor (EGFR) and anti-vascular endothelial growth factor (VEGF) therapy5,6. However, even within CMS subtypes extensive heterogeneity exists and despite stratification many patients benefit only from an initial treatment response with therapy resistance frequently observed. Interestingly, recent evidence suggests that resistance to the EGFR inhibitor cetuximab can be acquired by switching molecular subtype indicating non-genetic, transcriptional mechanisms may play an important role in modulating response to therapy7. Together, this outlines a pressing need to develop both novel therapeutic options and to also better understand disease complexity to enable better stratification of available treatments.
The majority of CRC cases are initiated by loss or inactivation of the APC tumour suppressor gene with accumulated mutations in other key pathways, such as MAPK, TP53 and TGF-β promoting tumour progression8,9,10. APC is a negative regulator of the WNT signalling pathway that when lost, allows β-catenin to accumulate in the nucleus and drive an oncogenic transcription programme leading to tumour formation11,12. Previous studies have identified the WNT target gene Myc as a key mediator of oncogenic WNT signalling and demonstrated reduced MYC levels perturbs intestinal tumorigenesis13,14. MYC has long been proposed as a therapeutic target for multiple cancer types but direct inhibition of the protein has proven difficult owing to a lack of defined ligand binding sites. However, a number of pathways regulated by MYC signalling have subsequently been shown to be important for efficient tumorigenesis following APC loss demonstrating alternative mechanisms by which the outputs of oncogenic WNT signalling may be targeted15,16,17,18,19,20.
We previously identified one such pathway, RAC1 signalling, as being critical for the expansion of intestinal stem cells and subsequent tumour formation following Apc deletion in the mouse19. Activation of RAC1 is achieved by binding of Rho-Guanine Exchange factors (GEFs) and we previously identified upregulation (and subsequent RAC1 activation) of a number of these following Apc loss19. An alternative mechanism via which RAC1 signalling can be activated is via the splice variant termed RAC1B, which is overexpressed in numerous tumour types21. RAC1B results from the inclusion of exon 4 (alternatively designated exon 3b) encoding an additional 19 amino acids which leads to constitutive activation22,23,24. It is believed that RAC1B has a distinctive, more restricted set of effector pathways than RAC1, but appears to be more critical for tissue transformation25,26,27,28,29. Despite some in vitro evidence supporting a tumorigenic role for RAC1B, its in vivo function and mechanism of action is poorly understood and to date no studies have assessed whether RAC1B is required for tumorigenesis in vivo and thus the potential benefits of its therapeutic targeting.
We therefore set out to determine the requirement for RAC1B during intestinal tumorigenesis. Here we find that RAC1B is overexpressed in CRC and high RAC1B expression correlates with high WNT activity and poor prognosis. We find that deletion of Rac1b in a mouse model of intestinal cancer significantly increases survival and reduces tumour number, tumour-cell proliferation and tumorigenic WNT signalling. Mechanistically, RAC1B interacts with a network of membrane-bound receptor tyrosine kinases (RTKs) including the important oncogenic mediators EGFR and ERBB2. RAC1B is required for efficient activation of EGFR signalling and organoid cultures lacking Rac1b are sensitised to EGFR inhibition. Importantly, cetuximab-resistant human tumour organoids treated with a novel inhibitor of RAC1B splicing show this same increased sensitivity to EGFR inhibition suggesting RAC1B may be a candidate therapeutic target for co-treatment with EGFR inhibitors in CRC.
Results
RAC1B overexpression correlates with high WNT activity and poor prognosis in human CRC
To investigate the expression of RAC1B in human CRC, we analysed previously determined percent spliced in (PSI) values of RAC1B exon 4 from TCGA RNAseq experiments30. In line with previous reports, this demonstrated a significant increase in RAC1B expression in human colorectal tumours (Fig. 1A). We confirmed this using isoform expression data obtained directly from TCGA RNAseq dataset (Fig. S1A). We also observed significantly increased RAC1B expression in advanced stage (TIII and TIV) compared to early stage (TI and TII) disease, in patients with lymphovascular tumour invasion and in patients with metastatic disease (Fig. 1B). Tumours were classified as high or low for RAC1 expression taking as reference the thresholds for PSI values described previously21. Using a stringent threshold we designated three groups: RAC1Bhigh (delta PSI > 0.2 above normal tissue average, ~18% of tumours), RAC1Blow (PSI value lower than normal tissue average, ~7% of tumours) and RAC1Bint (intermediate expression, ~75% of tumours) (Fig. S1B). Confirming our expression analysis, tumours in the RAC1Bhigh group were significantly more likely to be found at a more advanced stage and with presence of lymphovascular invasion than tumours in the RAC1Blow and RAC1Bint groups (Fig. S1C, D). Furthermore, high levels of RAC1B expression were correlated with significantly reduced disease-free (hazard ratio of 2.64) and lower overall survival (hazard ratio of 2.08) (Fig. 1C). These data demonstrate that elevated levels of RAC1B correlates with poorer disease outcome in CRC.

A Individual value plot of RAC1B exon 4 PSI values for normal and colorectal tumour tissue (TCGA dataset) (data are presented as mean ± SD; ****P = 2 × 10−15; two-tailed t-test, n = 41 vs 457 tumour samples). B Individual value plots of RAC1B exon 4 PSI values from TCGA CRC dataset comparing: early (TI&II) vs late (TIII&TIV) stage CRC (left panel) (data are presented as mean ± SD; **P = 0.0029; two-tailed t-test, n = 227 vs 179 tumour samples), absence vs presence of lymphovascular invasion (middle panel) (data are presented as mean ± SD; **P = 0.004; two-tailed t-test, n = 241 vs 162 tumour samples) and absence (M0) or presence (M1) of metastasis (right panel) (data are presented as mean ± SD; ***P = 0.0008; two-tailed t-test, n = 329 vs 136 tumour samples). C Disease-free (top panel) and overall (bottom panel) survival of human CRC patients separated by high vs low/intermediate RAC1B expression. Log-rank P value, hazard ratio (HR) and 95% confidence intervals (CI) presented. D Bar plots of Pearson’s correlation coefficients for RAC1B exon 4 PSI values and genes that identify different CRC CMSs. Each bar corresponds to the correlation of RAC1B and an individual gene, red bars indicate genes positively correlated with RAC1B and blue bars genes negatively correlated with RAC1B expression (P < 0.05). E Individual value plots of RAC1B exon 4 PSI values in tumours from each CMS compared to all other tumours. CMS1 vs all; data are presented as mean ± SD; ****P = 4.83 × 10−13; two-tailed t-test, n = 71 vs 299 tumour samples. CMS2 vs all; data are presented as mean ± SD; ****P = 5.49 × 10−6; two-tailed t-test, n = 145 vs 225 tumour samples. CMS3 vs all; data are presented as mean ± SD, n = 54 vs 316 tumour samples. CMS4 vs all; data are presented as mean ± SD, n = 100 vs 270 tumour samples. F Individual value plots of WNT target gene expression in tumours expressing low vs high RAC1B. AXIN2; data are presented as mean ± SD; ***P = 0.0005; two-tailed t-test, n = 22 vs 60 tumour samples. LGR5; data are presented as mean ± SD; ***P = 0.0004; two-tailed t-test, n = 22 vs 60 tumour samples. G Disease-free survival of human CRC patients with tumours from the CMS2 CRC subtype separated by high vs low/intermediate RAC1B expression. Survival of patients with CMS4 CRC subtype tumours included for comparison. Log-rank P value shown. Source data are provided as a Source Data file.
To determine whether high RAC1B expression was associated with a particular CRC CMS, we correlated expression of RAC1B with the gene classifier sets for each individual subtype. RAC1B expression was positively correlated with the majority of genes that classify the CMS2 subtype and negatively correlated with the majority of genes that classify the CMS1 subtype (Figs. 1D and S1E). Little enrichment for either the CMS3 or the CMS4 subtype was observed. Further analysis of the transcriptional profile of RAC1B in this dataset confirmed these observations with tumours classified as CMS1 expressing low levels of RAC1B and tumours classified as CMS2 expressing high levels of RAC1B (Figs. 1E and S1F). Additionally, we found that RAC1Bhigh tumours were most common in CMS2 tumours, although notably also present in CMS4 (Fig. S1G, H). Furthermore, GSEA carried out on RAC1Bhigh vs RAC1Blow expressing tumours confirmed the positive enrichment of genes associated with the CMS2 subtype, other gene sets associated with WNT signalling activation and a negative enrichment of genes associated with the CMS1 subtype (Fig. S1I). Validating these findings, we found expression of canonical WNT target genes AXIN2, LGR5, TCF7 and ASCL2 were elevated in tumours expressing high levels of RAC1B (Figs. 1F and S1J). The same analysis revealed significantly lower expression of RAC1B in MSI tumours and/or tumours carrying mutations in BRAF as hypothesised by the negative correlation with CMS1 (Fig. S1K). These data indicate RAC1B expression correlates with high levels of WNT signalling in human CRC. Apparently contradictory to these findings, the prognosis of patients with WNT-activated CMS2 tumours is relatively good with CMS4 tumours resulting in the worst overall survival1. As RAC1Bhigh tumours are also found in CMS4 it is possible that these tumours are responsible for the poor prognosis identified for RAC1Bhigh tumours. To investigate this further, we plotted the outcome of patients with CMS2 tumours only, split according to RAC1B-expression levels. This analysis demonstrated that patients with CMS2 tumours could be separated based on RAC1B expression with high levels of RAC1B conferring survival outcomes as poor as patients with the highly aggressive mesenchymal CMS4 subtype tumours (Fig. 1G). Therefore, a significant proportion of CMS2 tumours have poor prognosis and these are marked by high levels of RAC1B expression. Together, these data demonstrate that high RAC1B expression marks a subset of tumours with high WNT activity and poor patient outcome suggesting an important role in colorectal tumorigenesis.
Rac1b is expressed in mouse intestinal crypts and its expression is increased following Apc deletion
We next examined the expression of Rac1b in mouse intestinal tissue. To do so, we fractioned intestinal epithelium into villus and crypt / crypt base fractions (Fig. 2A). Expression analysis of stem and progenitor cell markers validated the fractionation protocol (Fig. S2A). Interestingly, whilst Rac1 was fairly uniformly expressed across all fractions, Rac1b showed significantly higher expression in the crypt fractions, indicative of high expression in proliferative intestinal cells (Figs. 2B and S2A). To investigate whether Rac1b expression is enriched in intestinal stem cells, we sorted Lgr5+ cells from mice carrying the Lgr5-EGFP-ires-CreERT2 transgene (Fig. S2B). Consistent with our fractionation experiments, we found increased Rac1b expression in Lgr5+ compared to Lgr5- cells (Fig. 1C). To confirm these observations we designed a RNA in situ hybridisation BaseScope probe to detect Rac1b expression (exon 4–5 junction) (Figs. 2D and S2C). We detected low Rac1b-expression levels in mouse small intestine (SI) and large intestine (LI) with significantly higher expression observed in the crypts than the villi of the small intestine (Figs. 2D, E and S2C, D). Positional scoring of Rac1b-positive signal along the crypt villus axis confirmed Rac1b expression was found primarily in the crypt region (Fig. S2E). However, expression was not restricted to the crypt base suggesting Rac1b expression is enriched in proliferative crypt cells but is not a specific marker of Lgr5+ stem cells (Fig. S2E). Tissue from induced VilCreERT2 Rac1bfl/fl mice (described later) was completely lacking hybridisation signal demonstrating the specificity of the BaseScope probe (Fig. S2C). To determine the effects of constitutive WNT signalling activation on Rac1b expression, we deleted Apc from the mouse small and large intestine (by tamoxifen induction of VilCreERT2 Apcfl/fl mice and analysis 5 days later) and stained for Rac1b. Rac1b signal was increased in crypts and villi in both tissues indicating increased Rac1b expression following Apc deletion (Figs. 2D–F and S2C, D). Immunohistochemical analysis of β-catenin indicated increased Rac1b expression in the villus of Apc-deleted intestines correlated with nuclear accumulation of β-catenin (Fig. S2F). Again, tissue from induced VilCreERT2 Apcfl/fl Rac1bfl/fl mice was almost completely negative for hybridisation signal (Fig. S2C). We also found significant overexpression of Rac1b following Apc deletion using qRT-PCR (Fig. S2G). These data demonstrate that Rac1b expression is increased following Apc deletion and correlates with WNT signalling activation, consistent with data from human tumours.

A Schematic outlining crypt extraction protocol. B qRT-PCR of Rac1b from villus and crypt fractions (data are presented as mean ± SD; **P = 0.0018, ***P = 0.0005; one-way ANOVA with Tukey multiple correction, n = 3 biologically independent mice). C qRT-PCR of Rac1b from sorted Lgr5+ and Lgr5- cells (data are presented as mean ± SD; *P = 0.0117; two-tailed t-test, n = 3 biologically independent mice). D Representative images of Rac1b basescope of WT and Apcfl/fl small intestinal tissue. Magnified areas are shown in right panels. Pink dots are positive probe detection. Scale bars are 100 µm (left panels) and 25 µm (magnified). E Quantification of Rac1b basescope probe counts comparing WT vs Apcfl/fl small intestine separated by villus and crypt regions (data are presented as mean ± SD; *P = 0.018, **P = 0.0034, ****P = 4 × 10−6 (WT vs Apcfl/fl villus), ****P = 8.62 × 10−6 WT vs Apcfl/fl crypt); two-way ANOVA with Tukey multiple correction, n = 3v3 biologically independent mice. For each mouse, at least five areas for crypt and villus regions were scored. In total, these areas incorporated at least 50 crypt/villus axes and covered ~2 mm2 total area). F Quantification of Rac1b basescope probe counts comparing WT vs Apcfl/fl across the entire small intestine (data are presented as mean ± SD; ****P = 5.47 × 10−6; two-tailed t-test, n = 3v3 biologically independent mice). Source data are provided as a Source Data file.
Rac1b deletion attenuates intestinal tumorigenesis and tumorigenic WNT signalling
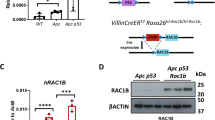
To test the functional significance of Rac1b expression in intestinal tumorigenesis we generated mice carrying a novel floxed allele permitting deletion of Rac1 exon 4 (Rac1bfl) (Figs. 3A and S3A) and bred these mice onto the tamoxifen-inducible VilCreERT2 Apcfl/+ intestinal cancer model31. In this model, tamoxifen induction leads to intestinal specific loss of a single copy of the Apc gene. When aged, stochastic loss of the WT Apc allele leads to tumour formation in the small and large intestines. We generated cohorts of VilCreERT2 Apcfl/+ (Apc) and VilCreERT2 Apcfl/+ Rac1bfl/fl (Apc Rac1b) mice, induced them with tamoxifen and aged them until clinical signs of intestinal tumorigenesis became apparent. Deletion of Rac1b significantly increased tumour-free survival compared to WT controls (median survival 138 vs 171 days) (Fig. 3B). Macroscopic and histological analysis of the intestines of these mice also revealed a significant reduction in average tumour number (54 vs 27) (Fig. 3C–E). Analysis in different regions of the intestine revealed a significant reduction in tumour number in the duodenum and jejunum, but not in the ileum or colon (Fig. S3B). qRT-PCR analysis demonstrated a significant increase in Rac1b expression in small intestinal tumours arising in Apc mice confirming our observations that WNT signalling activation increases Rac1b expression (Fig. S3C). Interestingly, analysis of tumours harvested from Apc Rac1b mice did not show the expected reduction in Rac1b expression, and analysis of individual tumours indicated around half of small intestinal and all colonic tumours expressed higher levels of Rac1b than normal intestinal tissue (Fig. S3C, D). This indicated a failure to efficiently delete Rac1b suggesting a positive selective pressure for maintaining Rac1b expression in tumour-initiating cells. Phenotypically, tumours from Apc and Apc Rac1b mice had comparable histological appearance with similar levels of apoptosis (as measured by Caspase 3 immunohistochemistry, IHC) and similar numbers of Paneth and Goblet cells (Fig. S3G, H). However, tumours from Apc Rac1b mice were significantly less proliferative than controls (Fig. 3F, G). To determine whether there is a direct association between RAC1B and tumour-cell proliferation, we used Rac1b Basescope to identify tumours from our Apc Rac1b mice that had not efficiently deleted Rac1b (Fig. S3E, F). By staining these tumours for BrdU incorporation, we found that these tumours proliferate significantly better than those that are Rac1b-negative and to a similar extent as tumours from Apc control mice (Fig. S3E, F). Thus, Rac1b expression is required for efficient tumour-cell proliferation. Importantly, Rac1b deletion in normal adjacent tissue was maintained for the duration of the experiment (~6 months) in all mice analysed (Fig. S3C, D), and cell proliferation, apoptosis, Paneth cell and goblet cell populations were unchanged suggesting Rac1b deletion is well tolerated in normal intestinal tissue (Fig. S3I, J). In addition, analysis of Rac1 expression indicated this was mostly unaffected by deletion of Rac1b exon 4 demonstrating the specificity of our knockout allele (Fig. S3K). To further investigate the tumorigenic phenotype, we utilised an ex vivo organoid clonogenic culture method to determine the tumour-initiating capacity of Apc-deficient cells in the absence of Rac1b expression (Fig. S3L, M). We generated intestinal organoid cultures from mice following acute homozygous deletion of Apc and Rac1b (Apcfl/fl and Apcfl/fl Rac1bfl/fl), digested them to single cells and determined their clonogenic capacity. Interestingly, the ability of Apc-deficient cells to form colonies from single cells was significantly reduced by Rac1b deletion (Fig. 3H). Together, these data demonstrate an important role for Rac1b expression during WNT-mediated intestinal adenoma formation.

A Schematic of Rac1b floxed allele. B Survival plot of Apc vs Apc Rac1b mice. Log-rank P value shown, n = 19v17 biologically independent mice. C Representative photographs of intestines from Apc and Apc Rac1b tumour-bearing mice. Black arrows indicate tumours. D H&E of whole intestinal rolls from Apc and Apc Rac1b mice. Box indicates magnified area. Black arrows indicate tumours. Scale bars are 2.5 mm (top panels) and 250 µm (magnified). E Quantification of total tumour number in the intestines of Apc vs Apc Rac1b mice (data are presented as mean ± SD; **P = 0.003; two-tailed Mann-Whitney test, n = 19v16 biologically independent mice). F BrdU IHC in tumours from Apc vs Apc Rac1b mice. Scale bars are 100 µm (top panels) and 50 µm (magnified). G Quantification of BrdU positivity in tumours from Apc vs Apc Rac1b mice (data are presented as mean ± SD; **P = 0.0079; two-tailed Mann-Whitney test, n = 5v5 biologically independent mice). H Clonogenicity assays of Apcfl/fl vs Apcfl/fl Rac1bfl/fl organoids (data are presented as mean ± SD; *P = 0.0228; two-tailed t-test, n = 3v4 biologically independent mice). Scale bars are 1000 µm. Source data are provided as a Source Data file.
To identify potential pathways mediated by Rac1b expression involved in tumorigenesis, we carried out RNAseq analysis on 4 vs 4 small intestinal tumours dissected from Apc and Apc Rac1b mice (confirmed negative for Rac1b by qRT-PCR). This analysis identified 506 mRNAs with significantly altered expression (fold change > 1.5, padj < 0.05). Of these, 265 had significantly increased expression and 241 had significantly decreased expression (Supplementary Data 1). Pathway analysis of our dataset identified regulation of immune response, antigen processing and viral response amongst the most upregulated pathways indicating a role for Rac1b in regulating these processes (Fig. 4A). Interestingly, amongst the most significantly downregulated pathways were the canonical WNT signalling pathway and WNT-protein binding (Fig. 4A). Furthermore, GSEA demonstrated significant enrichment of genes downregulated in our RNAseq dataset with gene sets associated with WNT signalling activation (Fig. 4B, C). We used qRT-PCR to validate the differential gene expression of some of the identified stem cell/ WNT target genes in Rac1b deficient (Fig. 4D). There was no significant expression changes in any of these target genes in normal tissue from the same mice suggesting the requirement for Rac1b is specific to tumour cells. Intestinal tumours arising in Apc Rac1b mice demonstrated strong, nuclear β-catenin positivity suggesting a role for RAC1B downstream of β-catenin nuclear localisation (Fig. S4A). This is in agreement with our previous findings that β-catenin operates upstream of RAC1 signalling in promoting intestinal tumorigenesis19. Alongside our analysis of human TCGA data, these findings suggest a key role for RAC1B in regulating tumorigenic WNT signalling in CRC.

A Bar plot showing the top 10 significantly enriched upregulated and downregulated pathways in tumours from Apc vs Apc Rac1b mice. B GSEA plots of WNT target gene sets comparing tumours from Apc vs Apc Rac1b mice. C Heatmap of RNAseq expression values of WNT target genes and intestinal stem cell markers comparing tumours from Apc vs Apc Rac1b mice. D qRT-PCR analysis of various WNT target genes comparing tumours from Apc vs Apc Rac1b mice. As controls, matched normal tissue from these mice (WT and Rac1b, respectively) are also included (data are presented as mean ± SD; two-tailed t-test; n = 6v6 mice). Source data are provided as a Source Data file.
RAC1B interacts with core components of the EGFR signalling pathway
To better understand the mechanism by which RAC1B mediates intestinal tumorigenesis, we sought to identify its interacting proteins using proximity-dependent biotin identification (BioID) (Fig. 5A). We overexpressed RAC1 and RAC1B proteins fused to the BirA ligase (and BirA-only controls) in the mouse rectal tumour cell line CMT93, treated with biotin followed by streptavidin capture and identified enriched proteins by mass spectrometry. Here, 60 enriched proteins were identified (Supplementary Data 2) with both RAC1 and RAC1B having highly similar interactomes (50 common interactors, and only 2 specific to RAC1 and 8 specific to RAC1B) (Fig. 5B). Pathway analysis of identified proteins demonstrated enrichment in adherens junctions, focal adhesions and localisation to the plasma membrane (Fig. S5A). We used NetworkAnalyist and the IMEx interactome database to visualise the protein–protein interactions of the identified proteins. This analysis identified 7 protein subnetworks with the major network clustered around membrane localised components of oncogenic signalling pathways (Fig. 5C). This network contained core members of the EGFR/ERBB (EGFR, ERBB2), IGF (IGF1R), SRC (YES1) and EPH (EFNB2) signalling pathways. Further analysis of identified proteins indicated other EGFR-related signalling components such as ASAP1 and PTPRJ32,33. A number of proteins with links to the WNT signalling pathway were also identified, such as CCNY, which activates WNT signal through LRP6 receptor phosphorylation34, and PROM1, a marker of intestinal stem/progenitor cells and colon cancer stem cells35,36,37. However, no members of the core receptor and/or destruction complex were identified indicating that, in our model, RAC1B may not function directly in the WNT signalling pathway. Interestingly, several reports have highlighted potential crosstalk between EGFR and WNT signalling pathways38,39. Additionally, the EGFR signalling pathway is a therapeutic target in CRC, so the involvement of RAC1B on its activity may have implications for the treatment of this disease. Therefore, we chose to investigate the relevance of the identified RAC1B–EGFR pathway interaction. We first validated the interaction between RAC1B and EGFR. The BioID experiment was repeated by treating BirA- and BirA-RAC1B-transfected CMT93 cells with biotin followed by streptavidin-dependent pull-down. Western blot analysis of the pull-down fractions confirmed efficient, specific biotinylation of EGFR by BirA-RAC1B and not BirA control (Fig. S5B). Thus, EGFR and RAC1B are either interacting or neighbouring proteins in CRC cells.

A Schematic of BioID protocol. B Venn diagram outlining RAC1 and RAC1B interacting proteins identified by MS and the overlap between the interactomes. C Network analysis of the RAC1/RAC1B interactome. D CMT93 cells were treated with NT or Rac1b PMO, serum-starved and then stimulated with EGF for 5, 10, 15 or 30 min before analysis by western blotting for EGFRpY1068, EGFR and Vinculin (sample control blot). Representative blots shown. E EGFRpY1068 band densitometry relative to EGFR (normalised to Vinculin) of 4 replicate time course experiments (data are presented as mean ± SEM; **P = 0.002; two-way ANOVA with Tukey multiple correction; n = 4v4). Source data are provided as a Source Data file.
RAC1B is required for efficient stimulation of EGFR signalling
We next determined whether RAC1B is required for EGFR and downstream signalling. We designed an antisense oligonucleotide to bind the putative exon splice enhancer of Rac1 exon 4 to disrupt the inclusion of this exon. Vivo-Morpholinos targeting exon 4 (Rac1b PMO) demonstrated efficient in vitro knockdown efficacy with ~95% reduction in Rac1b transcript compared to treatment with a non-targeting, control Vivo-Morpholino (NT PMO) (Fig. S5C). Total Rac1 transcript levels were not significantly altered following this treatment indicating specific inhibition of exon 4 inclusion without knockdown of total transcript (Fig. S5C). To determine the effects of Rac1b depletion on EGFR signalling, CMT93 cells were pre-treated with NT or Rac1b PMO for 2 days and then treated with 20 ng/ml EGF after serum starvation. Samples were collected for analysis 5, 10, 15 and 30 min after treatment and untreated cells grown in 10% serum or following starvation were used as controls. In control cells p-EGFR levels rapidly increased, reaching a peak at 10 min post treatment. Rac1b-deleted cells presented a reduction in the phosphorylated status of EGFR, implying lower levels of activation (Fig. 5D, E). This difference in p-EGFR was most apparent 10 min post stimulation and Rac1b PMO-treated cells never reached a level of phosphorylation as high as the NT PMO. To determine whether decreased EGFR phosphorylation translated into lower downstream EGFR pathway activation, we assessed ERK1/2 and AKT phosphorylation and found both proteins were less phosphorylated in Rac1b PMO-treated cells compared to controls (Fig. S5D, E). Replicate experiments performed in serum-starved cells with EGF treatment for 10 min confirmed these findings (Fig. S5F, G). Next, we sought to determine whether Rac1b regulates EGFR signalling in vivo in the Rac1b-deficient mice whose survival and tumorigenesis was diminished (Fig. 3). We analysed pEGFR and pERK levels in tumours derived from these mice and found that pEGFR levels are significantly lower in the Rac1b-deficient tumours but pERK levels are unchanged (Fig. S5H, I). This suggests that as tumours establish and develop, EGFR phosphorylation remains dependent on Rac1b expression but alternative mechanisms to activate pERK can be utilised. We also investigated the expression of known EGFR pathway target genes (Etv4 and Etv5). Consistent with the pEGFR reduction, expression of both is significantly lower in Rac1b deficient tumours (Fig. S5J).
To provide mechanistic insight into how RAC1B controls EGFR signalling we investigated EGFR trafficking using Alexa 555-labelled EGF (EGF-555). Confocal analysis of EGF-555-stimulated cells indicated that internalisation of EGF-555 was not significantly impaired in Rac1b-depleted cells (Fig. S6A). However, the localisation of internalised EGF-555 appeared altered. In control cells, we observed EGF-555 foci throughout the cell, whereas in Rac1b-depleted cells we observed them around the cell periphery (Fig. S6B, inset panels). To investigate this in more detail, we used live cell imaging to monitor overall endocytic trafficking of EGFR using EGF-555. Tracking the paths of EGF-555 vesicles revealed that the dynamic trafficking pattern in control cells was restricted following Rac1b depletion (Fig. S6A, C). We found that both total EGF-555 displacement and tracking velocity were significantly lower in Rac1b depleted cells suggesting altered EGFR trafficking (Fig. 6B, C). To investigate this further, we assessed the co-localisation between EGFR and the early endosome marker Rab5. In agreement with our previous observations that EGF-555 internalisation was unaffected by Rac1b depletion, EGFR co-localisation with Rab5 endosomes following EGF-555 stimulation was unchanged in Rac1b depleted cells (Fig. S6D). Together, this suggests Rac1b depletion does not prevent EGF internalisation into early endosomes but leads to an impairment of subcellular EGFR trafficking. Following internalisation, EGFR can be either recycled back to the cell surface to maintain signalling or directed towards multivesicular bodies for lysosomal targeting40. To investigate whether the absence of RAC1B promotes a shift in EGF-bound EGFR destination fate, NT and Rac1b PMO cells were stimulated with EGF-555 and treated with LysoTracker Green dye to identify lysosomal compartments. In control cells, co-localisation of labelled EGF and LysoTracker was observed from 30 min post EGF stimulation. However, Rac1b-depleted cells present significantly more EGF-555 Lysotracker clusters starting at earlier time points suggesting that deletion of Rac1b promotes EGFR lysosomal sorting and receptor degradation over the recycling route (Fig. 6D, E). Together, these data suggest RAC1B is required for efficient trafficking and recycling of EGFR, thus potentiating its activity.

A Live cell imaging of Alexa 555‐EGF tracking with time and velocity represented as colour spectrums. Tracking started after addition of EGF-555 and was carried out for the indicated time period. B Individual value plot of quantification of tracking distance of EGF-555-positive vesicles (median value indicated; ****P = 1.67 × 10−13; two-tailed t-test; n = 1213 vs 658 foci). C Individual value plot of quantification of tracking velocity of EGF-555-positive vesicles (median value indicated; **** P = 6.93 × 10−11; two-tailed t-test; n = 1213 vs 658 foci). D Representative images of live cell imaging of Alexa EGF-555 (magenta) and LysoTracker Green (green) at indicated time points post EGF-555 addition. White arrows indicate areas of EGF-555 LysoTracker co-localisation. Co-localised foci are observed as white dots. Scale bars are 10 μm. E Quantification of the percentage of EGF-555 foci co-localised with LysoTracker Green at indicated time points (data are presented as mean ± SD; *P = 0.0263, **P = 0.0094 (25 min), **P = 0.0094 (30 min), two-way ANOVA with Tukey multiple correction; n = 3v3 independent treatment experiments). Source data are provided as a Source Data file.
We next investigated how RAC1B modulates WNT signalling, in particular the potential link between these signalling pathways in intestinal cancer. Firstly, we investigated the impact of EGFR inhibition on WNT target gene expression in Apc-deficient organoids and found that, similar to following Rac1b deletion, a subset of targets were downregulated (Fig. S6F). In addition, we depleted Rac1b in Apc-deficient organoids carrying an additional activating mutation of Kras. In these organoids, where MAPK signalling is constitutively active, downstream of EGFR, the depletion of Rac1b had no effect on organoid clonogenic capacity or WNT target gene expression (Fig. S6G–J). We also investigated the possible involvement of NF-κB signalling and ROS activity but found these were unaltered in Rac1b deficient organoids (Fig. S6K, L). Interestingly, we have recently shown that deletion of a number of RacGEFs leads to reduced ROS activity in Apc-deficient tissue, suggesting that RacGEF-activated RAC1 controls ROS production but RAC1B does not (Pickering et al.41). Together, these data suggest there is crosstalk between WNT and EGFR signalling pathways in intestinal cancer and RAC1B mediated control of EGFR signalling activation is required for efficient WNT signalling activity.
RAC1B depletion sensitises EGFR inhibitor-resistant colorectal cancer liver metastatic organoids to cetuximab treatment
These results suggest that RAC1B modulates EGFR receptor and signalling activation. To determine whether RAC1B stimulation of EGFR signalling was an important mediator of tumorigenic growth, we grew Apcfl/fl and Apcfl/fl Rac1bfl/fl intestinal organoids in the presence or absence of EGF ligand. Whereas in the presence of EGF organoids from both lines grew to a comparable size, in its absence the growth of those lacking RAC1B was significantly perturbed (Fig. S7A). We found similar results when the same organoid lines were treated with the EGFR inhibitor PD153035, with Apcfl/fl Rac1bfl/fl organoids being significantly more sensitive to this treatment (Figs. 7A, B and S7B). To determine whether inhibition of RAC1B might cooperate with EGFR inhibition to increase the efficacy of treatment in human tumour organoids, we designed a Vivo-Morpholino to target human RAC1 exon 4 (hRAC1B PMO) (Fig. 7C). We cultured patient-derived organoids (PDOs) from a benign colonic polyp and a stage 3 invasive tumour (T2N1M0) from a familial adenomatous polyposis (FAP) patient undergoing tumour resection. PDOs treated with hRAC1B PMO demonstrated a robust (<95%) knockdown in RAC1B transcript and protein levels (Figs. 7D and S7C). As before, total RAC1 transcript was not significantly altered (Fig. 7D). To determine the effects of RAC1B knockdown on human tumour organoid growth, we pre-treated PDOs derived from the benign colonic polyp with hRAC1B or NT PMO, digested to single cells and carried out clonogenicity experiments in the continued presence of morpholino. In agreement with our mouse in vivo and in vitro data, RAC1B depletion led to a significant reduction in clonogenic capacity (Fig. S7D). Interestingly, organoids derived from the more advanced invasive tumour resected from the same patient did not show reduced clonogenicity suggesting depletion of RAC1B alone is unable to suppress proliferation of later stage, more aggressive tumours (Fig. S7E). However, removal of EGF ligand or addition of EGFR inhibitor led to a significant reduction in growth of tumour organoids when combined with RAC1B knockdown (Fig. S7E). Collectively, these results indicate a functional relevance for RAC1B-dependent EGFR regulation and suggest a potential beneficial therapeutic effect when both are simultaneously inhibited. To address this in a more clinically relevant model, we obtained previously described cetuximab-resistant human CRC liver metastatic organoids42. Two independent organoid lines were tested, C001 and C002, both are KRAS WT, both express high levels of RAC1B compared to the stage 3 invasive tumour utilised above (Fig. S7F) and both show resistance to cetuximab treatment in vitro. In addition, C002 was derived from a patient who acquired resistance to cetuximab upon treatment. In line with previous reports, treatment of both organoid lines with cetuximab did not affect organoid growth (Figs. 7E, F and S7G–J)42. However, combining cetuximab treatment with RAC1B depletion led to a significant inhibition of organoid growth (Figs. 7E, F and S7G–J). These results suggest the cooperation between RAC1B and EGFR signalling we identified in our mouse models is conserved in human CRC, and depletion of RAC1B can enhance the potency of EGFR inhibition in cetuximab-resistant tumours.

A Representative images of Apcfl/fl and Apcfl/fl Rac1bfl/fl organoids grown with or without the EGFR inhibitor PD153035. Scale bars are 1000 µm. B Quantification of organoid size 20 h after addition of PD153035 (data are presented as mean ± SD; **P = 0.0061; two-way ANOVA with Tukey multiple correction; n = organoids from 3v3 mice). C Schematic outlining design of RAC1B PMO. D qRT-PCR analysis of PDOs treated with control or RAC1B PMO (data are presented as mean ± SD; **P = 0.0019; two-way ANOVA with Tukey multiple correction; n = 3v3 independent treatment experiments). E Representative images (left panel) and quantification (right panel) of C001 organoid PDOs treated with control or RAC1B PMO in the presence or absence of cetuximab (left panel) (data are presented as mean ± SD; **P = 0.0011; two-way ANOVA with Tukey multiple correction; n = 4v4 independent treatment experiments). Scale bars are 1000 µm. F Representative images (left panel) and quantification (right panel) of C002 organoid PDOs treated with control or RAC1B PMO in the presence or absence of cetuximab (left panel) (data are presented as mean ± SD; **P = 0.0019; two-way ANOVA with Tukey multiple correction; n = 4v4 independent treatment experiments). Scale bars are 1000 µm. Source data are provided as a Source Data file.
Discussion
Here, we demonstrate that RAC1B mediates intestinal tumorigenesis. Using genetic mouse models we show that RAC1B is required for efficient formation of intestinal tumours with deletion of Rac1b leading to a reduction in tumour number, reduced tumour proliferation and decreased oncogenic WNT signalling. We find that RAC1B interacts with EGFR, and controls the activation of the EGFR signalling pathway by mediating its intracellular trafficking providing a plausible mechanism for its pro-tumorigenic functions. Using a novel inhibitor of RAC1B splicing, we demonstrate enhanced sensitivity to EGFR inhibition following RAC1B depletion in cetuximab-resistant human tumour organoids. We also find that in human CRC, high levels of RAC1B associate with elevated WNT signalling and poor prognosis. Together, these findings highlight an important oncogenic function of RAC1B and suggest therapeutic targeting of RAC1B may enhance the efficacy of EGFR inhibition.
Previous findings from in vivo models have shown that Rac1b overexpression alone is insufficient to drive tumorigenesis in both intestine and lung43,44, suggesting RAC1B function is important for tumour initiation and progression in cooperation with other oncogenic driver events. Our analysis of TCGA data is in agreement as it shows a significant association of high RAC1B expression with later tumour stage, presence of metastatic disease and poor prognosis. We also identified a significant correlation of elevated RAC1B and the WNT high, CMS2 CRC subtype and a negative correlation of RAC1B expression and the MSI high, BRAF-mutated CMS1 subtype. Apparently contradictory to the survival data, CMS2 subtype CRC is characterised by relatively good prognosis1. We subsequently found that survival of patients with CMS2 subtype tumour could be separated based on RAC1B expression, with the poorest prognosis in patients with high RAC1B. This subgroup of CMS2 patients perform as poorly as those with highly aggressive CMS4 subtype tumours suggesting a potential role for RAC1B in promoting tumorigenesis. It is possible that RAC1B is merely a marker of WNT-activated CRC, however, the suppression of WNT signalling in RAC1B-deficient tumours derived from our intestinal cancer model suggests that RAC1B is a functional mediator of WNT signalling. Another characteristic of CMS2 tumours is a favourable response to treatment with EGFR inhibitors. Interestingly, other CRC subtyping attempts have identified a WNT-activated subtype (termed TA) with relatively good prognosis45. However, the TA subtype contains two distinct populations that differ in their responsiveness to EGFR inhibitors termed TA-CS (cetuximab sensitive) and TA-CR (cetuximab resistant). Intriguingly, the TA-CR group displays high WNT activity and poor prognosis, similar to the RAC1B high, CMS2 group we have identified in this study. Therefore, it is tempting to speculate that high RAC1B expression plays a functional role in promoting the WNT high, CMS2 subtype and may promote characteristics of the cetuximab-resistant TA-CR subtype. If this is the case, inhibition of RAC1B could be of therapeutic benefit to patients with RAC1B high, CMS2 subtype tumours.
A number of previous studies have linked RAC1 and RAC1B to WNT signalling; so to define the mechanism by which RAC1B modulates tumorigenesis, we investigated its interactome using BioID. We found a number of proteins linked to WNT signalling, including CCNY and PROM1 but no core WNT receptor or signalling components were identified. The interactions with CCNY and PROM1 warrant future investigation but it is also possible that RAC1B modulates alternative pathways that cooperate with WNT signalling, to drive tumorigenesis. Of particular note was our identification of numerous RTKs as RAC1/RAC1B interacting proteins including the epidermal growth factor receptors EGFR and ERBB2. Both are well-defined drivers of oncogenesis, and inhibitors of EGFR are used clinically for the treatment of KRAS WT CRC. Interestingly, numerous reports have identified crosstalk between EGFR and WNT signalling. In particular, a recent study has shown that EGFR is required for WNT signalling via FZD9B in haematopoietic stem cells. This study shows that phosphorylation of FZD9B by EGFR promotes internalisation and signal transduction of the WNT9A–FZD9B–LRP complex providing evidence of a direct link between these pathways38. It is not clear if this mechanism is important for receptor-independent WNT activation such as following Apc loss, but our observations of changes to WNT target gene expression following EGFR inhibition suggest that links between these pathways are maintained under such conditions. In our study, we show that RAC1B depletion attenuates EGFR phosphorylation and downstream phosphorylation of AKT and ERK. Thus, efficient EGFR pathway activation requires RAC1B. Activation of EGFR signalling is controlled, in part, by the endocytic machinery, which directs receptors towards recycling or lysosomal sorting. Given the requirement for RAC1B to maintain EGFR phosphorylation after EGF stimulation, we speculated that RAC1B might interfere in EGFR intracellular dynamics. Indeed, using live cell imaging we observed altered EGFR trafficking upon Rac1b depletion and increased EGFR co-localisation with lysosomes. Therefore, it appears the depletion of Rac1b modifies EGFR trafficking, leading to increased lysosomal sorting and thus reduced activity. Our findings are in agreement with other studies that have demonstrated crosstalk between RAC1 and EGFR signalling pathways. For example, EGFR has been shown to activate RAC1 by accumulating the RacGEF TIAM1 at the membrane and promoting its GTP-bound state46. Additionally, EGF has been shown to promote RAC1B expression itself through modulation of the splicing factor HNRNPA1 although we did not find direct evidence of this in our model47. Together with our data, this suggests a complex network of interacting signalling pathways where WNT signalling can promote RAC1B expression, which in turn promotes efficient activation of EGFR, thus driving tumorigenesis.
In this study, we have developed a novel method of modulating RAC1B splicing in vitro using an antisense morpholino. Antisense morpholinos have previously been utilised to induce exon skipping, with the approach showing promise as a therapeutic option for Duchenne muscular dystrophy and Spinal muscular atrophy48. Targeting RAC1B in this way is extremely specific and efficient, therefore, it may be a viable way of inhibiting the splicing of RAC1B without disrupting the function of RAC1 itself. The effects of RAC1B depletion in human-derived tumour organoid cultures corresponded well with our in vivo findings. RAC1B depletion in a benign polyp was sufficient to significantly attenuate clonogenic capacity, but in organoid cultures derived from invasive and metastatic tumours it had little effect. This would likely rule out the possibility of targeting RAC1B as a single-agent therapeutic in late stage disease. However, treating RAC1B-depleted tumour cells with an EGFR inhibitor led to a significant reduction in the clonogenic growth of these cells. Importantly, we also found that depletion of RAC1B in cetuximab-resistant liver metastatic organoids sensitised them to cetuximab treatment. The EGFR inhibitors, cetuximab and gefitinib, are utilised for the treatment of late stage KRAS WT CRC. Although initial responses are observed in some patients, resistance rapidly emerges leading to disease progression. In some cases, resistance is driven by activation of MAPK pathway via mutation of genes such as KRAS, NRAS, NF1 or BRAF. In others, no obvious mutational drivers of resistance are present, suggesting non-genetic drivers of resistance. Intriguingly, a recent paper has suggested the acquisition of resistance to cetuximab can occur via the transcriptional switching from CMS2 to CMS4 subtypes7. CMS4 CRC is characterised by an EMT-like, TGF-β-activated transcriptome. We observed high RAC1B expression in a proportion of CMS4 tumours and it has previously been demonstrated that RAC1B can drive EMT, suggesting a potential functional role for RAC1B in this process49. In addition, activation of lncRNA MIR100HG-derived miR-100 and miR-125b have been observed in CRC cells that have acquired resistance to cetuximab50. These microRNAs promote WNT signalling activation via repression of multiple negative regulators of the pathway suggesting WNT signalling activation may be an important mechanism driving cetuximab resistance. Whilst our strategy of depleting RAC1B using morpholinos remains unproven in vivo, our proof-of-principle findings that depletion of RAC1B sensitises tumour organoids to EGFR inhibition in vitro suggest RAC1B may be considered as a potential therapeutic target for enhancing the efficacy of clinically used EGFR inhibitors.
Methods
Mouse experiments
All mouse experiments were performed under the UK Home Office regulations and all relevant ethical regulations were adhered to. The study protocols were approved by University of Edinburgh AWERB. Mice were bred at the animal facilities of the University of Edinburgh and the Beatson Institute, were kept in 12 h light–dark cycles and were given access to water and food ad libitum. Mice were maintained in a temperature- (20–26 °C) and humidity- (30–70%) controlled environment. Mice (of both genders) were induced with tamoxifen between the age of 6 and 12 weeks, once they had reached a minimum weight of 20 g. Mice colonies had a mixed background (50% C57Bl6J, 50% S129). The genetic alleles used for this study were as follows: vil-Cre-ERT2 51, Apcfl 52 and Rac1bfl. Mice were genotyped by Transnetyx (Cordoba, USA). To achieve deletion of RAC1B protein without affecting RAC1 expression, loxP sites were inserted on either side of exon 4 of the mouse Rac1b transcript (Rac1-202; ENSMUST00000100489.3; Chromosome 5: 143,509,597 – 143,509,653). Details are given in “Supplementary Materials and Methods”. For long-term Cre recombination in tumour cohorts, mice were induced with a single intraperitoneal (IP) injection of 80 mg/kg of tamoxifen and were aged until symptomatic of disease (rectal bleeding, weight loss, hunched position and/or pale feet). For short-term Cre recombination, two consecutive IP doses of 120 mg/kg and 80 mg/kg of tamoxifen were administrated to the mice and they were sacrificed at day 5 post induction. Mice were randomly distributed by sex and age and a minimum of 15 or 3 mice were used for long- and short-term experiments, respectively. Proliferation was evaluated by a single IP injection of 200 µl of BrdU cell proliferation label (GE, 1392253) 2 h prior to termination. In long-term tumour cohort mice, tumour number and burden were macroscopically quantified in situ after mouse termination.
Analysis of TCGA data
For isoform expression analysis of the TCGA COAD dataset, normal and matched tumour tissue expression data of RAC1 (uc003spx.3) and RAC1B (uc003spw.3) isoforms were downloaded from the TCGA Synapse group (accession doi:10.7303/syn300013) and the IlluminaHiSeq_RNASeqV2 study. For RAC1B PSI analysis, data from each tumour type containing normal tissue expression were downloaded from the TCGASpliceSeq web-based platform30. RAC1B high and low groups were categorised according to whether PSI value was above 0.2 of normal tissue PSI average (high) or below the average of normal tissue PSI (low). Information about patient survival, lymphovascular invasion and metastasis was extracted from CBioPortal (cbioportal.org), as well as the mutational data of TCGA tumours and their CMS classification. For CMS and RAC1B correlation analysis, RSEM-calculated reads of TCGA COAD tumours’ dataset were downloaded from Wang et al. study53 and genes of each CMS subtype were correlated to its RAC1B PSI value. Moreover, the expression of some genes of interest was grouped according to RAC1B high or low tumours. Tumour stage data, MSI status and the presence of BRAFV600E mutation were acquired from the clinical data published by the CRC Subtyping Consortium (CRCSC) (accession doi: 10.7303/syn2623706).
RNA sequencing and analysis
Following RNA isolation using Qiagen RNeasy Mini Kit from intestinal tumours, RNA integrity was evaluated on an Agilent 2200 Bioanalyser and a RIN over 8 was considered optimal for analysis. Illumina RNAseq and library preparation were performed by Edinburgh Genomics (UK). Transcript abundances were quantified using kallisto54 from a transcriptome index compiled from coding and non-coding cDNA sequences defined in GRCm38 (Ensembl 95). Differential expression was called using DESeq255 at the gene level after importing estimated counts per transcript from kallisto using tximport56. Significant genes were considered for pathway analyses using the Innate DB database (innatedb.com). Genes were pre-ranked and analysed for Gene Set Enrichment Analysis (GSEA, v3.0) to generate enrichment plots for Apc-, Wnt-, Lgr5- and β-catenin-related target genes’ datasets.
Organoid culture
For organoid culture, intestinal epithelial cells were isolated as detailed in the ‘Intestinal epithelial isolation and fractionation’ section below. During cell fractionation step, fractions 3 and 4 were combined, washed with 20 ml of Advanced DMEM/F-12 medium (ADF, Gibco) supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-Glutamine and 10 mM HEPES (Life Technologies, 15630080). This was centrifuged at 300g for 3 min, supernatant was discarded and the pellet was resuspended with 10 ml of supplemented ADF (from now on, ADF) and filtered through a 70 µm cell strainer to obtain isolated crypt fragments. Crypts were centrifuged at 700g for 10 min and pellet was carefully resuspended with 500 µl of Cultrex PathClear Reduced Growth Factor BME (Bio-Techne, 3533–010–02). In a pre-warmed 24-well plate, a volume of 20 µl/well of BME was plated. The plate was incubated for 10 min at 37 °C to allow BME solidification and 500 µl of growth medium was added. Growth medium was composed of ADF, 1X N2 (Gibco, 17502048), 1X B27 (Gibco, 17504044), 50 ng/ml EGF (Peprotech, 315–09–500) and 100 ng/ml Noggin (Peprotech, 250–38–500). PDOs were established and grown as previously described57. Ethical approval for human CRC organoid derivation was carried out under NHS Lothian Ethical Approval Scottish Colorectal Cancer Genetic Susceptibility Study 3 (SOCCS3) (REC reference: 11/SS/0109). Briefly, tumours were cut and washed with ADF in a 15 ml Falcon tube. Tumoral pieces were digested with 2 ml of digestion medium containing 1700 µl ADF, 1 mg/ml Collagenase II (Sigma, C1764), 0.5 mg/ml Hyaluronidase (Sigma, H3506) and 10 µM Y-27632 (Tocris, 1254). Digestion was performed at 37 °C for 90 min with vigorous pipetting every 15 min to facilitate digestion. Digestion was stopped by adding 100 µl of 1% BSA (bovine serum albumin). Cells were passed through a 40 µm cell strainer to obtain single cells and centrifuged for 3 min at 600g. Pellet was resuspended with 500 µl of BME and plated in a pre-warmed 24-well plate. PDOs were grown in ADF medium containing 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-Glutamine, 10 mM HEPES, 1X Primocin, 1X B27, 1.25 mM NAC, 10 mM Nicotinamide, 50 ng/ml EGF, 500 nM A83-01, 10 µM SB202190, 100 ng/ml Noggin, 1 µM PGE and 10 nM gastrin. The organoid cultures were passaged every 3–4 days.
Depletion of Rac1b with an antisense morpholino
Two antisense morpholinos were designed to specifically target the exon splice enhancer region of exon 4 in the mouse and human Rac1/RAC1 transcript:
mRac1b PMO - GTCTATCTTTACCACATGTGTCTCC
hRAC1B PMO - TCCTTACCGTACGTTTCTCCAACCT
Binding led to the skipping of the exon 4 and prevented translation of RAC1B protein. Morpholinos conjugated to an octa-guanidine dendrimer delivery moiety (Vivo-Morpholino, PMO) were purchased from GeneTools (USA) and a non-targeting morpholino (NT PMO) was used as control. For depletion of Rac1b in the CMT93 cells, 2.5 × 105 cells were seeded in a 6-well plate and the medium was supplemented with 2.5 µM of Rac1b PMO or NT PMO for 24 h. For depletion of RAC1B in human organoids, these were treated with 1 µM hRAC1B PMO or NT PMO for 96 h. Further experiments using 2D cells or 3D organoids deficient for RAC1B were conducted following this pre-treatment with morpholino. Morpholino was kept in the medium over the duration of the experiment.
Clonogenicity assays
Plated mouse organoids were washed and digested into single cells through incubation with 500 µl of StemPro Accutase solution (Life Technologies, A1110501) at 37 °C for 15 min. ADF was added to stop digestion and cells were filtered through a 40 µm cell strainer. Cells were spun down for 5 min at 600g, the pellet was resuspended with 2 ml of ADF and cells were automatedly counted using the Countess machine (Thermo Fisher). In all, 10,000 single cells were plated per 5 µl BME drop, and a minimum of 4 drops was plated per each genotype. Growth medium was added as usual and formed clones were counted after 4 days. The percentage of clonogenic capacity was calculated as the average percentage of spheres formed in each drop. This experiment was repeated with at least 3 biological replicates and statistical differences were calculated. For PDOs, organoids were digested with TripLE at 37 °C for 45 min with regular mixing. Single cells were filtered and counted as above and 2500 cells plated per 5 µl BME drop and a minimum of 4 drops was plated for each treatment group. Clone formation was scored 14 days later.
Data plotting and statistical analysis
All statistical analyses and graphs were performed using GraphPad Prism v7.0 software (La Jolla, USA) unless otherwise stated. Student’s t-test was used for the comparison of two groups following normality. In contrast, groups that did not follow normality were analysed with the non-parametric Mann-Whitney test. Survival curves and percentage of survival were calculated with a Log-rank (Mantel-Cox) test. Pearson correlation was used to evaluate the correlation between two datasets, while Fisher’s test was used to assess the association between two categories. All P values less than 0.05 were considered statistically significant.
Generation of Rac1b fl allele
A conditional allele of Rac1b (Ensembl ID: ENSMUSG00000001847 in mouse genome assembly GRCm38.p6) was created by inserting loxP sites on either side of exon 4 (ENSMUSE00000648127; Chromosome 5: 143,509,597 - 143,509,653) of the Rac1b transcript (Rac1-202; ENSMUST00000100489.3). Small targeting vectors spanning the insertion sites of the loxP sites on the 5ʹ and 3ʹ side of Rac1b exon 4 were synthesised (GeneArt). An F3-Neo cassette and an Frt-Hygro cassette were inserted into the 5ʹ and 3ʹ loxP synthetic DNA plasmids, respectively, by co-transfection into EL250 Escherichia coli58, which express Flp recombinase under arabinose induction. The loxP FRT Hygro DNA fragment bounded by the homology arms was excised and recombineered58 into a mouse genomic DNA BAC clone (Source Biosciences) carrying the mouse Rac1b gene in EL250 E. coli59. The Hygro cassette was then removed by arabinose-induced Flp expression. Subsequently, the loxP F3 Neo DNA fragment was introduced into the same Rac1b BAC clone by recombineering. A linearised retrieval plasmid was generated by PCR of a p15A vector backbone with oligos incorporating 70 bp Rac1b homology arms60. The modified Rac1b sequences were retrieved from the BAC clone in EL250 E. coli by recombineering. The retrieved plasmid represents the targeting vector with approximately 6.5 and 3.4 kb homology arms. The targeting vector was linearised and transfected into HM1 mESCs61. Cells were selected under G418 (250 µg/ml) and surviving colonies picked and screened for targeting by long range PCR (using the Roche Expand Long Template PCR System) from within F3-Neo cassette to sequences beyond the ends of the homology arms. Oligo sequences used to screen cells to ensure appropriate targeting of the Rac1b gene were ATGTGGTATAGCTGTTCCCTGGTC and CTAGAGCTTGCGGAACCCTTAATG (7.3 kb) for the 5ʹ side and CTACTTCCATTTGTCACGTCCTGC and GTTGAGATGTGGTCCATGCTAAGC (5.3 kb) for the 3ʹ side. The presence of the isolated loxP site was confirmed by PCR with TTGGAGACACATGTGGTAAAGATAG and ACAGAACACCAGAGTCAGAGAAGAG (424 bp) and confirmed by digestion of the PCR product with XbaI, which is within the linked FRT site. Following identification of correctly targeted clones, mouse lines were derived by injection of ES cells into C57BL/6J blastocysts according to standard protocols. After breeding of chimeras, germline offspring were identified by coat colour and the presence of the modified allele was confirmed with the 3ʹ loxP primers described above. Mice were subsequently crossed with a mouse line expressing Flpe (Tg(ACTFLPe)9205Dym) to delete the selectable marker by recombination at the FRT sites62. Deletion with the selectable marker was confirmed by PCR across the remaining F3 site with the oligos CCCACAGATGAAACCAGGAG and GCTCAGCGTTCAGAAAGTGG (495 bp).
Immunohistochemistry
Intestinal tissue was harvested, flushed with PBS solution and fixed as “Swiss-roll” sections in PFA for 24 h at 4 °C. For tumour scoring, intestines were fixed in Methacarn (60% methanol, 30% chloroform and 10% glacial acetic acid). Tissue was automatedly processed through the Tissue-TeK VIP infiltration Processor (Sakura) for paraffin embedding and cut into 5 µm sections with the microtome (Leica). Standard IHC techniques were conducted during this study. Antibodies used were as follows: BrdU, 1:500 (Bioss, bs-0489H), β-catenin, 1:50 (BD Biosciences, 610154), cleaved Caspase 3, 1:800 (R&D), Lyz1 (DAKO, A009), Muc2 (Genetex, GTX100664), EGFRpY1068, 1:25 (Cell Signalling, 3777S), EGFRpY1068, 1:400 (Abcam, ab40815) and ERK1/2pT202/Y204, 1:100 (Cell Signalling, 4370S). At least 3 different mice of each genotype were used as biological replicates in every IHC experiment. Scoring of the staining was done blinded for evaluation and representative images were selected. Images were digitalised using the Nanozoomer Digital slide scanner (Hamamatsu) and analysed with the viewer software NDP.view2 (Hamamatsu). Scoring of tumour sections was automatedly performed using the QuPath software (qupath.github.io), whilst proliferation scoring of normal intestine was carried out manually.
BaseScope™ in situ hybridisation
A specific BaseScope™ probe was designed against the exon 3–4 junction of the transcript variant 1 of Rac1 (NM_001347530.1), which included the following nucleotides: GTTGGAGACACATGTGGTAAAGATAGACCCTCCAGGGGCAAAGACAAGCCGATTGCCGACGTGTTC. The probe was purchased from Advanced Cell Diagnostics (ACD Bio-Techne, UK) and BaseScope™ in situ hybridisation was conducted on formalin-fixed intestine tissues embedded in paraffin blocks following company instructions. For control of RNA integrity in tissue sections, sections were also hybridised with a reference positive-control probe. Three different mice per each genotype were used for the experiment and positive dots per tissue area were scored manually by blind scoring. For false colour analysis, to enhance visibility of Basescope dots, the image was split into red, green and blue channels in ImageJ. A threshold (180/255) was set in the blue image, and the colour channels remerged.
Western blotting
Proteins from organoids and cellular pellets were extracted using RIPA buffer (Sigma, R0278) supplemented with 1% of phosphatase and protease inhibitors (Sigma, P0044 and P8340). BCA Protein Assay kit (Pierce) was used to determine protein concentration. Here, 10 µg of denatured protein lysate was separated by electrophoresis in 4–12% Bis-Tris protein gels (NuPage, Thermo Fisher) and wet transferred onto methanol pre-activated nitrocellulose membrane. Quick staining with Ponceau solution was used to evaluate transfer efficiency and following PBST washes (PBS with 0.1% Tween, Sigma), membrane was blocked for 1 h at RT with 5% milk/PBST (dried milk, Marvel) or 3% BSA/PBST (BSA, Sigma) when probing phosphorylated proteins. The membrane was incubated with primary antibody diluted in the blocking solution o/n at 4 °C and with anti-rabbit or anti-mouse IgG (HRP linked, Cell Signalling) for 1 h at RT. Primary antibodies and concentrations used were as follows: AKT, 1:2000 (Cell Signalling, 9272S); AKTp5473, 1:3000 (Cell Signalling, 4060S); EGFR, 1:1000 (Cell Signalling, 2232S); EGFRpY1068, 1:500 (Cell Signalling, 2234S); ERK1/2, 1:2000 (Cell Signalling, 4695S); ERK1/2pT202/Y204, 1:3000 (Cell Signalling, 4370S), NF-κB p65, 1:1000 (Abcam, ab7970), NF-κB p65pS536, 1:1000 (Cell Signalling, 3033S), Rac1b (Millipore, 1:1000), Vinculin (Abcam, 1:5000), Streptavidin-HRP, 1:10,000 (Abcam, ab7403), Myc-tag, 1:10,000 (Cell Signalling, 2276S) and β-actin, 1:5000 (Cell Signalling, 4970S). Antibody signal was detected by chemiluminescence using the ECL Plus Substrate (Thermo Scientific) following product instructions, and the membrane was developed at the darkroom with ECL hyperfilm (Amersham). Bands’ densitometry was assessed using FIJI (ImageJ). To allow subsequent use, membranes were stripped by incubation with a Stripping solution (Millipore, 2504) for 15 min at RT.
RNA isolation
Intestinal tissue and tumours preserved in RNAlater (Sigma, R0901) and pellets from tumour organoids or 2D cells were used to isolate RNA. RNA extraction was conducted using the Qiagen RNeasy Mini Kit (Qiagen) and the protocol was performed according to the manufacturer’s instructions. For tissue RNA extraction, a homogenisation pre-step with stainless-steel beads and the Qiagen Tissuelyser LT (Qiagen) was carried out. Genomic DNA contamination was removed with the DNA-free removal kit (Ambion/Applied Biosystems, AM1906) and RNA concentration was quantified with the Nanodrop ND-100 spectrophotometer (Thermo Fisher).
cDNA synthesis and quantitative qRT-PCR
Here, 1 µg of RNA was reverse transcribed to cDNA using 4 µl of qScript cDNA SuperMix reagent (Quanta Bioscience, 95048–100) in a final reaction volume of 20 µl. PCR cycling conditions were as follows: 5 min incubation at 25 °C, DNA polymerisation at 42 °C for 30 min and enzyme deactivation at 85 °C for 5 min. PCR product was diluted 1:10. For the qRT-PCR experiment, a reaction mixture of 20 µl containing 10 µl of SYBR Master mix (Life Technologies, A25742), 0.5 µM of each reverse and forward primer, 5 µl of cDNA template and 4 µl of RNase-free water was prepared. The list of primers used are shown in Table S1. Reactions were conducted in duplicate and β-actin was used as a reference gene for CT-value normalisation. Amplification was conducted using the CFX Connect Real-Time System machine (Bio-Rad) and cycling conditions were as follows: a pre-incubation step of 95 °C at 15 min, followed by an amplification step of 95 °C for 10 s, 60 °C for 30 s and 72 °C for 30 s repeated for 44 cycles, and a melting curve analysis from 65 °C to 95 °C in 0.5 °C intervals.
BioID protein interactome
A plasmid encoding the BirA* enzyme tagged to a myc tag at the C-terminus was purchased from Addgene (Myc-BioID2-MCS plasmid, 74223) and purified by Miniprep (Qiagen) according to the manufacturer’s protocol. Then, 5 µg of plasmid was digested with NheI-HF (NEB, R3131) and EcoRI-HF (NEB, R3101S) restriction enzymes to introduce the Myc-BioID2 sequence into the pLJM1-EGFP lentiviral vector (Addgene, 19319). Upon plasmid ligation and transformation, successful colonies were grown and purified by Miniprep. In parallel, Rac1b was purchased from GeneScript (OHu22224) and Rac1 was amplified from normal murine small intestine tissue. Primers containing the sequences of Eco-RI-HF and SacII-HF (NEB, R0175S) restriction enzymes were used for Rac1b and Rac1 amplification. Primer sequences were as follows: Rac1/Rac1b Forward (5ʹ-3ʹ): TTTTTGAATTCCAGGCCATCAAGTGTGTG and Rac1/Rac1b Reverse (5ʹ-3ʹ): TATATCCGCGGTTACAACAGCAGGCATTTTCTC. For amplification, Phusion DNA polymerase (NEB, M0530S) was used and the PCR protocol consisted on a pre-incubation step of 98 °C for 4 min, 27 amplification cycles composed of 45 s at 95 °C, 45 s at 59 °C, and 40 s at 72 °C and a final elongation step of 7 min at 72 °C. After an A-tailing reaction with the FastTaq polymerase (Roche, 12161508103) and 1 mM of dATP (PCR: 1 min at 95 °C and 30 min at 70 °C), the pGEM-T Easy Vector System (Promega, A1360) was utilised to insert Rac1 and Rac1b sequences into the Myc-BioID2-containing lentiviral vector. Blue-white screening with X-gal/IPTG plates was used to detected positive (white) pGEM-T transformed colonies, which were grown and purified. PGEM-T insert was digested and sequenced for verification. Lentiviral vector was digested with EcoRI-HF and SacII-HF too and ligated with the pGEM-T inserts. Successful colonies were sequenced and amplified by HiSpeed Plasmid Maxi kit (Qiagen). As a BioID interaction control, an insert-free vector was also amplified. For the BioID pull-down experiment, a total of 3 × 106 cells of the CMT93 cell line were seeded in 10 cm2 dish the day before transfection. Three plates were used per each condition (Rac1-BirA, Rac1b-BirA and BirA), 24 µg of DNA per plate was used for transfection, using 60 µg of Lipofectamine 2000 (Invitrogen, 11668030) and 40 h later, transfection medium was replaced with 50 µM of Biotin (1 mM biotin stock: 0.0122 g biotin dissolved in 50 ml DMEM) in 10 ml of cell medium (Sigma, B4501). Cells were incubated for 20 h and collected for protein extraction with 500 µl of RIPA buffer per dish. Upon protein clarification, the lysate was incubated with 15 µl of neutralised Streptavidin Sepharose matrix (GE Healthcare, 17–5113–01) for 6 h at 4 °C on a rotator. Following incubation, the lysate was spun down at 1000g for 5 min and supernatant was carefully removed. Beads were gently washed with 1 ml of wash buffer (50 mM TrisCl and 8 M Urea, pH 7.4) for 8 min at RT on a rotator and centrifuged at 1000g for 2 min. This washing step was repeated three times, the last wash with a urea-free buffer. Beads’ pellets were a digested with Trypsin and processed as previously described63. Mass spectrometry was done using a Lumos Fusion (Thermo) mass spectrometer coupled to a RLS-nano uHPLC (Thermo). Peptides were separated by a 40 min linear gradient from 5% to 30% acetonitrile, 0.05% acetic acid. Proteins were identified and quantified the MaxQuant software suite using label-free quantification and searching against the mouse Uniprot database. Proteins enriched in Rac1-BirA, Rac1b-BirA vs BirA Protein were determined by >2-fold enrichment and P value < 0.05. NetworkAnalyst platform with the IMEx database was used for the interactome analysis of the resultant protein hits (networkanalyst.ca). To validate EGFR as a RAC1B-interacting protein, washed beads’ pellets were resuspended with RIPA and 10X DTT, run on a western blot gel and probed against Streptavidin-HRP and EGFR. Lysate input was used as a positive control.
Intestinal epithelial isolation and fractionation
Harvested small intestine was washed with cold PBS, opened longitudinally and scrapped off with a coverslip to discard villi. The intestine was cut into small pieces and transferred to a 50 ml Falcon tube. Pieces were washed up and down several times with cold PBS, discarding supernatant with debris, and these were incubated with 25 ml of 2 mM EDTA for 30 min at 4 °C with agitation to allow crypt separation from surrounding tissue. EDTA was discarded and intestinal pieces were gently washed with cold PBS. Intestinal fractionation was initiated by adding 10 ml of cold PBS and thoroughly pipetting up and down several times. These 10 ml cell solutions were collected, which contained loosened intestinal cells, and corresponded to fraction number 1. This step was repeated three more times, the latter fractions being enriched in crypt/stem cells. For fractionation and transcriptomic analysis, fractions were independently spun down and pellets were used for RNA extraction and qRT-PCR experiments. However, when evaluation of overall epithelial intestinal cells was carried out, the four fractions were jointly collected and the cellular pellet was used for either RNA or protein extraction.
Cells and cell culture
The cell line used in this study was the mouse rectal carcinoma cell line CMT9364. A stock of 3–6 × 106 cells/ml was maintained over the study in T25 or T75 cell flasks (Corning) in Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma) supplemented with 10% foetal calf serum and 1% of penicillin and streptomycin (Life Technologies, 15140). When cells reached 80% confluency, standard cell culture techniques were used for cell passaging and maintenance.
EGF stimulation experiment
In a 6-well plate, 2.5 × 105 CMT93 cells per well were seeded. The day after, a medium solution with 20 ng/ml EGF was prepared and was added to the cells following 4 h serum starvation. Cells were collected after 5, 10, 15 and 30 min of EGF stimulation for protein extraction. A non-starved well and a non-EGF-stimulated well were used as controls in each experimental group.
EGF uptake assay
CMT93 cells were subjected to knockdown of Rac1b using mouse-specific PMOs targeting Rac1b, and a non-targeting control for 96 h. They were serum-starved for 6 h before being treated with EGF-AlexFluor555 (20 ng/ml) for the indicated times. Cells were then fixed in 4% PFA at room temperature (RT) for 15 min, before being quenched with 100 mM Glycine-PBS for 10 min. Following three washes with PBS, cells were permeabilised (0.2% saponin, 5% BSA, PBS pH 7.5) for 20 min at RT. Following 1 wash with IF buffer (0.2% Triton-X100, 0.05% Tween-20, PBS pH 7.5). Cells were then blocked in 3% BSA in IF buffer for 30 min, before being stained with α-EGFR (ab52894) in 1% BSA in IF buffer overnight at 4 °C. Following three washes in IF buffer, cells were stained with α-rabbit AlexaFluor488™ in 1% blocking solution for 45 min at RT. Cells were washed three times in IF buffer, before stained in Fluoromount-G® with DAPI. Images were taken on Nikon A1R confocal microscope. Images were analysed using ImageJ.
EGFR-Rab5 co-localisation assay
CMT93 cells stably expressing pBabe-mCherry-Rab5 construct (a gift from Dr. Noor Gammoh) were generated by retroviral transduction in DMEM high-glucose media containing (8 µg/ml polybrene). Two days post transduction, cells were selected for with puromycin (2 µg/ml) for 7 days. CMT93 cells were subjected to knockdown of Rac1b using mouse-specific PMOs targeting Rac1b, and a non-targeting control for 96 h. Cells were serum-starved for 6 h before being treated with EGF (20 ng/ml) for the indicated times. Cells were then fixed in 4% PFA at RT for 15 min, before being quenched with 100 mM Glycine-PBS for 10 min. Following three washes with PBS, cells were permeabilised (0.5% Triton X-100, PBS pH 7.5) for 20 min at RT. Following 1 wash with IF buffer (0.2% Triton-X100, 0.05% Tween-20, PBS pH 7.5). Cells were then blocked in 3% BSA in IF buffer for 30 min, before being stained with α-EGFR (ab52894) in 1% BSA in IF buffer overnight at 4 °C. Following three washes in IF buffer, cells were stained with α-rabbit AlexaFluor488™ in 1% blocking solution for 45 min at RT. Cells were washed three times in IF buffer, before being stained in Fluoromount-G® with DAPI. Images were taken on Nikon A1R confocal microscope. Co-localisation was measured using Coloc2 plugin on ImageJ.
EGF tracking and lysosome-stained live cell imaging
CMT93 cells were subjected to knockdown of Rac1b using mouse-specific PMOs targeting Rac1b, and a non-targeting control for 96 h. They were serum-starved for 6 h before treatment. Cells were stained with 25 nM LysoTracker™ Green DND-26 (Thermo Fisher Scientific) for 30 min at 37 °C, followed by two washes in Live Cell Imaging Solution (Invitrogen). Cell were then treated with 20 ng/ml EGF-AlexFluor555 and imaged every 30 s for the indicated times using an Andor spinning disk confocal microscope. EGF tracking was performed using TrackMate plugin on ImageJ. Co-localisation was performed by manual scoring of EGF-555/LysoTracker double-positive foci at each indicated time point. The total number of EGF-555 foci was determined using ImageJ “Find Maxima” at setting 100. Percentage EGF-555 foci co-localised with LysoTracker was calculated using these two values.
Modulation of EGFR signalling in tumour organoids
To inhibit EGFR with the PD153035 small inhibitor (Sigma, SML0564), organoids were washed and split as fragments by mechanically breaking the organoids with a P1000 instead of a P200. A density of 100 fragments/µl of BME was plated in 15 µl BME drops. Following 24 h of plating, growth media was replaced with media containing either 1 µM of PD15303 or 1% DMSO (Sigma) as control. Brightfield organoid pictures were taken following 20 h of treatment. To assess RAC1B dependency for EGFR signalling activation, organoids plated as fragments were grown either with EGF-free medium or with medium supplemented with EGF. Pictures were taken 72 h post plating. For both experimental setups, images were used to assess the organoid area using ImageJ.
Cetuximab treatment of patient-derived organoids
Human colorectal organoids derived from liver metastases (C001 and C002) were cultured in ADF medium containing 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-Glutamine, 10 mM HEPES, 1X Primocin, 1X B27, 1.25 mM NAC, 10 mM Nicotinamide, 50 ng/ml EGF, 500 nM A83-01, 10 µM SB202190, 100 ng/ml Noggin, 1 µM PGE and 10 nM gastrin. Organoids were passaged by breaking into small fragments and replating. RAC1B was depleted by treatment with 1 µM hRAC1B PMO or NT PMO for 96 h. After 96 h, the media was replaced with fresh media containing 1 µM hRAC1B PMO or NT PMO plus or minus 30 μg/ml cetuximab (Selleck, A2000). Pictures were taken 96 h post treatment. For all four experimental setups, images were used to assess the organoid area using ImageJ.
Software
The following software was used for data analysis: Microsoft Office Excel 365, GraphPad Prism v7.0 and v8.3.1, RNAseq analysis - TrimGalore 0.6.5, cutadapt 3.2, tophat 2.1.1 and cuffdiff 2.2.1, ImageJ 1.52p, kallisto 0.45, and DESeq2 v1.30.1.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability statement
RNAseq data that support the findings in this manuscript have been deposited at Gene Expression Omnibus (NCMI) with the study accession code: GSE167876. Source data are provided with this paper.
References
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Zhao, B. et al. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 8, 3980–4000 (2017).
Gong, J., Cho, M. & Fakih, M. RAS and BRAF in metastatic colorectal cancer management. J. Gastrointest. Oncol. 7, 687–704 (2016).
Isella, C. et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat. Commun. 8, 15107 (2017).
Lenz, H. J. et al. Impact of consensus molecular subtype on survival in patients with metastatic colorectal cancer: results from CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 37, 1876–1885 (2019).
Mooi, J. K. et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 29, 2240–2246 (2018).
Woolston, A. et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-EGFR treatment in colorectal cancer. Cancer Cell 36, 35–50 e39 (2019).
Drost, J. et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47 (2015).
Fearon, E. R. & Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990).
Matano, M. et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 21, 256–262 (2015).
Kinzler, K. W. & Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 87, 159–170 (1996).
Korinek, V. et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 275, 1784–1787 (1997).
Athineos, D. & Sansom, O. J. Myc heterozygosity attenuates the phenotypes of APC deficiency in the small intestine. Oncogene 29, 2585–2590 (2010).
Sansom, O. J. et al. Myc deletion rescues Apc deficiency in the small intestine. Nature 446, 676–679 (2007).
Ashton, G. H. et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev. Cell 19, 259–269 (2010).
Cole, A. M. et al. Cyclin D2-cyclin-dependent kinase 4/6 is required for efficient proliferation and tumorigenesis following Apc loss. Cancer Res. 70, 8149–8158 (2010).
Faller, W. J. et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517, 497–500 (2015).
Gay, D. M. et al. Loss of BCL9/9l suppresses Wnt driven tumourigenesis in models that recapitulate human cancer. Nat. Commun. 10, 723 (2019).
Myant, K. B. et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 12, 761–773 (2013).
Myant, K. B. et al. Rac1 drives intestinal stem cell proliferation and regeneration. Cell Cycle 12, 2973–2977 (2013).
Climente-Gonzalez, H., Porta-Pardo, E., Godzik, A. & Eyras, E. The functional impact of alternative splicing in cancer. Cell Rep. 20, 2215–2226 (2017).
Fiegen, D. et al. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 279, 4743–4749 (2004).
Matos, P., Collard, J. G. & Jordan, P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J. Biol. Chem. 278, 50442–50448 (2003).
Singh, A. et al. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 23, 9369–9380 (2004).
Ungefroren, H. et al. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 5, 277–290 (2014).
Ungefroren, H. et al. RAC1B suppresses TGF-beta1-dependent cell migration in pancreatic carcinoma cells through Inhibition of the TGF-beta type I receptor ALK5. Cancers 11, 691 (2019).
Matos, P. & Jordan, P. Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp. Cell Res. 305, 292–299 (2005).
Li, G. et al. Rac1b enhances cell survival through activation of the JNK2/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways. Oncotarget 7, 17970–17985 (2016).
Esufali, S., Charames, G. S., Pethe, V. V., Buongiorno, P. & Bapat, B. Activation of tumor-specific splice variant Rac1b by dishevelled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res. 67, 2469–2479 (2007).
Ryan, M. et al. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res. 44, D1018–D1022 (2016).
Myant, K. B. et al. HUWE1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO Mol. Med. 9, 181–197 (2017).
Muller, T. et al. ASAP1 promotes tumor cell motility and invasiveness, stimulates metastasis formation in vivo, and correlates with poor survival in colorectal cancer patients. Oncogene 29, 2393–2403 (2010).
Tarcic, G. et al. An unbiased screen identifies DEP-1 tumor suppressor as a phosphatase controlling EGFR endocytosis. Curr. Biol. 19, 1788–1798 (2009).
Davidson, G. et al. Cell cycle control of wnt receptor activation. Dev. Cell 17, 788–799 (2009).
O’Brien, C. A., Pollett, A., Gallinger, S. & Dick, J. E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, 106–110 (2007).
Ricci-Vitiani, L. et al. Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111–115 (2007).
Snippert, H. J. et al. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology 136, 2187–2194 e2181 (2009).
Grainger, S. et al. EGFR is required for Wnt9a-Fzd9b signalling specificity in haematopoietic stem cells. Nat. Cell Biol. 21, 721–730 (2019).
Wei, G. et al. Erk and MAPK signaling is essential for intestinal development through Wnt pathway modulation. Development 147, dev185678 (2020).
Tomas, A., Futter, C. E. & Eden, E. R. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24, 26–34 (2014).
Pickering, K. A. et al. A RAC-GEF network critical for early intestinal tumourigenesis. Nat. Commun. 12, 56 (2021).
Vlachogiannis, G. et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018).
Kotelevets, L. et al. The Rac1 splice form Rac1b favors mouse colonic mucosa regeneration and contributes to intestinal cancer progression. Oncogene 37, 6054–6068 (2018).
Zhou, C. et al. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene 32, 903–909 (2013).
Sadanandam, A. et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 19, 619–625 (2013).
Zhu, G. et al. An EGFR/PI3K/AKT axis promotes accumulation of the Rac1-GEF Tiam1 that is critical in EGFR-driven tumorigenesis. Oncogene 34, 5971–5982 (2015).
Wang, F. et al. SPSB1-mediated HnRNP A1 ubiquitylation regulates alternative splicing and cell migration in EGF signaling. Cell Res. 27, 540–558 (2017).
Hua, Y. et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 24, 1634–1644 (2010).
Radisky, D. C. et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436, 123–127 (2005).
Lu, Y. et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat. Med. 23, 1331–1341 (2017).
el Marjou, F. et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 (2004).
Shibata, H. et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278, 120–123 (1997).
Wang, Q. et al. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data 5, 180061 (2018).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4, 1521 (2015).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Liu, P., Jenkins, N. A. & Copeland, N. G. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13, 476–484 (2003).
Adams, D. J. et al. A genome-wide, end-sequenced 129Sv BAC library resource for targeting vector construction. Genomics 86, 753–758 (2005).
Fu, J., Teucher, M., Anastassiadis, K., Skarnes, W. & Stewart, A. F. A recombineering pipeline to make conditional targeting constructs. Methods Enzymol. 477, 125–144 (2010).
Magin, T. M., McWhir, J. & Melton, D. W. A new mouse embryonic stem cell line with good germ line contribution and gene targeting frequency. Nucleic Acids Res. 20, 3795–3796 (1992).
Rodriguez, C. I. et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 25, 139–140 (2000).
Turriziani, B. et al. On-beads digestion in conjunction with data-dependent mass spectrometry: a shortcut to quantitative and dynamic interaction proteomics. Biology 3, 320–332 (2014).
Kitamura, T. et al. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc. Natl Acad. Sci. USA 107, 13063–13068 (2010).
Acknowledgements
We thank the core technical services at the Institute of Genetics and Molecular Medicine. In particular the staff of the Bioresearch & Veterinary Services and Dr. Ann Wheeler and Dr. Laura Murphy from the Advanced Imaging Resource (AIR) for their assistance. We thank the patient / recruiting nursing team at the Western General Hospital for providing the tumour material used in this study. Patient material was obtained under ethical consent SOCCS3 (REC: 11/SS/0109). K.B.M, V.G., C.B., P.C. and A.E.H. are supported by a CRUK Career Development Fellowship A19166 and an ERC Starting Grant 715782. O.J.S. is supported by CRUK grants A26825, A28233, A23390, A21139, A12481 and A17196 and an ERC Starting Grant 311301. F.V.N.D. was supported by Clinical Scientist Fellowships from CRUK (C26031/A11378) and Chief Scientist Office (SCAF/16/01). A.B. and A.v.K. carried out Mass Spectrometry analysis supported by the Wellcome Trust (Multiuser Equipment Grant, 208402/Z/17/Z). Fig. S8 was created with BioRender.
Author information
Authors and Affiliations
Contributions
Study concept and design: K.B.M. Acquisition of data: V.G., C.V.B., P.C., A.B., S.O.P., C.N., A.E.H., A.W., L.M. and K.B.M. Analysis and interpretation of data: V.G., C.V.B., P.C., A.B., S.A., S.O.P., M.A., J.C., A.v.K., A.W., L.M. and K.B.M. Drafting of manuscript: K.B.M., V.G., S.O.P., P.C. and A.E.H. Material support: D. Ste, D. Str, P.F., M.D., F.D., L.P., G.D. and O.J.S. Design of PMO: L.P. and G.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gudiño, V., Pohl, S.ÖG., Billard, C.V. et al. RAC1B modulates intestinal tumourigenesis via modulation of WNT and EGFR signalling pathways. Nat Commun 12, 2335 (2021). https://doi.org/10.1038/s41467-021-22531-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-22531-3
This article is cited by
-
Elevated galectin-3 levels detected in women with hyperglycemia during early and mid-pregnancy antagonizes high glucose − induced trophoblast cells apoptosis via galectin-3/foxc1 pathway
Molecular Medicine (2023)
-
RAC1B function is essential for breast cancer stem cell maintenance and chemoresistance of breast tumor cells
Oncogene (2023)
-
RHO GTPase family in hepatocellular carcinoma
Experimental Hematology & Oncology (2022)
-
T-box transcription factor 19 promotes hepatocellular carcinoma metastasis through upregulating EGFR and RAC1
Oncogene (2022)
-
RNA splicing is a key mediator of tumour cell plasticity and a therapeutic vulnerability in colorectal cancer
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
