
Abstract
With increasing age of the population, countries across the globe are facing a substantial increase in osteoporotic fractures. Genetic association signals for fractures have been reported at the RSPO3 locus, but the causal gene and the underlying mechanism are unknown. Here we show that the fracture reducing allele at the RSPO3 locus associate with increased RSPO3 expression both at the mRNA and protein levels, increased trabecular bone mineral density and reduced risk mainly of distal forearm fractures in humans. We also demonstrate that RSPO3 is expressed in osteoprogenitor cells and osteoblasts and that osteoblast-derived RSPO3 is the principal source of RSPO3 in bone and an important regulator of vertebral trabecular bone mass and bone strength in adult mice. Mechanistic studies revealed that RSPO3 in a cell-autonomous manner increases osteoblast proliferation and differentiation. In conclusion, RSPO3 regulates vertebral trabecular bone mass and bone strength in mice and fracture risk in humans.
Similar content being viewed by others

Introduction
Osteoporosis is a common skeletal disease, leading to a reduction in bone density and quality, and an increased fracture risk. One in two elderly women and one in four elderly men will at some point suffer from an osteoporotic fracture1,2. It is predicted that the number of people aged 65 years and older will triple globally by year 21003,4 as compared to those aged 12–64 years, and the incidence of fractures increases exponentially with age5. Therefore, the identification and treatment of patients at high fracture risk is an important public health goal.
Fracture risk is a moderately heritable trait6,7, and two recent large-scale genome-wide association studies (GWAS) have identified 15 loci associated with fractures at any bone site3,8. The identified fracture loci were all also associated with BMD, supporting the notion that BMD is a major regulator of fracture risk3,8. These previous large-scale human genetic studies reported that the strongest genetic determinant for fractures is located at the WNT16 locus and detailed translational studies revealed that osteoblast-derived WNT16 increases cortical bone mass9,10,11. The second strongest genetic determinant for fractures at any bone site (P = 8.9 × 10−65)8 is located at the RSPO3 locus. RSPO3 is a known WNT-signaling modulator12,13, and WNT-signaling is a major determinant of bone density and strength. We therefore hypothesized in this study that RSPO3 is the causal gene for the prominent fracture signal at the RSPO3 locus.
WNT proteins belong to a family of secreted cysteine-rich glycoproteins that signal through both the WNT–β-catenin pathway, also termed the canonical WNT pathway, and noncanonical WNT pathways13,14,15,16. WNT ligands increase bone mass by targeting the trabecular and/or cortical bone compartments10,17. WNT16 is the major known cortical bone-specific WNT10, while WNT10b protects against age-dependent trabecular bone loss18,19. WNT binding to its Frizzled receptors leads to nuclear accumulation of the transcriptional co-activator β-catenin and, thereby, increased WNT target gene expression12. There are a number of secreted extracellular proteins including sclerostin, DKK1 and NOTUM that inhibit WNT-signaling and thereby reduce bone mass12,20. Another group of secreted proteins, R-spondins (RSPO 1–4), are described to amplify WNT-signaling21,22,23,24,25,26. They are ligands of the leucine-rich repeat-containing G-protein coupled receptors (LGRs), including LGR4, -5, and -627,28,29, and reduce degradation of Frizzled receptors and consequently increase WNT-signaling23,28,30,31. The in vivo role of RSPO3 for adult bone metabolism is not known as global Rspo3−/− mice display early embryonic lethality due to impaired formation of the placenta29.
Previous human genetic studies have reported that the fracture reducing C-allele of rs7741021, the strongest genetic determinant for fractures at the RSPO3 locus, also is robustly associated with increased BMD (Table 1a)8. However, the causal gene(s) and the underlying mechanism are unknown. In addition, it is unknown if its associations with fractures at different bone sites differ and if the association with BMD is mediated via trabecular and/or cortical bone parameters. We, herein, used a combination of human genetic studies and extensive functional mechanistic studies to identify the causal gene at the RSPO3 locus and its mechanism to explain its effect upon fracture.
Results
Human genetic studies indicate that increased RSPO3 expression is associated with increased trabecular bone mineral density and reduced risk of distal forearm fractures
Previous large-scale human genetic studies have reported that the strongest genetic determinant at the RSPO3 locus for fractures at any bone site is located at the intronic SNP rs7741021 (Table 1a; P = 8.9 × 10−65)8. A substantial part of the SNPs associated with evaluated phenotypes in GWAS is located in intronic regions and may directly or via highly linked SNPs affect regulatory regions and thereby gene transcription32. We, therefore, evaluated the association between rs7741021 and mRNA levels of RSPO3 using the GTEx resource. We observed that the C-allele of rs7741021, previously reported to associate with reduced fracture risk at any bone site and increased BMD (estimated BMD as determinated by ultrasound in the heel; Table 1a), was associated with increased RSPO3 mRNA levels in both subcutaneous adipose tissue (P = 2.4 × 10−4; n = 581) and cultured fibroblasts (P = 1.8 × 10−4; n = 483; Table 1a). Unfortunately, no bone-related tissue was available in the GTEx resource. From human genetic association studies, it is not possible to determine if a factor such as RSPO3 exerts local effects within the bone tissue. However, it is possible to determine if the genetic signals that associate with bone health parameters also associate with circulating levels of RSPO3. It should be emphasized that such association does not show that the effect on bone is mediated via circulating levels of RSPO3 as the circulating levels may only reflect the local levels, without being of biological importance as a systemic regulator of local bone metabolism.
We observed that SNP rs3734626, which is highly linked with SNP rs7741021 (r2 = 0.84; coefficient of linkage disequilibrium measure, D′ = 0.99) at the RSPO3 locus, was strongly associated with circulating protein levels of RSPO3 (P = 1.8 × 10−45; Table 1b)33. The association for SNP rs3734626 with the circulating protein levels of RSPO3 was derived from a previous report, demonstrating that this SNP was the most significant cis-protein quantitative trait loci (cis-pQTLs) for RSPO333. The T-allele of rs3734626, associated with reduced fracture risk at any bone site and increased BMD, was associated with increased circulating levels of RSPO3 (Table 1b). Collectively, these human genetic studies indicate that the main fracture signal at the RSPO3 locus regulates RSPO3 expression and that the fracture-reducing allele associates with both increased RSPO3 expression and BMD.
We next evaluated the impact of rs7741021 on trabecular and cortical bone parameters analyzed separately by pQCT in the tibia in humans9,34. The fracture reducing C-allele of rs7741021 was significantly associated with increased trabecular volumetric BMD, while no significant association was observed with cortical volumetric BMD or cortical bone thickness (Table 1a).
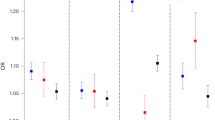
As the distal radius is a bone compartment with relatively high proportion of trabecular bone, we next extended the fracture association studies by evaluating the associations for rs7741021 specifically with distal forearm fractures, defined by ICD codes, in the large UK Biobank cohort. The C-allele of rs7741021 was robustly associated with reduced risk of forearm fractures (OR 0.88, 95% confidence intervals (CI) 0.86–0.91 per C-allele; P = 9.6 × 10−14; Table 1a) in age, age2, sex, weight and height adjusted analyses. We also evaluated the association between rs7741021 and hip fractures, defined by ICD codes. The C-allele of rs7741021 was associated with reduced risk of hip fractures (OR 0.94, 95% CI 0.90–0.99 per C-allele; P = 4.4 × 10−3; Table 1a). The effect size for the association of rs7741021 with distal forearm fractures was significantly larger than the effect sizes for the association of rs7741021 with fractures at any bone site (Table 1a, OR 0.95, 95% CI 0.94–0.96 per C-allele; P = 1.6 × 10−5; Z-test) and hip fractures (P = 3.5 × 10−2; Z-test).
To estimate the impact of RSPO3 on bone parameters and fracture risk, we performed two-sample inverse variance-weighted (IVW) Mendelian randomization (MR) studies. We used either the above described strong cis-pQTL rs3734626 for circulating RSPO3 identified by Emilsson et al. 33 (Table 1b) or the linked SNP rs2489623 (r2 = 0.74, D′ = 0.88; Table 1c), a cis-pQTL for circulating RSPO3 (P = 3.6 × 10−28) identified by Sun et al. in an independent dataset35, as genetic instruments for circulating protein levels of RSPO3. These MR analyses indicated that increased circulating RSPO3 was strongly associated with increased trabecular vBMD, and reduced risk of distal forearm fractures, and as expected, with very similar effect sizes for the two genetic instruments (Table 2). However, it should be emphasized that this association might be completely driven by circulating RSPO3 being associated with local RSPO3 levels, and from these human associations studies it is, therefore, not possible to determine possible local effects of factors acting on bone remodeling. Thus, to determine the relative role of circulating RSPO3 and local RSPO3, functional studies in mice with cell-specific inactivation of RSPO3 is required.
To exclude reverse causality, wherein BMD influences levels of RSPO3, we performed a bidirectional MR using BMD as the exposure and circulating RSPO3 as the outcome. These studies revealed no evidence of reverse causality (P = 0.80).
Colocalization analyses may be used to determine if there is a shared genetic signal between two traits at a specific gene locus. Traditional colocalization analyses require that only one independent association signal exists at a specific locus. Because colocalization inference are very sensitive to small linkage disequilibrium (LD) changes in the tested populations, multiple causal signals presented in the same region may introduce substantial bias36. As only one independent association signal was observed for trabecular volumetric BMD at the RSPO3 locus (Supplementary Fig. 1), we used colocalization analyses to determine if there is a shared genetic signal for circulating RSPO3 and trabecular volumetric BMD at this locus. Bayesian colocalization analyses using COLOC37 found that the plasma RSPO3 signal identified by Sun et al. was reasonably well colocalized with the single signal observed for trabecular volumetric BMD at the RSPO3 locus with a posterior probability of a single shared signal of 72% (posterior probability of the hypothesis 4 that the association with trait 1 and trait 2 has one shared SNP37, PP.H4 = 0.72), indicating that there is a single shared genetic signal in the 1MB locus of the RSPO3 cis-pQTL SNP (rs2489623), affecting both circulating RSPO3 and trabecular volumetric BMD (Supplementary Fig. 1). Since there are multiple independent signals for both estimated BMD (eBMD) and forearm fractures at the RSPO3 locus, and since we do not have access to any individual level data to assume accurate LD, we did not undertake colocalization analyses for these parameters.
These human genetic studies demonstrate that the strong fracture signal at the RSPO3 locus has the capacity to regulate RSPO3 expression, and that increased circulating RSPO3 levels associate with increased trabecular BMD, substantially reduced risk of distal forearm fractures and moderately reduced risk of hip fractures. Based on these human genetic data, we hypothesized that RSPO3 increases trabecular bone mass and thereby reduces fracture risk. To test this hypothesis, we performed extensive mechanistic studies using several conditional Rspo3-inactivated mouse models and cultured bone-derived cells.
RSPO3 is expressed in osteoprogenitor cells and osteoblasts but not in osteocytes or osteoclasts
We observed Rspo3 mRNA expression in several mouse tissues, revealing the highest Rspo3 expression in brain cortex and diaphyseal cortical bone with intermediate expression in the vertebral body and modest expression in flushed bone marrow (Fig. 1a). Rspo3 mRNA levels were high in primary calvarial osteoblasts after prolonged culture in osteogenic media (Fig. 1b; Supplementary Fig. 2a). In contrast, no Rspo3 expression was observed in bone marrow-derived macrophages cultured with M-CSF or osteoclasts differentiated from bone marrow-derived macrophages stimulated with RANKL (Fig. 1b; Supplementary Fig. 2b).

a Rspo3 mRNA expression pattern in various tissues of male, wildtype mice (n = 6). Mean of individual values are presented as horizontal lines and ±SEM as vertical lines. Data are presented as % of expression in cortical diaphyseal bone. b Rspo3 is expressed in primary calvarial osteoblasts (OBL), but not in bone marrow-derived macrophages (BMM) or RANKL-differentiated osteoclasts (OCL). Rspo3 gene expression increase from day 2 (d2) to day 7 (d7) in primary calvarial osteoblasts cultured in osteogenic media. Presented as % of expression in osteoblasts day 2. ND=not detectable. Mean of individual values are presented as horizontal lines and ±SEM as vertical lines. Difference in expression between day 2 and 7 was analyzed by two-sided Student’s t test, n = 4 wells per group. c–f Representative in situ hybridization images in mice. Transverse sections across lumbar vertebra 5 in mouse showing the mRNA expression pattern (red) of Rspo3 (c–f, upper) and Runx2 (c, lower), Col1a1 (d, lower), Dmp1 (e, lower), and Acp5/Trap (f, lower). The lower images are consecutive to the upper images. Rspo3 mRNA could be observed in Runx2-expressing (c), Col1a1-expressing (d), and Dmp1-expressing (e) osteoblast lineage cells (OBL) on the bone surface. In contrast, Rspo3 was not detectable in osteocytes (Ocy; e), or osteoclasts (OCL; f). Scale bar 50 µm. g–i Representative in situ hybridization images in humans. Sections from human proximal femur (g) and iliac crest (h, i) confirmed moderate RSPO3 mRNA expression. Staining for RSPO3 mRNA (red) were observed in immature osteoblast lineage cells (OBL) on eroded lining surfaces (reversal cells), and on quiescent bone surfaces (bone lining cells), as well as in a subset of cells close to blood vessels (bv; g, h). No RSPO3 mRNA expression was observed in osteocytes (Ocy) or TRAP-immunostained osteoclasts (OCL, black; i). Scale bar 20 µm. Experiments were repeated one (a), two (g–i) or at least three (b–f) times. Source data are provided as a Source Data file.
Next, the RSPO3 expression in mouse and human bone sections was evaluated using chromogenic in situ hybridization (Fig. 1c-i). Assessment of Rspo3 expression in mouse vertebral bone revealed that Rspo3 mRNA is expressed in Runx2-expressing (Fig. 1c), Col1a1-expressing (Fig. 1d) and Dmp1-expressing (Fig. 1e) osteoblast-lineage cells on bone surface. In contrast, Rspo3 expression was not detectable in osteocytes (Fig. 1e) or osteoclasts (Fig. 1f). Rspo3 expression was also observed in a subset of cells close to blood vessels and adipocytes in the bone marrow (Supplementary Fig. 2c,d). Analyses of human bone sections (Fig. 1g–i) confirmed moderate RSPO3 mRNA expression in immature osteoblast lineage cells, on eroded lining surfaces (reversal cells) and quiescent bone surfaces (bone lining cells) (Fig. 1g), and in a subset of cells close to blood vessels and adipocytes (Fig. 1h). No staining for RSPO3 mRNA expression was observed in osteocytes or osteoclasts in human sections (Fig. 1i).
Thus, in addition to expression in osteoblast-lineage cells on bone surfaces, RSPO3 expression was found in a subset of cells close to blood vessels, where mesenchymal skeletal stem cells are located within the bone marrow38. To determine Rspo3 expression specifically in mesenchymal stem cells in the bone marrow, we analyzed two independent single cell RNA-sequencing (scRNA seq) data sets of bone marrow cells. First, scRNA seq analysis of Cxcl12 expressing bone marrow cells showed that Rspo3 was expressed in stromal cells expressing both adipogenic and osteoblastic markers (Fig. 2a–c)39. These cell clusters also expressed Lepr (Supplementary Fig. 3), often used in combination with Cxcl12 as a marker for mesenchymal stem cells, supporting the notion that Rspo3 is expressed also in mesenchymal stem cells. No Rspo3 expression was found in Cd45-expressing hematopoietic cell clusters (Fig. 2a–c).

Single cell RNA sequencing analysis of Cxcl12 expressing bone marrow cells. a Uniform Manifold Approximation and Projection (UMAP)-based visualization of major cell clusters (Cluster 0–7) and cluster expressed genes (right). Feature plot (b) and violin plot (c) of Rspo3 expression.
The expression of Rspo3 in Cxcl12+Lepr+ mesenchymal stem cells was further confirmed in a second recently published scRNA seq dataset (Supplementary Fig. 4)40. This dataset includes scRNA seq analysis of cells expressing Cdh5 (found in vasculature), Lepr (perivascular stromal stem cells) and Col1a1 (osteoblastic cells). Using this dataset, we identified Rspo3 expression in stromal cells expressing both adipogenic and osteogenic markers (Supplementary Fig. 4). Rspo3 mRNA was expressed also in pro-osteogenic Lepr+ cells expressing Alpl and in the more mature osteoblast lineage Col1a1-expressing cells. No Rspo3 expression was found in the vascular cell clusters. Collectively, these two scRNA seq data sets of bone marrow cells reveal robust Rspo3 expression in mesenchymal stem cells expressing both osteogenic and adipogenic markers.
Osteoblast-derived RSPO3 is the principal source of RSPO3 in bone and is a major regulator of trabecular bone mass
As global Rspo3−/− mice display early embryonic lethality29, and since we observed robust Rspo3 expression in osteoblast-lineage cells, we first evaluated the impact of osteoblast-derived RSPO3 for trabecular and cortical bone parameters. To this end, we generated a conditional Rspo3-inactivated mouse model targeting osteoblasts and osteocytes. A mouse model with exons 2–4 of Rspo3 flanked by LoxP sites (Rspo3flox/flox, Fig. 3a) was used41. To achieve inactivation of Rspo3 early in the osteoblast lineage, we generated Runx2-creRspo3flox/flox mice, which express Cre recombinase driven by the Runx2 promoter, a promoter expressed in early osteoblast lineage cells but not in osteoclasts (Fig. 3a)42. Runx2-creRspo3flox/flox mice had more than 90% reduced Rspo3 mRNA levels in both vertebral trabecular bone and tibial cortical diaphyseal bone with no reduction in brain cortex or liver compared to Rspo3flox/flox mice (Fig. 3b). Thus, osteoblast-derived RSPO3 is the principal source of RSPO3 in both trabecular and cortical bones.

a Schematic figure of the conditional osteoblast-lineage specific Rspo3-inactivated mouse model. b mRNA expression analyses of Rspo3 in vertebral trabecular bone, cortical diaphyseal bone, liver, and brain cortex in male Runx2-creRspo3flox/flox mice, compared to Rspo3flox/flox mice. c Body weight in Runx2-creRspo3flox/flox mice compared to Rspo3flox/flox mice at 5, 9, and 13 weeks-of-age. d Weight of liver, gonadal fat, kidneys, and spleen, per body weight (BW) in Runx2-creRspo3flox/flox mice, and Rspo3flox/flox mice. e Representative 3D µCT images of vertebra L5 in Runx2-creRspo3flox/flox mouse (left) and Rspo3flox/flox mouse (right). f–i Trabecular bone volume over total volume (BV/TV; f), trabecular thickness (Tb.Th; g), trabecular number (Tb.N; h), and trabecular separation (Tb.Sp; i) in vertebra L5 from Runx2-creRspo3flox/flox mice, compared to Rspo3flox/flox mice, as measured by µCT. j Correlation between Rspo3 mRNA levels in vertebral body (relative quantity, x-axis) and trabecular BV/TV (%, y-axis) in male Runx2-creRspo3flox/flox and Rspo3flox/flox mice. Variance explained (r2) is given in the figure. k Maximal load at failure (N) of vertebra L5 as measured by compression test in 14-week-old male Runx2-creRspo3flox/flox (n = 10) mice and Rspo3flox/flox (n = 9) mice. l, m Cortical volumetric bone mineral density (Ct.vBMD; l) and cortical thickness (Ct.Th; m) in femur from Runx2-creRspo3flox/flox mice, compared to Rspo3flox/flox mice. n mRNA expression of sclerostin (Sost), Dickkopf-1 (Dkk1), and R-spondins 1, -2, and -4 (Rspo1, Rspo2, and Rspo4) in vertebral body in male Runx2-creRspo3flox/flox mice, compared to Rspo3flox/flox mice. o Levels of procollagen type I N-terminal propeptide (P1NP, left) and C-terminal type I collagen (CTX, right) in male Runx2-creRspo3flox/flox mice, compared to Rspo3flox/flox mice. Unless otherwise stated, the results refer to 13-week-old Runx2-creRspo3flox/flox mice, males n = 14; females n = 12, and Rspo3flox/flox mice, males n = 15; females n = 12. Individual values are presented in all graphs with the mean presented as horizontal lines and ±SEM as vertical lines. Statistical analyses were performed using two-sided Student’s t test. Source data are provided as a Source Data file.
Runx2-creRspo3flox/flox mice were born apparently healthy and had a normal body weight growth at termination at 12 weeks of age and displayed normal weights of several visceral organs (Fig. 3c, d). Consistent with the human genetic findings, CT analyses revealed reduced trabecular bone mass reflected by reduced trabecular bone volume fraction (BV/TV; Fig. 3e, f), trabecular thickness (Fig. 3g), trabecular number (Fig. 3h) and increased trabecular separation (Fig. 3i) in the lumbar vertebra L5 of Runx2-creRspo3flox/flox in both female and male mice compared to Rspo3flox/flox mice. Reduced trabecular bone mass in the vertebrae was confirmed by histomorphometric analyses, revealing a 27% (P < 0.001) reduction in trabecular BV/TV in Runx2-creRspo3flox/flox mice (Supplementary Fig. 5a–d). Furthermore, the trabecular BV/TV was positively correlated with Rspo3 mRNA levels in the vertebral body (r2 = 0.76, P < 0.001; Fig. 3j), supporting the notion that local RSPO3 expression regulates trabecular bone mass. To evaluate whether the decreased trabecular bone mass in mice with Rspo3 inactivation in osteoblast-lineage cells resulted in decreased bone strength, lumbar vertebra L5 was evaluated using a compression test, demonstrating that the maximal load at failure was significantly decreased in Runx2-creRspo3flox/flox mice compared with Rspo3flox/flox mice (Fig. 3k). No significant effect of osteoblast-lineage specific Rspo3 inactivation was observed on cortical volumetric BMD or cortical thickness in the femur diaphysis (Fig. 3l, m).
Expression analyses in the vertebral body revealed reduced mRNA levels of the two WNT signaling inhibitors Sost and Dkk1 (Fig. 3n), suggesting a compensatory mechanism to counteract additional trabecular bone loss in the adult Runx2-creRspo3flox/flox mice with life-long inactivation of Rspo3 in osteoblast-lineage cells. No compensatory regulation of Rspo1, -2 or -4 was observed (Fig. 3n).
Circulating levels of the bone formation marker procollagen type I N-terminal propeptide (P1NP; Fig. 3o, left) were reduced while the levels of the bone resorption marker C-terminal type I collagen fragments (CTX; Fig. 3n, right) were unchanged in Runx2-creRspo3flox/flox mice compared to Rspo3flox/flox mice. However, neither static nor dynamic histomorphometry of trabecular bone in vertebra L5 or gene expression analyses of vertebral trabecular bone revealed any significant effects on other parameters reflecting bone remodeling in the adult Runx2-creRspo3flox/flox mice with life-long inactivation of RSPO3, suggesting that a new steady state of bone remodeling had been reached (Supplementary Fig. 5e,f).
We next determined if RSPO3 with an impact on trabecular bone is derived from osteoblast precursors/early osteoblasts or from osteocytes/late osteoblasts. To this end, we compared the phenotype of Runx2-creRspo3flox/flox mice, having inactivation of Rspo3 in all osteoblast-lineage cells, with the phenotype of a mouse model with Rspo3 inactivated specifically in Dmp1-expressing osteocytes and late osteoblasts. Rspo3flox/flox mice were mated with Dmp1-cre mice expressing Cre recombinase specifically in late osteoblasts and osteocytes43, hereafter called Dmp1cre-Rspo3flox/flox mice (Fig. 4a). Dmp1cre-Rspo3flox/flox mice displayed a moderate reduction of Rspo3 mRNA levels in the vertebral body compared with Rspo3flox/flox mice (−32%; P < 0.001; Fig. 4b). The substantially less pronounced reduction of Rspo3 mRNA levels in the vertebral body of Dmp1cre-Rspo3flox/flox mice compared with the substantial reduction (>90%, P < 0.001) in Runx2-creRspo3flox/flox mice demonstrates that osteoblasts, and not osteocytes, are the major source of RSPO3 in vertebral trabecular bone (Figs. 3b and 4b).

a Schematic image of the conditional osteocyte/late osteoblast-specific Rspo3-inactivated mouse model. b mRNA expression analysis of Rspo3 in vertebral trabecular bone in male Dmp1cre-Rspo3flox/flox mice, compared to Rspo3flox/flox mice. c Body weight in Dmp1cre-Rspo3flox/flox mice, and Rspo3flox/flox mice, at 9 and 15 weeks-of-age. d Weight of liver, gonadal fat, kidneys, and spleen per body weight (BW) in Dmp1-creRspo3flox/flox mice, and Rspo3flox/flox mice. e–h Trabecular bone volume over total volume (BV/TV; e), trabecular thickness (Tb.Th; f), trabecular number (Tb.N; g), and trabecular separation (Tb.Sp; h) in vertebra L5 from Dmp1-creRspo3flox/flox mice, compared to Rspo3flox/flox mice. i Representative 3D µCT images of vertebra L5 in Rspo3flox/flox mouse (left) and Dmp1creRspo3flox/flox mouse (right). j and k Cortical volumetric bone mineral density (Ct.vBMD, j) and cortical thickness (Ct.Th, k) in femur from Dmp1-creRspo3flox/flox mice, compared to Rspo3flox/flox mice. l Schematic image of the global, tamoxifen-inducible Rspo3-inactivated mouse model. m mRNA expression analysis of Rspo3 in vertebral trabecular bone in CAGGCre-ER-Rspo3flox/flox (n = 10) mice, compared to Rspo3flox/flox (n = 14) mice. (n) Body weight in male CAGGCre-ER-Rspo3flox/flox (n = 11) mice, and Rspo3flox/flox (n = 17) mice, at 8, 9, and 13 weeks-of-age. o Weight of liver, gonadal fat, kidneys, and spleen per body weight in CAGGCre-ER-Rspo3flox/flox (n = 10) mice, and Rspo3flox/flox mice (n = 14). p–s Trabecular BV/TV (p), trabecular thickness (Tb.Th; q), trabecular number (Tb.N; r), and trabecular separation (Tb.Sp; s) in vertebra L5 from CAGGCre-ER-Rspo3flox/flox (n = 11) mice, compared to Rspo3flox/flox (n = 16) mice. t, u Cortical volumetric BMD (Ct.vBMD; t) and cortical thickness (Ct.Th; u) in femur from CAGGCre-ER-Rspo3flox/flox (n = 11) mice, compared to Rspo3flox/flox (n = 17) mice. v mRNA expression of sclerostin (Sost), Dickkopf-1 (Dkk1), and R-spondins 1, -2, and -4 (Rspo1, Rspo2, and Rspo4) in vertebral body in CAGGCre-ER-Rspo3flox/flox (n = 10) mice, compared to Rspo3flox/flox (n = 14) mice. For Dmp1-creRspo3flox/flox mice, males n=14; females n = 16, and littermate control Rspo3flox/flox mice, males n = 14; females n = 14. Individual values are presented in all graphs with the mean presented as horizontal lines and ±SEM as vertical lines. Statistical analyses were performed using two-sided Student’s t test. Source data are provided as a Source Data file.
Dmp1cre-Rspo3flox/flox mice were born apparently healthy and had a normal body weight growth at termination at 15 weeks of age and displayed normal weights of several visceral organs (Fig. 4c, d). CT analyses revealed modestly reduced trabecular bone volume fraction (−9.3%; BV/TV, Fig. 4e–i) in Dmp1cre-Rspo3flox/flox mice compared with Rspo3flox/flox mice, reaching statistical significance in a two-way ANOVA considering genotype and sex (P = 0.027 for genotype). The reduction of trabecular bone volume fraction, however, was substantially more pronounced in the Runx2-creRspo3flox/flox mice compared with the reduction observed in Dmp1cre-Rspo3flox/flox mice (Figs. 3f, 4e), demonstrating that RSPO3 with an impact on trabecular bone is mainly osteoblast- and not osteocyte-derived. As expected, no effect on cortical bone density or cortical bone thickness was observed in the Dmp1cre-Rspo3flox/flox mice (Fig. 4j, k).
Inducible inactivation of Rspo3 reduces trabecular bone volume fraction
Although very informative, the studies using life-long osteoblast-lineage specific Rspo3 inactivation cannot separate between early developmental effects of RSPO3 and its effects on adult bone metabolism. To evaluate the effect of RSPO3 specifically on adult bone homeostasis and the underlying mechanism, before a compensatory new steady state of bone remodeling has been reached, we developed a mouse model with tamoxifen-inducible Rspo3 inactivation in adult mice. To this end, we bred Rspo3flox/flox mice with CAGGCre-ER transgenic mice expressing a tamoxifen-inducible Cre-mediated recombination system (Fig. 4l)44. We have previously shown that CAGGCre-ER mice do not have a skeletal phenotype45. All mice evaluated were treated with the same tamoxifen regime and the phenotype of CAGGCre-ER-Rspo3flox/flox and Rspo3flox/flox mice were compared (Fig. 4l). The Rspo3 mRNA levels were lower (−38%, P = 0.05) in the vertebral body of tamoxifen-treated CAGGCre-ER-Rspo3flox/flox mice compared with tamoxifen-treated Rspo3flox/flox mice (Fig. 4m). To determine the skeletal phenotype in the inducible Rspo3 knockout mice, 8-week-old Rspo3flox/flox and CAGGCre-ER-Rspo3flox/flox mice were treated with tamoxifen and the phenotype was evaluated 38 days after the first tamoxifen dose. The inducible inactivation of Rspo3 did not affect body weight or weights of several visceral organs (Fig. 4n,o). Analyses of the vertebral trabecular bone volume fraction revealed a significant reduction both when evaluated using CT (BV/TV; −12.3%, P = 0.009, Fig. 4p–s) and histomorphometry (−18.0%, P = 0.038; Supplementary Fig. 6a–d) in tamoxifen-treated CAGGCre-ER-Rspo3flox/flox mice compared with tamoxifen-treated Rspo3flox/flox mice. Inducible Rspo3 inactivation did not affect cortical bone density or cortical bone thickness (Fig. 4t, u). Expression analyses in the vertebral body revealed that inducible Rspo3 inactivation reduced Dkk1 mRNA levels (−32%, P = 0.049; Fig. 4v), suggesting a compensatory mechanism to counteract additional trabecular bone loss. Dynamic histomorphometry of vertebral trabecular bone demonstrated a 31% reduction of trabecular bone formation rate per tissue volume (P = 0.047, Supplementary Fig. 6e) and similar effect sizes were observed for number of osteoblasts per trabecular bone perimeter (−32%, P = 0.088; Supplementary Fig. 6f) and for levels of the circulating bone formation marker P1NP (−20%, P = 0.067; Supplementary Fig. 6g) in tamoxifen-treated CAGGCre-ER-Rspo3flox/flox mice compared with tamoxifen-treated Rspo3flox/flox mice. In contrast, there was no indication of bone resorption being affected in mice with inducible Rspo3 inactivation (Supplementary Fig. 6f, g). To further explore the role of osteoblast-derived RSPO3 on bone formation, we performed extensive studies using different osteoblast cell cultures.
Osteoblast-derived RSPO3 increases osteoblast proliferation and differentiation
In our first attempts to assess the role of cell-autonomous RSPO3 for osteoblast differentiation we used calvarial osteoblasts isolated from newborn Rspo3flox/flox and Runx2-creRspo3flox/flox mice. The cells were incubated for 4 days in culture flasks to expand cell numbers and subsequently seeded in multiwells for 3 days. Rspo3 mRNA was moderately lower (−59%; P < 0.01) in cells from Runx2-creRspo3flox/flox mice compared with cells from Rspo3flox/flox mice and this was associated with a significant decrease of Alpl mRNA (−61%, P = 0.011; Supplementary Fig. 7a).
To achieve a more efficient Rspo3 inactivation in calvarial osteoblasts, we used cells isolated from CAGGCre-ER-Rspo3flox/flox mice treated with tamoxifen in vitro, resulting in an almost complete inactivation of Rspo3 mRNA expression (Fig. 5a), whereas tamoxifen-treated cells from Rspo3flox/flox mice had unaltered Rspo3 mRNA levels (Supplementary Fig. 7b). When these calvarial osteoblasts were cultured in osteogenic medium it was observed that inactivation of Rspo3 resulted in decreased osteoblast differentiation as assessed by substantially decreased ALP staining in culture using cells from tamoxifen-treated CAGGCre-ER-Rspo3flox/flox mice as compared to genotypically identical cells not treated with tamoxifen, or with cells from Rspo3flox/flox mice incubated in the absence or presence of tamoxifen (Fig. 5b). Importantly, osteoblasts from tamoxifen-treated CAGGCre-ER-Rspo3flox/flox mice had almost completely lost the capability to form mineralized noduli, in contrast to cells from the same genotype not treated with tamoxifen (Fig. 5c, d), or cells from Rspo3flox/flox mice with or without tamoxifen (Fig. 5c, Supplementary Fig. 7c). The effects on ALP activity and bone noduli formation were associated with lower expression of Alpl and Col1a1 mRNA in tamoxifen-treated CAGGCre-ER-Rspo3flox/flox cells (Fig. 5e,f) as compared to the absence of effect by tamoxifen in cells from Rspo3flox/flox mice (Supplementary Fig. 7d, e).

a Rspo3 mRNA expression in primary calvarial osteoblasts (cOBL) isolated from CAGGCre-ER-Rspo3flox/flox mice cultured for 2 and 6 days (d) with or without prior Rspo3 inactivation using tamoxifen (TAM). Veh=vehicle. b Representative photos of alkaline phosphatase staining of CAGGCre-ER-Rspo3flox/flox and Rspo3flox/flox cOBL cultured for 6 days after veh treatment or Rspo3 inactivation. c, d Representative photos (c) and quantification (d) of mineralized noduli in cultures of CAGGCre-ER-Rspo3flox/flox and Rspo3flox/flox cOBL cultured for 14 days after veh treatment or Rspo3 inactivation. e, f mRNA expression analysis of Alpl (e) and Col1a1 (f) in CAGGCre-ER-Rspo3flox/flox cOBL cultured for 2 and 6 days with or without prior Rspo3 inactivation. g Haematoxylin (Htx) and Dapi staining of CAGGCre-ER-Rspo3flox/flox cOBL cultured for 3 days with or without prior Rspo3 inactivation. Scale bar 100 μm. h Amount of DNA per well in CAGGCre-ER-Rspo3flox/flox cOBL at the time of TAM addition (day −1), directly after removal of TAM (day 0) and after culture for 1, 3, 6, and 10 days. i Mki67 mRNA expression in CAGGCre-ER-Rspo3flox/flox cOBL cultured for 2 days after Rspo3 inactivation. j, k mRNA expression of Tnfsf11 (j) and Tnfrsf11b (k) in CAGGCre-ER-Rspo3flox/flox cOBL cultured with or without PTH and PGE2 for 5 days with or without prior Rspo3 inactivation. n = 3–4 wells. l–n TRAP staining (l), counting (m), and Acp5 mRNA expression (n) of bone marrow macrophages cultured in M-CSF (M) or in M-CSF and RANKL (RL) to induce osteoclastogenesis in the absence or presence of recombinant RSPO3 (rRSPO3). Scale bar 200 μm. Individual values are presented in all graphs with the mean presented as horizontal lines and ±SEM as vertical lines. If not otherwise stated, n = 4 wells per group. Experiments were repeated two (g, j–n) or at least three (a–f, h, i) times. For a, e, f, h, two-way ANOVA was used to determine the overall effect of Rspo3 inactivation by treatment of TAM or veh, time, as well as their interaction. For j, k, two-way ANOVA was used to determine the overall effect of PTH/PGE2 treatment, Rspo3 inactivation by treatment of TAM or veh, as well as their interaction. When only the effect of Rspo3 inactivation was evaluated, two-sided Student’s t was used. Source data are provided as a Source Data file.
Deletion of Rspo3 in tamoxifen-treated cells from CAGGCre-ER-Rspo3flox/flox mice resulted in less dense cell cultures due to decreased cell numbers as demonstrated by decreased amounts of DNA, an effect absent in tamoxifen-treated cells from Rspo3flox/flox mice or cells from Rspo3flox/flox mice with or without tamoxifen (Fig. 5g, h, Supplementary Fig. 7f). Reduced cell numbers were due to an effect on cell proliferation as demonstrated by lower mRNA expression of Mki67, a well-established marker for cell proliferation (Fig. 5i, Supplementary Fig. 7g). Collectively, these studies demonstrate that RSPO3 enhances osteoblast proliferation and differentiation in a cell-autonomous manner.
In agreement with the in vivo observations, RSPO3 did not affect osteoclast formation as demonstrated by the observations that the osteoclastogenic response to PTH in the calvarial osteoblasts (increased Tnfsf11 and decreased Tnfrsf11b mRNA) was not affected by deletion of Rspo3 (Fig. 5j, k, Supplementary Fig. 7h, i), nor did addition of recombinant RSPO3 protein affect osteoclast formation induced by RANKL in bone marrow macrophage cultures (Fig. 5l–n).
RSPO3 increases osteoblast proliferation and differentiation through WNT-β-catenin signaling
Through their Furin-like domain 1, RSPOs bind to extracellular domains of the E3 ligases ZNRF3/RNF43, which are important for ubiquitination and proteasomal degradation of LRP6 and Frizzleds46. When the ternary complex of RSPOs, LGRs and ZNRF3/RNF43 is formed, ZNRF3/RNF43 is endocytosed and degraded, resulting in decreased breakdown of Frizzleds and LRP6, and consequently to increased sensitivity to WNT canonical signaling47,48. We found that deletion of Rspo3 in tamoxifen-treated osteoblasts from CAGGCre-ER-Rspo3flox/flox mice resulted in decreased amounts of total LRP6 as well as pLRP6 (Fig. 6a–d). In contrast, tamoxifen treatment did not affect total amounts of LRP6 in osteoblasts from Rspo3flox/flox mice (Supplementary Fig. 8a, b). Increased WNT canonical signaling is associated with enhanced mRNA expression of negative regulators such as the ligases Znrf3 and Rnf4347,48. We noticed that the mRNA expression of Znrf3 and Rnf43 was decreased in tamoxifen-treated osteoblasts from CAGGCre-ER-Rspo3flox/flox mice (Fig. 6e, f), whereas tamoxifen did not affect their expression in osteoblasts from Rspo3flox/flox mice (Supplementary Fig. 8c, d). These observations suggest that deletion of Rspo3 in the osteoblasts results in decreased WNT canonical signaling. To directly assess if downregulation of LRP6 and decreased phosphorylation of LRP6 reduced WNT canonical intracellular signaling, we analyzed the mRNA expression of Tcf7 and Lef1, two well-known WNT canonical target genes49. It was found that the mRNA expressions of both Tcf7 and Lef1 were significantly decreased in osteoblasts from CAGGCre-ER-Rspo3flox/flox mice treated with tamoxifen (Fig. 6g, h). In contrast, tamoxifen treatment of osteoblasts from Rspo3flox/flox mice did not affect Tcf7 or Lef1 mRNA expression (Supplementary Fig. 8e, f).

a, b LRP6 protein analysis in primary calvarial osteoblasts isolated from CAGGCre-ER-Rspo3flox/flox mice cultured in osteogenic media for 1 day with or without prior Rspo3 inactivation using tamoxifen (TAM). Lane view from the software in the capillary-based electrophoresis immunodetection system (a) and protein levels (b) normalized to total protein. c, d pLRP6 protein analysis in primary calvarial osteoblasts isolated from CAGGCre-ER-Rspo3flox/flox mice cultured in osteogenic media for 1 day with or without prior Rspo3 inactivation using tamoxifen (TAM). Lane view from the software in the capillary-based electrophoresis immunodetection system (c) and protein levels (d) normalized to total protein. e–h mRNA expression analyses of Znrf3 (e), Rnf43 (f), Tcf7 (g) and Lef1 (h) in primary calvarial osteoblasts isolated from CAGGCre-ER-Rspo3flox/flox mice cultured in osteogenic media for 6 days with or without prior Rspo3 inactivation using TAM. Veh=vehicle. Individual values are presented in all graphs with the mean presented as horizontal lines and ±SEM as vertical lines. n = 4 wells per group. Experiments were repeated one (e, f), two (a, b, g, h) or at least three (c, d) times. Statistical analyses were performed using two-sided Student’s t test. Source data are provided as a Source Data file.
Discussion
Bone fractures are a major public health concern. Recent large-scale human genetic studies have identified 15 loci associated with fractures at any bone site and the understanding of the causal genes and underlying mechanisms for these fracture signals may result in improved prevention and/or treatment of patients with high fracture risk3,8. A major genetic determinant for fractures at any bone site is located at the RSPO3 locus. We, herein, used a combination of human genetic studies and extensive functional mechanistic studies to identify the causal gene and its mechanism for the fracture signal at the RSPO3 locus. Human genetic studies revealed that the strong fracture association signal at the RSPO3 locus has the capacity to regulate RSPO3 expression, and that increased circulating RSPO3 levels associate with increased trabecular BMD and substantially reduced risk of distal forearm fractures. Studies using several different conditional Rspo3-inactivated mouse models revealed that osteoblast-derived RSPO3 is the principal source of RSPO3 in bone and a major regulator of trabecular bone volume fraction in mice. Further functional studies revealed that osteoblast-derived RSPO3 improves bone strength in vertebrae of mice. Mechanistic studies showed that osteoblast-derived RSPO3 increases osteoblast proliferation and differentiation via cell-autonomous effects, involving WNT canonical signaling. Given that the RSPO3 locus confers amongst the largest effects on fracture risk in the human genome, these findings point to a protein, whose effects may be a major contributor to fracture risk in humans.
Several previous studies have reported that genetic determinants at the RSPO3 locus are robustly associated with fractures at any bone site, BMD as measured by DXA at the hip and/or spine, and estimated BMD as determined by ultrasound in the heel3,8,50,51,52,53,54,55. Peripheral CT, but not DXA or ultrasound, can differentiate between the cortical and trabecular bone compartments. We extended previous human genetic studies by showing that the most prominent fracture signal at the RSPO3 locus was significantly associated with trabecular volumetric BMD as measured by peripheral CT in the distal metaphyseal region of tibia, while no clear association was observed with cortical bone parameters in the diaphyseal region of tibia. These findings suggest that the main fracture signal at the RSPO3 locus has an impact on fracture risk mainly via an effect on the trabecular bone mass9,34. An effect mediated via trabecular bone mass was supported by our findings from larg -scale bone site-specific fracture association analyses, revealing a pronounced association for the genetic signal at the RSPO3 locus with fractures at the distal forearm, a bone site dependent on trabecular bone.
These human genetic studies, however, did not determine if RSPO3 is the causal gene. Our first evidence of RSPO3 being the causal gene was the observations that the prominent fracture association signal at the RSPO3 locus was associated with RSPO3 mRNA levels in both subcutaneous adipose tissue and cultured fibroblasts and circulating RSPO3 protein levels in two independent cohorts33,35. Importantly, the fracture reducing allele at the RSPO3 locus was associated with increased RSPO3 expression both at the mRNA and protein levels and increased trabecular volumetric BMD in the distal tibia. Based on these human genetic data, we hypothesized that RSPO3 increases trabecular bone mass and thereby reduces fracture risk. To test this hypothesis, we performed extensive mechanistic studies using several conditional Rspo3-inactivated mouse models and cultured bone-derived cells.
In descriptive studies, we first demonstrated that substantial RSPO3 expression was observed in both cortical and trabecular bone. Detailed cell-specific expression analyses using chromogenic in situ hybridization, single cell RNA sequencing of bone marrow cells and cultured bone-derived cells, revealed RSPO3 expression in osteoprogenitor cells and osteoblasts but not in osteocytes or osteoclasts in both humans and mice. The single cell RNA sequencing data showed RSPO3 expression in mesenchymal stem cells in the bone marrow, supporting previous studies describing RSPO3 expression in different types of stem cells56. These descriptive findings were verified by functional studies using mouse models with inactivation of Rspo3 in osteoblast-lineage cells. To determine the source of RSPO3 in bone, we developed and compared conditional Rspo3-inactivated mouse models with inactivation of Rspo3 early in both osteoblasts and osteocytes (Runx2-cre) or in late osteoblasts and osteocytes (Dmp1-cre). Rspo3 mRNA levels in bone were almost completely abolished in the mice with inactivation of Rspo3 early in both osteoblasts and osteocytes while only marginal reduction was observed in mice with inactivation only in late osteoblasts and osteocytes. These findings demonstrate that osteoblasts and not osteocytes or osteoclasts are the major source of RSPO3 in both trabecular and cortical bone.
Importantly, inactivation of Rspo3 in both early osteoblasts and osteocytes resulted in a marked reduction of the trabecular bone volume fraction in the vertebrae, whereas inactivation of Rspo3 in late osteoblasts and osteocytes only marginally reduced the trabecular bone volume fraction, demonstrating that osteoblast-derived, but not osteocyte-derived RSPO3, is a major regulator of trabecular bone mass. Furthermore, the trabecular bone volume fraction was strongly directly correlated with Rspo3 mRNA levels in the vertebral body, supporting the notion that local Rspo3 expression regulates trabecular bone mass in mice.
Although informative, the studies using life-long osteoblast-specific RSPO3 inactivation could not exclude early developmental effects, confounding the adult phenotype. Therefore, to evaluate the effect of RSPO3 on adult trabecular bone homeostasis, we developed a mouse model with tamoxifen-inducible Rspo3 inactivation in adult mice. A reduction of trabecular bone volume fraction was observed 5.5 weeks after the inducible Rspo3 inactivation in adult mice, excluding confounding developmental effects and demonstrating that endogenous RSPO3 exerts crucial effects on trabecular bone homeostasis in adult mice. In contrast, no effect on cortical bone thickness or cortical volumetric BMD was observed in mice with life-long osteoblast-lineage specific Rspo3 inactivation or global adult inducible inactivation of Rspo3. These findings are in accordance with the human genetic finding in the present study, demonstrating that the most prominent fracture signal at the RSPO3 locus was associated with trabecular volumetric BMD, but not with cortical bone thickness or cortical volumetric BMD. Collectively, these findings indicate that RSPO3 mainly affects trabecular bone mass in both humans and mice.
It is well established that trabecular and cortical bone may respond differently to treatments1, and that WNT signaling has the capacity to regulate both the trabecular and the cortical bone compartments10,17,18,19,57,58. Previous studies have demonstrated that osteoblast-derived WNT16 is the major known cortical bone-specific WNT and we recently reported that the secreted WNT lipase NOTUM is a major regulator specifically of cortical bone mass with no effect on trabecular bone10,20,45,59. In contrast, we, herein, report that the extracellular WNT-signaling modulator RSPO3 mainly regulates trabecular bone mass. Similarly, WNT10b protects against age-dependent trabecular bone loss19. Thus, the data presented in the present study corroborate the clinical findings and suggest that the cortical and trabecular compartments are indeed biologically different, their homeostasis being independently regulated by different signaling molecules, all of which are involved in the regulation of bone mass10.
Mechanistic studies using a mouse model with inducible inactivation of Rspo3 in adult mice, and inducible Rspo3 inactivation in cultured osteoblasts, collectively revealed that osteoblast-derived RSPO3 increases osteoblast proliferation and differentiation in a cell-autonomous manner. A stimulatory effect in vivo of RSPO3 on bone formation is supported by a recent phase 1 clinical trial, evaluating the safety of different doses of an anti-RSPO3 antibody (OMP-131R10/rosmantuzumab) in patients with advanced solid tumors. High doses of the anti-RSPO3 antibody reduced serum levels of the bone formation marker P1NP in a majority of the evaluated subjects, indicating that RSPO3 enhances bone formation also in humans (https://publications.oncomed.com/Ph-1ab-OMP-131R10-anti-RSPO3-adv-solid-tumor-prev-treat-met-CRC-2016-EORTC-NCI-AACR.pdfDownloaded 2020.11.24).
RSPOs are potent enhancers of WNT signaling by making up a ternary complex with LGR receptors 4–6 and the E3 ubiquitin ligases ZNRF3/RNF43, through their furin-like domains 2 and 1, respectively30,31,46. A role of LGR4 for bone mass is suggested by the observations that a rare nonsense mutation within the LGR4 gene was reported to be strongly associated with low BMD and with osteoporotic fractures in Icelandic individuals60, and that global deletion of Lgr4 in mice results in reduced vertebral trabecular bone volume due to reduced bone formation61, suggesting that RSPO3 might influence bone formation in trabecular bone via activation of LGR4. Formation of the ternary complex results in membrane clearance of ZNRF3/RNF43 and thereby decreased proteasomal degradation of the WNT receptors Frizzleds and the Frizzled co-receptor LRP647,48, known to be important in WNT canonical signaling49. In a feedback loop, the mRNA expressions of Znrf3 and Rnf43 are increased47,48. We found that LRP6 protein was substantially decreased, and that the mRNA expressions of Znrf3 and Rnf43 were downregulated in osteoblasts in which Rspo3 had been deleted, suggesting that WNT canonical signaling was decreased as a consequence of Rspo3 deletion. Interestingly, LRP6 has previously been reported to be important for trabecular bone and osteoblast differentiation62. The fact that the mRNA expressions of Tcf7 and Lef1, two classical signature genes induced by WNT canonical signaling49, were decreased in osteoblasts with deletion of Rspo3, provides direct evidence that WNT canonical signaling is enhanced by RSPO3. These findings indicate that RSPO3 stimulates osteoblast proliferation and differentiation through enhanced WNT canonical signaling.
Similar to our present observations for RSPO3, it has been reported that also RSPO1 and RSPO2 can stimulate osteoblast differentiation in vitro and that deletion of Rspo2 in osteocalcin-expressing osteoblasts reduces trabecular bone mass due to decreased bone formation22,63. However, human genetic studies have not demonstrated any strong association between genetic variants at the RSPO1 or RSPO2 loci and bone mass or fracture susceptibility, suggesting that RSPO3 is the major RSPO-regulating bone mass and fracture susceptibility in humans. A previous report claimed that RSPO3 regulates the differentiation of human adipose-derived stem cells via an activation of LGR464. However, one effect of RSPO3-LGR4 activation in adipose-derived stem cells in vitro in that study was to inhibit their osteogenic potential while a robust stimulatory effect of RSPO3 on osteoblast differentiation and proliferation of osteoprogenitors/osteoblasts both in in vivo and in vitro was observed in the present study. Differential effects of RSPO3 with capacity to both increase and reduce adipogenesis, depending on the origin of adipose progenitors, have recently been described for abdominal-derived and gluteal-derived adipose progenitors65. Thus, RSPO3 may either enhance or reduce proliferation and/or differentiation in a cell-type specific and differentiation stage-dependent manner. In the present study, we observed RSPO3 expression in adipocytes within the bone marrow, in both mouse and human, as well as in scRNA seq of bone marrow adipogenic stromal cells. However, the functional role of RSPO3 in bone marrow adipocytes needs to be evaluated in future studies.
In conclusion, using a combination of human genetic studies and extensive functional mechanistic studies, we identified the causal gene and its mechanism for the prominent fracture association signal at the RSPO3 locus. We provide human genetic evidence that the fracture reducing allele at that locus increases RSPO3 expression, circulating RSPO3 level and trabecular bone mass and that it mainly reduces the risk of distal forearm fractures. The underlying mechanism includes a cell-autonomous effect of RSPO3 on osteoblast proliferation and differentiation.
Methods
Human genetic association studies
Look ups of fracture signals at the RSPO3 locus in different available data sets
Fractures at any bone site: Association with fractures at any bone site for the most significant SNP at the RSPO3 locus (rs7741021) is available in Table S4 from Morris et al.8 while the associations for the other evaluated SNPs at this locus (rs3734626 and rs2489623) are taken from the GWAS summary statistics for fractures at any bone site, available at the GEFOS website http://www.gefos.org.
Estimated BMD (eBMD) at the heel analyzed by ultrasound: Associations with eBMD for the evaluated SNPs at the RSPO3 locus are taken from the GWAS summary statistics from Morris et al.8, available at the GEFOS website http://www.gefos.org.
Trabecular vBMD, cortical vBMD and cortical thickness as analyzed by peripheral quantitative computed tomography (pQCT): Associations for the evaluated SNPs at the RSPO3 locus with trabecular vBMD in the distal metaphyseal region of tibia and cortical vBMD in the tibia diaphyseal region are taken from the GWAS meta-analyses summary statistics from Paternoster et al.34, while the association with cortical bone thickness in the tibia diaphyseal region are taken from Zheng et al.9.
eQTLs: Associations with eQTLs for the three SNPs at the RSPO3 locus were evaluated using the GTEx Portal https://www.gtexportal.org/home/
cis-pQTL: The associations for the most significant cis-protein QTLs (cis-pQTLs) for RSPO3 were taken from two independent data sets, including rs3734626 from Emilsson et al. (Supplemental Table 13)33 and rs2489623 from Sun et al. (Supplementary Table 4)35.
New associations analyses of candidate SNPs at the RSPO3 locus with distal forearm fractures and hip fractures
Our new analyses of associations between candidate SNPs at the RSPO3 locus with distal forearm fractures (defined by ICD10 codes S52.5 and S52.6; N = 7324 distal forearm fracture cases) and hip fractures (defined by ICD10 codes S72.0, S72.1 and S72.2; N = 4035 hip fracture cases) were performed using data from the UK Biobank study (http://www.ukbiobank.ac.uk). Briefly, the UK Biobank is a large prospective cohort study of approximately a half-million adult (ages 40–69 years) participants living in the United Kingdom, recruited from 22 centers across the United Kingdom in 2006–201066. We included 438,756 participants of white European descent with valid data on the candidate SNPs at the RSPO3 locus, the two fracture outcomes (distal forearm fractures and hip fractures) and relevant covariates (age, sex, height, weight). The UK Biobank has ethical approval from the North West Multi-Centre Research Ethics Committee (application no. 16/NW/0274), and informed consent was obtained from all participants. The present research was approved by the UK Biobank Research and Access Committee (application no. 51784).
Associations between the three candidate SNPs at the RSPO3 locus and the two fracture outcomes (distal forearm fractures and hip fractures) were performed using BOLT-LMM67. Briefly, this method uses a linear mixed model to account for relatedness and population structure. Fracture risk was corrected for age, age2, height, weight, sex, genotyping array and PC1 + … + PC20 in logistic regression models. Individuals were excluded based on unusually high heterozygosity or >5% missing genotype rate, a mismatch between self-reported and genetically inferred sex.
Mendelian randomization (MR)
We performed MR analyses to determine the causal associations of RSPO3 on fracture risk and bone parameters using inverse variance-weighted (IVW) MR. The most significant cis-pQTL, either rs3734626 from Emilsson et al.33, or rs2489623 from the independent data set of Sun et al.35, was used as genetic instrument for circulating RSPO3. Genetic associations with the exposure (RSPO3) and outcomes (fractures and bone parameter) used in the Mendelian randomization are presented in Table 1b (rs3734626) and Table 1c (rs2489623). As genetic variants are randomly distributed at birth, they are unaffected by confounders. We then regressed the association of the cis-pQTL SNP on the outcome measure, weighing its effect by the magnitude of its effect upon the corresponding exposure.
In order to reduce potential horizontal pleiotropy, we used only cis-SNPs for serum RSPO3 levels. These cis-SNPs for RSPO3 were defined in the published GWAS as the lead SNPs within 300 kB, in the data set of Emilsson et al.33, or 1 MB, in the independent data set of Sun et al.35, of the RSPO3 gene. Neither of the cis-SNPs at the RSPO3 locus from the two different sources were associated with any other circulating protein levels included in the extensive circulating proteomic analyses, reducing the risk of potential horizontal pleiotropy. To assess the possibility of reverse causality that bone mass regulates circulating RSPO3, a bidirectional MR using eBMD as the exposure and circulating RSPO3 from Sun et al.35 as the outcome was performed, using 1107 LD-independent SNPs from Morris et al.8 as instrumental variables for eBMD, followed by IVW to meta-analyze their combined effect on the RSPO3 levels.
Colocalization
We tested for colocalization of the genetic signal for circulating RSPO3 and trabecular volumetric eBMD using colocalization analyses, which assesses potential confounding by linkage disequilibrium. A stringent Bayesian analysis implemented in COLOC R package was performed to estimate the posterior probability (PP) that the same causal signal in the 1 MB genomic locus centered on the cis-SNP affects both circulating RSPO3 and trabecular volumetric BMD37. Summary statistics for the RSPO3 locus were available for the cis-pQTL identified in the study by Sun et al. but not for the cis-pQTL identified in the study by Emilsson et al.33 and consequently the analysis was done only for the cis-pQTL identified by Sun et al.35.
Animal experiments
Cell-specific and inducible Rspo3-inactivated mouse models were generated by breeding Rspo3flox/flox mice (Rspo3tm1.1Jcob/J, JAX stock #027313, Jackson Laboratories)41 with mice expressing cre recombinase under the control of different promoters. In the Rspo3flox mice, LoxP sites are introduced upstream from Rspo3 exon 2 and downstream from Rspo3 exon 4 (Fig. 3a) and in the presence of an active cre recombinase, the DNA fragment including exons 2–4 of the Rspo3 allele is excised.
All procedures involving animals were approved by the Ethics Committee in Gothenburg, Västra Götaland, and the care of the animals was in compliance with all relevant ethical regulations for animal testing and research. The mice were housed in a standard animal facility with a 12 h dark/light period, at standard temperature and humidity. Food and water were available ad libitum. At termination, the animals were anesthetized with Ketador/Dexdomitor (Richter Pharma/Orion Pharma), bled, and euthanized by cervical dislocation. Long bones and vertebrae were dissected and stored for future analyses, soft tissues were collected, weighed, and snap-frozen in liquid nitrogen.
Generation of Runx2-creRspo3flox/flox mice
To generate specific inactivation of Rspo3 in the osteoblast-lineage, Rspo3flox/flox mice were bred with Runx2-cre mice10,42. The Runx2-creRspo3flox/flox offspring display early osteoblast-specific cre expression and have the capacity to recombine LoxP-flanked DNA sequences in an early osteoblast-specific manner and recombination occurs at all sites of endochondral and intramembranous bone formation, particularly in periosteal cells, osteoblasts and osteocytes but not osteoclasts, when Runx2-cre mice were crossed to a Rosa26 reporter strain42. The littermate control mice were Rspo3flox/flox. Runx2-cre mice have an unchanged skeletal phenotype compared to WT mice10.
Generation of Dmp1cre-Rspo3flox/flox mice
To generate mice depleted of Rspo3 in late osteoblasts and osteocytes, Rspo3flox/flox female mice were crossed with heterozygous Rspo3+/flox male mice expressing cre recombinase driven by the 10-kb Dmp1 promoter specifically expressed in late osteoblasts and osteocytes (Tg(Dmp1-cre)1Jqfe mice) (Dmp1-cre mice)43. The littermate control mice were Rspo3flox/flox. We have previously demonstrated that efficient recombination occurs in osteocytes but not osteoblasts or osteoclasts when Dmp1-cre mice were crossed to a Rosa26 reporter strain68.
Generation of mice with inducible Rspo3 inactivation
In order to study the acute adult effects of Rspo3, avoiding confounding developmental effects, inducible Rspo3 knockout mice were created by breeding the Rspo3flox/flox mice with the CAGGCre-ER (B6.Cg-Tg(CAG-cre/Esr1*)5Amc/J, JAX stock #004682, Jackson Laboratories) transgenic mice44,69. The CAGGCre-ER-Rspo3flox/flox offspring express a tamoxifen-inducible cre-mediated recombinase. To study the effects of Rspo3 on the adult bone tissue, a long-term study using adult CAGGCre-ER-Rspo3flox/flox mice were performed. Male 8-week-old CAGGCre-ER-Rspo3flox/flox and Rspo3flox/flox littermate control mice were injected with tamoxifen (50 mg kg−1; Sigma-Aldrich) for three consecutive days to activate the cre construct. The mice were terminated 38 days after the first tamoxifen injection at the age of 13.5 weeks.
The different mouse models were genotyped using the primers listed in Supplementary Table 1.
In situ hybridization
The chromogenic in situ hybridization was performed on vertebra L5 from 5-month-old mice, on 8 G needle biopsies from the posterior iliac crest of human healthy volunteers, as well as proximal femur specimens obtained from coxa valga patients undergoing corrective surgery. The bone specimens were fixated in 4% formaldehyde in phosphate buffered saline, followed by demineralization in 15% EDTA with 0.4% PFA for 3 weeks, and paraffin embedded. All human subjects have given their written informed consent, and the study was conducted according to the World Medical Association Declaration of Helsinki and approved by the Danish National Committee on Biomedical Research Ethics, journal no. S-20110112 and S-20120193. 3.5-µm-thick (human) and 6-µm-thick (mouse) consecutive dewaxed sections were in situ hybridized using a modified protocol of the RNAscope 2.5 HD manual protocol (322300, Advanced Cell Diagnostics, Bio-Techne Ltd., Abingdon, UK)70. The following probes were used: mouse R-spondin 3 (Rspo3, 402011, target sequence 731–2164), human R-spondin 3 (RSPO3, 490581, target sequence 38–1546), mouse Runt-related transcription factor 2 (Runx2, 414021, target sequence 3838-4821), mouse collagen 1a1 (Col1a1, 319371, target sequence 1686–4669), mouse dentin matrix protein 1 (Dmp1, 441171, target sequence 689–1543), and mouse tartrate resistant acid phosphatase 5 (Trap (Acp5), 465001, target sequence 200–1414). Briefly, after pre-treatment, the sections were hybridized with the probe over night at 40 °C, followed by six steps of amplification according to the manufacturer’s instructions. To further amplify the signal, the DAB staining step was omitted and the signal was amplified with digoxigenin-labeled tyramide signal amplification (NEL 748001KT, Akoya Biosciences, Marlborough, MA, USA), detected by alkaline phosphatase-conjugated anti-digoxigenin Fab fragments (11093274910, Sigma-Aldrich), and visualized with Liquid Permanent Red (K0640, DAKO, Carpinteria, CA, USA). The human sections were subsequently immunostained with mouse anti-TRAP antibodies (Clone ZY-9C5, Zymed), labeled with horseradish peroxidase–conjugated anti-mouse IgG polymers (BrightVision, Immunologic, Duiven, the Netherlands), visualized with Deep Space Black (Biocare Medical, Concord, CA, USA). Finally, both mouse and human sections were counterstained with Mayer’s Hematoxylin (MHS1, Sigma-Aldrich) and mounted with Aquatex (1085620050, Sigma-Aldrich).
Real-time quantitative PCR
Total mRNA was prepared from mouse cortical bone, vertebral body, brain cortex, flushed bone marrow, liver, lung, gonadal fat, spleen, kidney, thymus, muscle, testis, and heart using TRIzol reagent (15596018; Thermo Fischer Scientific) and/or RNeasy Mini Kit (74106; Qiagen). For preparation of total mRNA from cultured cells, the RNeasy Micro Kit (74004; Qiagen) was used.
The mRNA was reversed transcribed to cDNA (4368814; Thermo Fischer Scientific) and real-time PCR analyses were performed using the StepOnePlus Real-Time PCR System (version 2.3, Thermo Fischer Scientific) and the following Assay-on-Demand primer and probe sets: R-spondin 3 (Rspo3), Mm01188251_m1; Sclerostin (Sost), Mm00470479_m1; Dickkopf-1 (Dkk1), Mm00438422_m1; R-spondin 1 (Rspo1), Mm00507077_m1; R-spondin 2 (Rspo2), Mm00555790_m1; R-spondin 4 (Rspo4), Mm00615419_m1; Alkaline phosphatase (Alpl), Mm00475834_m1; Acid phosphatase 5 (Acp5), Mm00475698_m1; Collagen type 1 alpha 1 (Col1a1), Mm00801666_g1; Tumor necrosis factor receptor superfamily, member 11b (Tnfrsf11b, Opg), Mm00435452_m1; Tumor necrosis factor superfamily member 11 (Tnfsf11, Rankl), Mm00441908_m1; Ki67 (Mki67), Mm01278617_m1; Tcf7 (Tcf1), Mm00493445_m1; Lef1, Mm01310389; Znrf3, Mm01191456_m1; and Rnf43, Mm00552558_m1. The expressions of each gene were normalized to 18S ribosomal subunit (4310893E; Thermo Fischer Scientific). The 2−∆∆Ct method was used to calculate the relative gene expression.
Serum analyses
ELISA RatLaps Kit (AC-06F1, Immunodiagnostic Systems, East Boldon, United Kingdom) were used to measure serum levels of C-terminal type I collagen (CTX) fragments to assess bone resorption. To assess bone formation, serum levels of procollagen type I N-terminal propeptide (P1NP) were measured using a Rat/Mouse EIA Kit (AC-33F1, Immunodiagnostic Systems).
Single cell RNA sequencing (scRNA seq)
The recently published single cell RNA seq data from Cxcl12-eGFP expressing cells was used to study Rspo3 expression in bone marrow stromal cells39. Both data sets (GSM4064136 Cxcl12GFPCxcl12CE#1 and GSM4064137 Cxcl12GFPCxcl12CE#2) was downloaded from GEO project GSE136970 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136970), imported into R and analyzed using Seurat39. Cells with less than 1000 genes per cell were filtered out and the two datasets were merged, resulting in 8026 cells from the two datasets. We further filtered out cells with more than 15 % mitochondrial read content, resulting in 7332 cells remaining for analysis. Further analysis was performed after normalization of the data using the logNormalize method and uniform manifold approxiamtion and projection (UMAP) cluster analysis was done using the first 10 principal components (1:10 dimensions). We obtained 8 different cell clusters (scRNA seq Fig. 1 and scRNA seq Supplementary Fig. 1). Heat map and feature plots of selected genes identified the clusters as four osteogenic/adipogenic (cluster 0, 1, 4 and 7), one mitotic (cluster 6), one endothelial (cluster 5) and two haematopoetic (cluster 2 and 3) (scRNA seq Fig. 1). In contrast to Matsushita et al we kept the haematopoetic clusters in our subsequent Rspo3 analysis. Rspo3 expression was in addition analyzed in the nicheExplorer made available by Iannis Aifantis lab (http://aifantislab.com/niche) in an interactive data browser as a supplement to their published scRNAseq data of mouse bone marrow vascular, perivascular, and osteoblast cell populations40.
Assessment of bone parameters
Bone histomorphometry
For dynamic histomorphometry, the mice were intraperitoneally injected with the fluorochromes calcein and alizarin (Merck GmbH, Darmstadt, Germany) 9 and 2 days prior to termination, respectively10. Upon dissection, vertebra L5 were fixated in 4% formaldehyde, dehydrated in EtOH, and imbedded in methyl methacrylate. Static trabecular bone parameters were determined in a 4-μm-thick plastic sections stained in Masson-Goldner Trichrome and dynamic trabecular bone parameters were determined in unstained 8-μm-thick sections. All parameters were analyzed using the OsteoMeasure7 histomorphometry system (OsteoMetrics, Atlanta, GA, USA), following the guidelines of the American Society for Bone and Mineral Research71.
High resolution micro-computed tomography
High-resolution micro-computed tomography (µCT) were used to analyze lumbar vertebra 5 (Skyscan, 1172; Bruker MicroCT, Aartselaar, Belgium)10. The lumbar vertebra were imaged with an X-ray tube voltage of 50 kV and a current of 201 µA, with a 0.5 mm aluminum filter, and the scanning angular rotation was 180°, and the angular increment was 0.70°. NRecon (version 1.6.9.8, Bruker MicroCT) was used to perform reconstruction after scans. The trabecular bone was analyzed 235 µm from the lower end of the pedicles and continued for approximately 229 µm. The data was analyzed using the CTAn software (version 1.13.2.1, Bruker MicroCT).
Peripheral quantitative computed tomography
Computed tomography (pQCT) scans were performed using the peripheral quantitative computed tomography XCT Research M (v.4,5B; Norland Stratec, Pforzheim, Germany), at a resolution of 70 µm72. The cortical bone parameters were analyzed in the mid-diaphyseal region of the left femur to determine cortical volumetric bone mineral density (BMD) and the cortical thickness73.
Mechanical strength
Intact lumbar vertebra 5 was axially loaded with a press head of 2 mm in diameter, with a 1 mm-thick holder through the vertebral foramen, at a speed of 0.155 mm s−1 using a mechanical testing machine (Instron 3366, Instron)74. The results were calculated by a custom Excel macro using the computer recorded load deformation raw data curves produced by Bluehill 2 software v2.6 (Instron).
Cell culture media
Primary murine osteoblasts and osteoclasts were cultured in complete α-MEM medium (MEM alpha medium (Gibco, 22561-021) supplemented with 10% heat-inactivated FBS (Sigma, F7524), 2 mM GlutaMAX (Gibco, 35050-038), 50 μg ml−1 gentamicin (Gibco, 15750-037), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Gibco, 15140-148)). For osteoblast differentiation and mineralization assays, mouse calvarial osteoblasts were cultured in complete osteogenic α-MEM supplemented as described above and with 4 mM β-glycerophosphate disodium salt hydrate (BGP; Sigma, G9422) and 0.28 mM l-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Asc-2P; Sigma, A8960). For osteoclast differentiation assays, bone marrow macrophages were cultured in complete α-MEM supplemented with 30 ng ml−1 M-CSF (R&D Systems, 416-ML-050) and 4 ng ml−1 RANKL (R&D Systems, 462-TEC-010).
Murine primary calvarial osteoblast culture
Murine primary calvarial osteoblasts were isolated from 4 to 6 days old mice by sequential collagenase treatments75. Cells in collagenase fractions 6-10 were cultured in complete α-MEM in T75 flasks for 4 days prior to re-seeding the cells at 20,000 cells cm−2 in 48-well plates. Osteoblasts were cultured in complete osteogenic α-MEM for indicated times with change of half of the culture media volume every 2–3 days. For gene expression analyses, cells were lysed in RNeasy lysis (RLT) buffer with β-mercaptoethanol (Qiagen), followed by RNeasy micro RNA purification (Qiagen), cDNA synthesis and real-time PCR as described above. DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen) following the manufacturer’s instruction for animal cells. For ALP staining, cells were fixed in citrate buffered acetone and stained according to the commercial kit protocol (Sigma, 85L2-1KT). Mineralized nodules were visualized by fixation in 2.5% glutaraldehyde in 70% ethanol for 5 min, followed by three washes in 70% ethanol. For quantification of mineralization, images were taken using a Jenoptik Gryphax camera connected to a Nikon Eclipse 80i microscope and mineralized surface per well was quantified using the Bioquant OSTEO software (version 20.8.6 volume 1993).
In vitro ablation of Rspo3 in cultured calvarial osteoblasts
Primary calvarial osteoblasts from CAGGCre-ER-Rspo3flox/flox and Rspo3flox/flox control mice were isolated as above and cultured in complete α-MEM for 2–3 days prior to re-seeding in multiwell plates and culture in complete osteogenic media. Cells seeded in 48-well plates were cultured in osteogenic media for 24 h, followed by addition of tamoxifen (Sigma, ReadyMade solution, SML1666) for additional 24 h, before complete media change and continued culture in complete osteogenic media without tamoxifen. 50 or 100 nM tamoxifen was used and resulted in similar results. Osteoblastic differentiation was followed and analyzed as described above with the time of tamoxifen removal designated day 0. PTH (10 nM, Bachem, H-1660) and PGE2 (1 µM, R&D Systems, 2296) was added the day after tamoxifen removal (day 1) and continued until day 6, when cells were harvested for gene expression analysis as described above.
Murine bone marrow macrophages and osteoclasts cultures
Mouse bone marrow derived macrophages (BMM) were obtained from 10 to 12 weeks old male wild type C57Bl/6N mice by culture of bone marrow cells in suspension culture dishes in complete α-MEM supplemented with 30 ng ml−1 M-CSF (R&D Systems, 416-;L-050)76,77. BMMs were spot seeded in 48-well plates (20,000 cells in 20 μl complete α-MEM) and allowed to attach for 5 min before addition of complete α-MEM supplemented with 30 ng ml−1 M-CSF, with or without 4 ng ml−1 RANKL (R&D Systems, 462-TEC-010) and 100 ng ml−1 recombinant RSPO3 (R&D Systems, 4120-RS-025) for 3 days to induce osteoclast differentiation. Cells were stained for tartrate resistant acid phosphatase (TRAP) after 3 days using a commercial kit (Sigma, 387A-KT1). TRAP-positive cells with at least three nuclei were defined as osteoclasts and counted using the Bioquant OSTEO software. For gene expression analyses, cells were lysed in RLT buffer with β-mercaptoethanol (Qiagen), followed by RNeasy micro RNA purification (Qiagen), cDNA synthesis and real-time PCR as described above.
Protein preparation and analyses
Primary calvarial osteoblasts from CAGGCre-ER-Rspo3flox/flox and Rspo3flox/flox control mice were isolated as above and cultured in complete α-MEM for 2–3 days prior to re-seeding in 12-well plates at 20,000 cells cm−2. Cells were then cultured in osteogenic media for 24 h, after which 100 nM tamoxifen was added for 24 h, followed by complete media change to osteogenic media without tamoxifen. Protein lysates were prepared 24 h after removal of tamoxifen by washing the cells once with cold PBS followed by addition and lysis in RIPA buffer (Sigma, R0278) with protease inhibitors (complete Mini EDTA-free; RocheDiagnostics, 5892970001) and phosphatase inhibitors (PhosSTOP; Roche Diagnostics, 04906845001). Lysates were transferred to tubes, centrifuged for 10 min at 11,000×g, +4 °C, and the supernatant was used for protein analysis. Protein concentration was quantified using the DC Protein Assay Kit (Bio-Rad, 500-0112). Levels of LRP6 and pLRP6 were analyzed by capillary-based electrophoresis and immunodetection using the JESS ProteinSimple system and the Compass for SW Software (version 5.0.0, Protein Simple) as described by the manufacturer. Protein lysates were analyzed with anti-LRP6 (clone C47E12, Cell Signaling Technology, 3395, dilution 1:250) or anti-pLRP6 (Ser1490, Cell Signaling Technology, 2568, dilution 1:10) together with the Protein Normalization Assay Module (ProteinSimple, AM-PN01) and the Anti-rabbit Detection Module (ProteinSimple, DM-001) on 12–230 kDa capillary separation cassettes (ProteinSimple, SM-W004). LRP6 and pLRP6 are visually presented in the lane view of the Compass Software and quantified by normalization to total protein levels according to the SimpleWestern protocol and following the guidelines for presentation and quantification of protein data by Journal of Biological Chemistry78.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
For the human association studies we have used data from UK Biobank. Access to the UKB Resource is available by application (http://www.ukbiobank.ac.uk/). Access to data from the UK Biobank can be obtained at https://www.ukbiobank.ac.uk/enable-your-research. The UK Biobank resource is available to bona fide researchers for health-related research in the public interest. All researchers who wish to access the research resource must register with UK Biobank by completing the registration form in the Access Management System. Registrations are according to UK Biobank reviewed within 10 working days of submission (https://www.ukbiobank.ac.uk/enable-your-research/register). After registration is approved, applications on access to the UK Biobank research resource can be submitted. Research applications to access the data are performed via the Access Management System (https://bbams.ndph.ox.ac.uk/ams/).
The datasets used for the look ups of fracture signals at the RSPO3 locus are detailed in the “Methods” section, and have been published by Morris et al.8, GWAS summary statistics are available at the GEFOS website http://www.gefos.org, associations with eQTLs for the three SNPs at the RSPO3 locus were evaluated using the GTEx Portal https://gtexportal.org/home/, and the associations for the cis-pQTLs for RSPO3 were taken from two independent datasets, including rs3734626 from Emilsson et al.33 and rs2489623 from Sun et al.35.
Single cell RNA sequencing (scRNA seq) data: scRNA seq data have previously been deposited in GEO, accession number GSE13697039 and GSE10889240.
Source data are provided with this paper.
Change history
06 September 2021
In the original version of this Article, the given and family names of author Maha EL Shahawy were incorrectly tagged, leading to incorrect indexing. This error has now been corrected. The PDF and HTML versions displayed the name correctly at the time of publication.
References
Baron, R. & Hesse, E. Update on bone anabolics in osteoporosis treatment: rationale, current status, and perspectives. J. Clin. Endocrinol. Metab. 97, 311–325 (2012).
Johnell, O. & Kanis, J. Epidemiology of osteoporotic fractures. Osteoporos. Int. 16, S3–S7 (2005).
Trajanoska, K. et al. Assessment of the genetic and clinical determinants of fracture risk: genome wide association and mendelian randomisation study. BMJ (Clin. Res. ed.) 362, k3225 (2018).
United Nations, D.o.E.a.S.A., Population Division. World Population Prospects: The 2015 Revision, Key Findings and Advance Tables. Working Paper No. ESA/P/WP.241 (2015).
Melton, L.J., 3rd & Cooper, C. Magnitude and impact of osteoporosis and fractures. In Osteoporosis (eds. Marcus, R. Feldman, D. & Kelsey, J.) 2nd edn (Academic Press, 2001).
Andrew, T., Antioniades, L., Scurrah, K. J., Macgregor, A. J. & Spector, T. D. Risk of wrist fracture in women is heritable and is influenced by genes that are largely independent of those influencing BMD. J. Bone Miner. Res. 20, 67–74 (2005).
Michaelsson, K., Melhus, H., Ferm, H., Ahlbom, A. & Pedersen, N. L. Genetic liability to fractures in the elderly. Arch. Intern. Med. 165, 1825–1830 (2005).
Morris, J. A. et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 51, 258–266 (2019).
Zheng, H. F. et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk.f PLoS Genet. 8, e1002745 (2012).
Moverare-Skrtic, S. et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 20, 1279–1288 (2014).
Wergedal, J. E., Kesavan, C., Brommage, R., Das, S. & Mohan, S. Role of WNT16 in the regulation of periosteal bone formation in female mice. Endocrinology 156, 1023–1032 (2015).
Lerner, U. H. & Ohlsson, C. The WNT system: background and its role in bone. J. Intern. Med. 277, 630–649 (2015).
Baron, R. & Kneissel, M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 19, 179–192 (2013).
Cadigan, K. M. & Peifer, M. Wnt signaling from development to disease: insights from model systems. Cold Spring Harb. Perspect. Biol. 1, a002881 (2009).
Jenny, A. Planar cell polarity signaling in the Drosophila eye. Curr. Top. Dev. Biol. 93, 189–227 (2010).
Kohn, A. D. & Moon, R. T. Wnt and calcium signaling: beta-catenin-independent pathways. Cell calcium 38, 439–446 (2005).
Zhong, Z. et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc. Natl. Acad. Sci. USA 109, E2197–E2204 (2012).
Bennett, C. N. et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 102, 3324–3329 (2005).
Stevens, J. R. et al. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J. Bone Miner. Res. 25, 2138–2147 (2010).
Moverare-Skrtic, S. et al. Osteoblast-derived NOTUM reduces cortical bone mass in mice and the NOTUM locus is associated with bone mineral density in humans. FASEB J. 33, 11163–11179 (2019).
Chen, P. H., Chen, X., Lin, Z., Fang, D. & He, X. The structural basis of R-spondin recognition by LGR5 and RNF43. Genes Dev. 27, 1345–1350 (2013).
Knight, M. N. et al. R-spondin-2 is a Wnt agonist that regulates osteoblast activity and bone mass. Bone Res. 6, 24 (2018).
Szenker-Ravi, E. et al. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature 557, 564–569 (2018).
Wei, Q. et al. R-spondin1 is a high affinity ligand for LRP6 and induces LRP6 phosphorylation and beta-catenin signaling. J. Biol. Chem. 282, 15903–15911 (2007).
Zhu, C. et al. LGR4 acts as a key receptor for R-spondin 2 to promote osteogenesis through Wnt signaling pathway. Cell. Signal. 28, 989–1000 (2016).
Cambier, L., Plate, M., Sucov, H. M. & Pashmforoush, M. Nkx2-5 regulates cardiac growth through modulation of Wnt signaling by R-spondin3. Development 141, 2959–2971 (2014).
Marie, P. J. & Miraoui, H. & Severe, N. FGF/FGFR signaling in bone formation: progress and perspectives. Growth Factors 30, 117–123 (2012).
Carmon, K. S., Gong, X., Lin, Q., Thomas, A. & Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 108, 11452–11457 (2011).
Aoki, M. et al. R-spondin3 is required for mouse placental development. Dev. Biol. 301, 218–226 (2007).
de Lau, W. et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297 (2011).
Glinka, A. et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 12, 1055–1061 (2011).
Trajanoska, K. et al. Genetic basis of falling risk susceptibility in the UK Biobank Study. Commun. Biol. 3, 543 (2020).
Emilsson, V. et al. Co-regulatory networks of human serum proteins link genetics to disease. Science (N. Y., N. Y.) 361, 769–773 (2018).
Paternoster, L. et al. Genetic determinants of trabecular and cortical volumetric bone mineral densities and bone microstructure. PLoS Genet. 9, e1003247 (2013).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Zheng, J. et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 52, 1122–1131 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Bianco, P. et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat. Med. 19, 35–42 (2013).
Matsushita, Y. et al. A Wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat. Commun. 11, 332 (2020).
Tikhonova, A. N. et al. The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 (2019).
Neufeld, S. et al. A conditional allele of Rspo3 reveals redundant function of R-spondins during mouse limb development. Genes. (N. Y., N. Y.: 2000) 50, 741–749 (2012).
Rauch, A. et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 11, 517–531 (2010).
Lu, Y. et al. DMP1-targeted Cre expression in odontoblasts and osteocytes. J. Dent. Res. 86, 320–325 (2007).
Hayashi, S. & McMahon, A. P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318 (2002).
Ohlsson, C. et al. Inducible Wnt16 inactivation: WNT16 regulates cortical bone thickness in adult mice. J. Endocrinol. 237, 113–122 (2018).
Zebisch, M. et al. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 4, 2787 (2013).
Hao, H. X. et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485, 195–200 (2012).
Koo, B. K. et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488, 665–669 (2012).
Wiese, K.E., Nusse, R. & van Amerongen, R. Wnt signalling: conquering complexity. Development 145, dev165902, (2018).
Correa-Rodriguez, M., Schmidt Rio-Valle, J. & Rueda-Medina, B. The RSPO3 gene as genetic markers for bone mass assessed by quantitative ultrasound in a population of young adults. Ann. Hum. Genet. 82, 143–149 (2018).
Mullin, B. H. et al. Genome-wide association study meta-analysis for quantitative ultrasound parameters of bone identifies five novel loci for broadband ultrasound attenuation. Hum. Mol. Genet. 26, 2791–2802 (2017).
Styrkarsdottir, U. et al. Sequence variants in the PTCH1 gene associate with spine bone mineral density and osteoporotic fractures. Nat. Commun. 7, 10129 (2016).
Moayyeri, A. et al. Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum. Mol. Genet. 23, 3054–3068 (2014).
Duncan, E. L. et al. Genome-wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet. 7, e1001372 (2011).
Estrada, K. et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 44, 491–501 (2012).
Da Silva, F. et al. Myocardial-specific R-spondin3 drives proliferation of the coronary stems primarily through the Leucine Rich Repeat G Protein coupled receptor LGR4. Dev. Biol. 441, 42–51 (2018).
Maeda, K. et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 18, 405–412 (2012).
Yu, B. et al. Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat. Med. 20, 1009–1017 (2014).
Brommage, R. et al. NOTUM inhibition increases endocortical bone formation and bone strength. Bone Res. 7, 2 (2019).
Styrkarsdottir, U. et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 497, 517–520 (2013).
Luo, J. et al. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 136, 2747–2756 (2009).
Riddle, R. C. et al. Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS ONE 8, e63323 (2013).
Kronke, G. et al. R-spondin 1 protects against inflammatory bone damage during murine arthritis by modulating the Wnt pathway. Arthritis Rheum. 62, 2303–2312 (2010).
Zhang, M. et al. RSPO3-LGR4 regulates osteogenic differentiation of human adipose-derived stem cells via ERK/FGF signalling. Sci. Rep. 7, 42841 (2017).
Loh, N. Y. et al. RSPO3 impacts body fat distribution and regulates adipose cell biology in vitro. Nat. Commun. 11, 2797 (2020).
Collins, R. What makes UK Biobank special? Lancet 379, 1173–1174 (2012).
Loh, P. R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Windahl, S. H. et al. Estrogen receptor-alpha in osteocytes is important for trabecular bone formation in male mice. Proc. Natl. Acad. Sci. USA 110, 2294–2299 (2013).
Ohlsson, C. et al. WNT16 overexpression partly protects against glucocorticoid-induced bone loss. Am. J. Physiol. Endocrinol. Metab. 314, E597–E604 (2018).
Lassen, N. E. et al. Coupling of bone resorption and formation in real time: new knowledge gained from human Haversian BMUs. J. Bone Miner. Res. 32, 1395–1405 (2017).
Dempster, D. W. et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 (2013).
Windahl, S. H., Vidal, O., Andersson, G., Gustafsson, J. A. & Ohlsson, C. Increased cortical bone mineral content but unchanged trabecular bone mineral density in female ERbeta(-/-) mice. J. Clin. Investig. 104, 895–901 (1999).
Vidal, O. et al. Estrogen receptor specificity in the regulation of skeletal growth and maturation in male mice. Proc. Natl. Acad. Sci. USA 97, 5474–5479 (2000).
Moverare-Skrtic, S. et al. The bone-sparing effects of estrogen and WNT16 are independent of each other. Proc. Natl. Acad. Sci. USA 112, 14972–14977 (2015).
Granholm, S., Henning, P., Lindholm, C. & Lerner, U. H. Osteoclast progenitor cells present in significant amounts in mouse calvarial osteoblast isolations and osteoclastogenesis increased by BMP-2. Bone 52, 83–92 (2013).
Granholm, S., Lundberg, P. & Lerner, U. H. Calcitonin inhibits osteoclast formation in mouse haematopoetic cells independently of transcriptional regulation by receptor activator of NF-{kappa}B and c-Fms. J. Endocrinol. 195, 415–427 (2007).
Takeshita, S., Kaji, K. & Kudo, A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J. Bone Miner. Res. 15, 1477–1488 (2000).
Fosang, A. J. & Colbran, R. J. Transparency is the key to quality. J. Biol. Chem. 290, 29692–29694 (2015).
Acknowledgements
We thank Anna Westerlund and Ulrika Björklund at the University of Gothenburg for excellent technical assistance. Single cell data analysis was performed by Sanna Abrahamsson from the Bioinformatics Core Facility at the Sahlgrenska Academy. We thank Jian Q Feng from the Department of Biomedical Sciences at the Texas A&M University for providing the Dmp1-cre mice. This study was supported by the Swedish Research Council, the Swedish Foundation of Strategic Research, the Swedish state under the agreement between the Swedish government and the county councils, the ALF agreement in Gothenburg, the IngaBritt and Arne Lundberg Foundation, the Torsten and Ragnar Söderberg’s Foundation, the Knut and Alice Wallenberg Foundation, the Novo Nordisk Foundation, and the Adlerbertska Research Foundation. The Richards research group is supported by the Canadian Institutes of Health Research (CIHR: 365825; 409511), the Lady Davis Institute of the Jewish General Hospital, the Canadian Foundation for Innovation, the NIH Foundation, Cancer Research UK, Genome Québec, the Public Health Agency of Canada and the Fonds de Recherche Québec Santé (FRQS). J.B.R. is supported by a FRQS Clinical Research Scholarship. Support from Calcul Québec and Compute Canada is acknowledged. TwinsUK is funded by the Welcome Trust, Medical Research Council, European Union, the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. These funding agencies had no role in the design, implementation or interpretation of this study. T.L.’s group has been financially supported by the Academy of Finland: grants 3322098, 286284, 117787; Competitive State Research Financing of the Expert Responsibility area of Tampere (grant X51001); Tampere University Hospital Supporting Foundation and Finnish Society of Clinical Chemistry.
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
K.H.N., P.H., M.E.S., J.W., K.L.G., and S.M.-S. performed the animal experiments, T.L.A., C.E., L.E.R., and M.E.S. performed the in situ hybridization; A.K. and J. Tuukkanen performed the vertebral compression test; P.H., P.P.C.S., and U.H.L performed the in vitro experiments; J.Tuckermann provided the Runx2-cre mice; M.L., T.L., J.H.T., S.Z., J.B.R., M.N., and C.O. performed the human genetic studies; K.H.N., P.H., U.H.L, S.M-S., and C.O. interpreted the results; K.H.N, P.H., U.H.L, S.M.-S., and C.O. wrote the manuscript. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.B.R. has served as an advisor to GlaxoSmithKline and Deerfield Capital. He has received investigator-initiated grant funding from Eli Lilly, GlaxoSmithKline and Biogen for projects unrelated to this paper. M.L. has received lecture fees from Amgen, Astellas, Lilly, Meda, Renapharma, UCB Pharma, and consulting fees from Amgen, Radius Health, UCB Pharma, Renapharma, and Consilient Health. The other authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Neil Davies, David Karasik, Stavros Manolagas and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nilsson, K.H., Henning, P., El Shahawy, M. et al. RSPO3 is important for trabecular bone and fracture risk in mice and humans. Nat Commun 12, 4923 (2021). https://doi.org/10.1038/s41467-021-25124-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25124-2
This article is cited by
-
An atlas of genetic determinants of forearm fracture
Nature Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
