
Abstract
The temporal and spatial coarseness of megafaunal fossil records complicates attempts to to disentangle the relative impacts of climate change, ecosystem restructuring, and human activities associated with the Late Quaternary extinctions. Advances in the extraction and identification of ancient DNA that was shed into the environment and preserved for millennia in sediment now provides a way to augment discontinuous palaeontological assemblages. Here, we present a 30,000-year sedimentary ancient DNA (sedaDNA) record derived from loessal permafrost silts in the Klondike region of Yukon, Canada. We observe a substantial turnover in ecosystem composition between 13,500 and 10,000 calendar years ago with the rise of woody shrubs and the disappearance of the mammoth-steppe (steppe-tundra) ecosystem. We also identify a lingering signal of Equus sp. (North American horse) and Mammuthus primigenius (woolly mammoth) at multiple sites persisting thousands of years after their supposed extinction from the fossil record.
Similar content being viewed by others

Introduction
Humans evolved and dispersed throughout the continents in an epoch dominated by giant terrestrial mammals. Megafauna (body mass ≥ 44 kg) only exist in comparable densities today within small refugia (mainly Africa) where most of their populations are in states of decline, and many of these species are threatened or endangered1,2. The ecological reverberations associated with the Late Pleistocene (130,000–11,700 years before present [BP]) loss of approximately 101 of 150 genera3 of Earth’s largest terrestrial animals is thought to have restructured the terrestrial biosphere, impacting vegetation composition and diversity, biogeochemistry, and climate feedback systems4,5,6,7,8,9,10,11,12,13. This rearrangement of terrestrial ecosystems, including massive biogeographic range shifts, local extirpations, and widespread extinctions, is argued by some to be the direct result of rapid climate change and attendant environmental feedbacks during the late Pleistocene14,15,16,17. Others contend18,19,20,21,22,23 that factors unique to the last glacial period are to blame, such as the coincident dispersal of a new predator—Homo sapiens. It is likely that no single factor can account for the staggered magnitude of such losses globally, but rather that each ecosystem experienced a variable set of locally compounding pressures17,24,25. Taphonomic processes challenge attempts to tease apart the palaeoecological nuances of the late Quaternary extinctions (LQE), necessitating relatively precise estimates for megafaunal population declines and last appearance dates26,27, for timings of ecological shifts (e.g. changes in plant community structure), as well as for robust archaeological evidence of anthropogenic impacts.
In the case of eastern Beringia (unglaciated regions of Yukon, Canada and Alaska, U.S.A.), Guthrie16, Mann et al.28,29, and Rabanus-Wallace et al.30 argue that the expansion of woody shrubs and peatlands following an increased moisture regime during the late Pleistocene was the leading contributor to the loss of megafaunal grazers, including mammoth, horse, and bison. By contrast, Zimov et al.23,31 contend that megafaunal extirpations preceded a rise in woody shrubs, with the loss of keystone megaherbivores having led to the disappearance of the graminoid and forb dominated, mammoth-steppe biome5,31,32,33. Disentangling the relative timings of ecological restructuring versus megafaunal population declines often exceeds the resolution34 of Quaternary records.
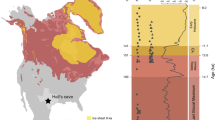
Here, we present hybridization capture enriched sedimentary ancient DNA (sedaDNA) data derived from loessal silts preserved in permafrost (Fig. 1, Table 1) and recovered from four sites in the Klondike goldfields—an unglaciated region of west-central Yukon Territory, Canada35—dating to ca. 30,000–4000 calibrated (calendar) years before present (cal BP). This work builds on the methodological results reported in Murchie et al.36 in which North American horse (Equus caballus) and woolly mammoth (Mammuthus primigenius) DNA was unexpectedly identified in a permafrost sample dating to ~9700 cal BP. This post-dates the last macrofossil evidence (such as bones, teeth, and soft-tissues) of these animals in Alaska by some 3300 years. Such a late date is indicative of a substantial ghost range (cryptic population)—an extended spatio-temporal range derived from palaeoecological proxies that post-date the last macrofossil remains37.

Base map data retrieved from GeoYukon (hosted by the Government of Yukon); contours elevation unit: meters above sea level. See Supplementary Fig. 2 for a description of inset map data sources.
Our aims here are two-fold. The first is to assess how our permafrost sedaDNA dataset can be used to assess the plausibility of two competing explanatory models of Beringian extinctions. These are the bottom-up “shrub/peatland expansion” model of Guthrie16 and Mann et al.28,29,30, and the top-down “keystone megaherbivore decline” model of Zimov et al.5,23. The shrub/peatland expansion model argues that climate change drove ecosystems into new equilibrium states that were unfavourable for megafaunal herbivores in eastern Beringia25. In contrast, the keystone-decline model posits that megaherbivores were active ecological engineers. When their populations began to decline (from anthropogenic or environmental pressures), it triggered numerous knock-on effects including the disappearance of mammoth-steppe vegetation and its replacement by shrublands and wet tundra. Our second objective is to replicate and test the extent of the sedaDNA ghost ranges for Equus sp. and Mammuthus as identified in Murchie et al.36.
Background
Late Quaternary losses on the eastern mammoth-steppe
Megafaunal extinctions and extirpations after 40,000 cal BP in the Holarctic (i.e., northern Eurasia, Beringia, and North America) arguably followed a two-stage pattern suggested by dated macrofossils27 (Supplementary Table 1). The first wave seems to have occurred prior to or during the Last Glacial Maximum (LGM, 26,500–19,000 cal BP)38 which in eastern Beringia included the loss of Homotherium latidens (scimitar-toothed cat) and Arctodus simus (short-faced bear). The second wave occurred between 15,000–10,000 cal BP, which in mainland Beringia included Equidae (caballine and stilt-legged horses), Mammuthus primigenius (woolly mammoth), Panthera spelaea (cave lion), and Saiga tatarica (saiga antelope), while at least 20 genera went extinct or were extirpated in North America south of the continental ice sheets. Other species such as Bison priscus (steppe bison) survived until at least ~6000 years ago in southern Yukon39, with genetic data supporting a B. priscus persistence until as recently as a few hundred years ago40. M. primigenius survived on Wrangel (Russia) and St. Paul (Alaska) islands until about 4000 and 5500 cal BP, respectively41,42. Holarctic faunal assemblages of Pleistocene predators, species with small populations, and taxa living in understudied regions are expectedly sparse. This has led some to argue that these two late Pleistocene extinction/extirpation pulses are an artifact of an inadequate record and the Signor-Lipps effect43,44, and that the true wave of losses occurred during or after the Younger Dryas, ca. 12,900 cal BP25,37,45.
Beringia’s environment during the late Pleistocene has been characterized as a graminoid and forb-dominated steppe-tundra mosaic generally referred to as the mammoth-steppe31,33,35,46,47,48,49,50. It is thought to have been the most extensive terrestrial biome on Earth during the late Pleistocene, stretching from the Iberian Peninsula eastward across Eurasia and into Canada31,50,51,52, although the extent and character of this ecosystem remains controversial for some53. This paradoxically productive54,55 high-latitude mosaic biome supported a diverse abundance of large bodied fauna31,50,52, facilitating higher biotic productivity (energy and nutrient turnover) than many habitats existing at high latitudes today23,56.
Owen-Smith32 proposed the keystone herbivore hypothesis based on the ecology of extant African megafauna to explain the role of megaherbivores in transforming vegetation structure and composition57,58,59,60,61,62,63. Nutrients locked in leaves and stems are liberated when used by fauna, accelerating biogeochemical cycling11. In this top-down model, megafauna are critical for maintaining and promoting biodiversity in open-mosaic environments, and for controlling the abundance of woody vegetation that can limit biodiversity5. A severe reduction of these megafaunal engineers64,65,66 is proposed to have resulted in the conversion of mosaic, steppe grasslands and wood pastures to more uniform forests and prairies, shrinking mosaic ecotones—high productivity transition areas between biological communities67—thereby reducing the carrying capacity of terminal Pleistocene environments. This in turn could have led to a positive-feedback response wherein diminishing populations of megafauna were increasingly unable to control woody shrub expansion, further reducing the biotic productivity of the mammoth-steppe.
Alternatively, an increasing moisture regime during the Bølling–Allerød interstadial (ca. 14,690–12,890 cal BP)68 is argued to have caused the rise of mesic-adapted woody shrubs that were highly defended against herbivory, replacing the diet of Pleistocene grazers (woolly mammoth, steppe bison, and horse)16,28,29,30, along with the paludification of Beringia (the spread of peatlands). In this line of bottom-up arguments, climate change and attendant environmental feedbacks led to the disappearance of the mammoth-steppe in eastern Beringia, along with the megafauna it supported.
Power and limits of sedimentary ancient DNA
Much of the LQE debate has been limited by the inability of dated macrofossils (primarily from detrital contexts) to convey the spatio-temporal resolution necessary to untangle the causative versus correlative ecological transformations associated with the Pleistocene-Holocene transition. Molecular (micro) methods69 are increasingly able to augment discontinuous macrofossil records. This can aid in identifying cryptic populations (or ghost ranges)37,70,71, independently assess population declines, and estimate the timings of functional extinctions/extripations15,72—the point at which undercrowding and inbreeding depression lead to a loss of fitness through Allee effects73,74,75 to the degree that a species no longer significantly contributes to ecosystem functioning, becoming a trace presence in records before completely disappearing.
Ancient environmental DNA (eDNA)76 is a powerful method for directly assessing the local77 presence of animals, plants, fungi, and microbiota through time33,36,37,41,78,79,80,81,82. Most eDNA is quickly metabolized by bacteria or otherwise degraded through a variety of chemical and physical processes. SedaDNA (referring to a subset of ancient eDNA sample types83) can survive these degradative processes, even in the absence of visible fossils, because cellular material can bind to sedimentary minerals84,85,86,87,88,89,90,91, protecting these molecular fragments for millennia, especially when perennially frozen92,93. Sediment samples as small as 100 mg can contain tens of billions of DNA fragments from all forms of life in a local ecosystem. However, there are several sedaDNA challenges to be aware of: (1) determining whether the recovered sedaDNA is stratigraphically accurate91; (2) whether the wet-lab recovery and targeting strategy, genetic reference databases, and taxon assignment approach (bioinformatic parameters) can accurately assess the breadth of eDNA or is prone to false-positive/negative assignments36,94; (3) assessing whether eDNA abundance retains a correlation with a population’s living biomass95,96; and (4) determining the degree to which sedimentary inhibitors or differential degradation may bias the sedaDNA signal36. Of particular importance for the kinds of permafrost sedaDNA analyzed in this report are the factors of reworked sedaDNA and leaching.
Perennially frozen sedaDNA has the potential to undergo erosion and redeposition while remaining chemically intact. Arnold et al.91 found evidence of reworked periglacial sediments in high-energy fluvial contexts within large catchments and local thermokarst deposists. They caution against sampling from settings where DNA can be readily reworked and redeposited within younger materials. In this study, we targeted loessal silts to mitigate the potential for fluvial reworking, although aeolian processes are certainly capable of reworking nanoscale sedaDNA complexes. Further, there is evidence throughout the Klondike of an early Holocene thaw unconformity97,98,99 that Mahony100 identified at Upper Goldbottom and Upper Quartz (among other sites) that presents the possibility for the localized reworking of sedaDNA during our early Holocene core samples. The Lucky Lady II section by contrast is continuous from >16,500– 8500 cal BP, sits toward the middle of a broad valley, and shows no evidence of erosion or redeposition by slope wash or thermokarst-induced slumping—suggesting that reworked early Holocene sedaDNA is of less concern at the Lucky Lady II site.
The vertical movement of free DNA has been found to be negligible in perennial frozen settings91. Studies have found synchronous palaeoecological shifts when comparing palynological and macrofossil evidence with sedaDNA reconstructions101,102, in addition to age-dependent DNA damage patterns103,104,105. Leaching is not considered to be a problem in perennially frozen sediment because there is minimal movement of liquid water33,37,102,106,107,108.
Results
Age-modelling and palynology
Age-depth models were first reported for cores used in this study by Sadoway109 and Mahony100. To refine the chronologies of these records we developed Bayesian age-depth models for each site using Oxcal v.4.4.2110 and the IntCal20 calibration curve111 (Supplementary Figs. 5–7 and Supplementary Table 3). Some outlier dates from the model were observed (Supplementary Figs. 6–7). As such, remodelled ages are reported here as 2σ maximum/minimum ranges to account for this uncertainty for comparing samples within and between sites (Table 1). A selection of subsamples were also analysed with palynology as a secondary proxy to the eDNA data. Most samples were nearly barren of palynomorphs, with only the Holocene-aged samples having preserved large numbers of pollen grains—in this case predominantly spruce (Picea) (Supplementary Table 4).
SedaDNA palaeoecology
Through the targeted capture36 of organelle eDNA preserved in loessal permafrost silts (Supplementary Fig. 4), Bison priscus (steppe bison), Mammuthus primigenius (woolly mammoth), Equus sp. (specifically limited to caballine horse), and Lagopus lagopus (willow ptarmigan) constitute most of the identifiable DNA reads of direct interest within Animalia (Fig. 2). This is in addition to less abundant organisms such as Rangifer tarandus (caribou/reindeer), and Ovis sp. (likely Dall sheep). Many reads expectedly lack taxonomic specificity at species and genus ranks (as many regions of the mitochondrial genome are variably conserved), and as such a large portion of reads could only be confidently assigned to higher ranks such as Caprinae, Pecora, Perissodactyla, and Elephantidae. In some cases, such as with hits to order Perissodactyla and superorder Afrotheria, we can be confident that they represent Equus (or its familial relative, Haringtonhippus [stilt-legged horse]), and Mammuthus, respectively, as unique members of their clades in the late Quaternary record of this region. There is also a low biomolecular signal from predators in this dataset including Canis lupus (grey wolf) and Martes sp. (marten). A variety of rodents were identified, including Urocitellus sp. (likely arctic ground squirrel), Microtus xanthognathus (taiga vole), and Dicrostonyx groenlandicus (northern collared lemming). Human DNA was identified despite not being targeted with the PalaeoChip baits. However, human DNA was also observed in the negative controls. We do not consider this human signal to be reliable without further investigation.

Values indicate unique reads assigned to that taxon node. Source data are provided as a Source Data file.
Overall, this taxonomically rich sedaDNA dataset reflects a gradual decline in the megafauna signal through time (Fig. 3). Elephantidae is one of the first to decrease in sedaDNA abundance after ~20,000 cal BP. This is followed by declining signals for Bovidae and Equidae (Fig. 2), until a punctuated decrease occurs near the Pleistocene-Holocene transition as their sedaDNA signals almost disappear while those for Alces alces (moose) and Cervus sp. (likely Cervus canadensis [elk/wapiti]) enter the dataset. Our admittedly temporally coarse set of permafrost samples suggests a delay in the disappearance of megafaunal grazing species in the Klondike to between ca. 13,000–10,000 cal BP, but we also observe a lag in the final appearance of Equus and Mammuthus sedaDNA as late as ca. 6000 cal BP. Cores from Lucky Lady II, Upper Goldbottom, and Upper Quartz retain 100+ DNA sequences assignable to those taxa well beyond their last dated macrofossils (Fig. 2).

Stacked normalized reads assigned to the rank ‘family’ in (A) Animalia (insects excluded) and (B) Viridiplantae. See Supplemental Figs. 10–12 for a metagenomic breakdown of samples by site. Source data are provided as a Source Data file. Note: sedaDNA abundance is likely affected by variable taphonomic intensity through time (such as temperature, moisture, and acidity) among other known and unknown micro-environmental conditions and recovery biases. While eDNA abundance and biomass arguably retain some correlation upon initial release, many other factors influence this relationship. As such, relative abundances with sedaDNA must be interpreted cautiously pending further research.
Plant sedaDNA clearly reflects a major environmental turnover between 13,500–10,000 cal BP (Figs. 3–4). Pleistocene graminoids (grass-like, herbaceous [non-woody] plants) such as Poaceae (grasses), Cyperaceae (sedges), along with a variety of forbs (herbaceous flowering plants) such as Artemesia (sagebrush), Lupinus (lupine), Saxifraga (rockfoil), Papaver (poppy), and Ranunculus (buttercup) were identified in relative sedaDNA abundance from 30,000–13,500 cal BP. Woody taxa such as Salix (willow), Populus (poplar), Betula (birch), Rhododendron, Arctous (bearberry), and Picea (spruce) were identified with increasing relative sedaDNA abundances after ~13,500 cal BP, along with Equisetum (horsetail), Gymnocarpium (oak ferns), and Sphagnum (peat moss). Our data suggests that forbs and graminoids (in this case Poaceae and Asteraceae) were dominant from ~30,000–13,500 cal BP while woody shrubs were comparatively rare (Fig. 3), indicating that the conventional idea of the mammoth-steppe holds until at least this late.

Values indicate unique reads assigned to that taxon node. Select taxon nodes depicted. Source data are provided as a Source Data file.
Assessing aDNA authenticity
All 13 negative controls had negligible library adapted molecules prior to indexing (Supplementary Fig. 8) and had minuscule molarities after targeted enrichment (Supplementary Fig. 9). Despite using the entire post-enrichment eluate for each of the controls during equimolar pooling, these blanks received minimal sequenced reads (102,440) and even fewer reads that could be taxonomically binned (81, 0.08%). Shotgun sequencing blanks processed with a subset of these libraries in Murchie et al.36 were almost entirely (>95%) adaptemers (adapter chimeric DNA), and likewise contained no signal of the ecologically relevant organisms under investigation here or in that previous work. As all 13 negative controls were processed identically in parallel with the permafrost subsample replicates (Supplementary Figs. 34–54), and yet contain none of the same sedaDNA signal (Figs. 2, 4, Supplementary Fig. 55), we can conclude that the trends observed here originate from the sediments themselves and are not the result of contamination.
We observe ancient DNA characteristic damage profiles where sufficient reads could be mapped to MEGAN and PIA identified references (Fig. 5, Supplementary Figs. 13–28), confirming that these molecules are ancient and not modern contaminants. The taxonomic constituents of these samples are also correlated more with age than site (Fig. 6), with 90% of the variance being explained by principal components 1/2 as driven by the proportions of Pinaceae, Asteraceae, and Salicaceae. These libraries also exhibit age- and climate-related damage patterns (Supplementary Figs. 29–33), suggesting that reworking and leaching have contributed minimally (if at all) to the ecological reconstructions.


Discussion
We observe four main trends within this dataset. (1) There is a surprising taxonomic richness and spatio-temporal consistency in the metagenomic signal across all sampling locations, suggesting that these reconstructions are representative of palaeoecological trends in the Klondike region. (2) Megafaunal sedaDNA declines gradually after the LGM with the Mammuthus primigenius signal being the first to drop out, followed by Bison priscus and Equus. (3) Signal dominance for forbs and graminoids is coeval with grazing megafauna, whereas the local transition towards woody shrubs is associated with a diminished faunal signal. Megafaunal sedaDNA reduces substantially after the Younger Dryas, by which time grazing megafauna had become functionally extirpated in the Klondike. (4) Despite this turnover, a low megafaunal sedaDNA signal persists into the Holocene. This signal—identified as a ghost range here—is suggestive of a late persistence of megafauna in a high latitude refugium, apparently outliving the functional extinction and complete loss of other continental populations.
Ecological turnover and collapse of the mammoth-steppe
Mammuthus, Equus, and Bison presence are closely associated in our dataset with forbs and graminoids characteristic of the mammoth-steppe biome. During the relative sedaDNA rise in Salicaceae (likely willow shrubs) ca. 13,500 and 10,000 cal BP, Asteraceae and Poaceae correspondingly decline in relative sedaDNA abundance while grazing megafaunal DNA largely disappears from our dataset. This suggests that, at least in the Klondike, Mammuthus primigenius may have been the first megafaunal species to undergo a local population reduction after ~20,000 cal BP.
It is difficult to say whether sedaDNA signal decay reflects an actual reduction in the regional abundance of animals or is reflective of other stochastic and unknown34 factors unique to this proxy such as variable eDNA release and turnover95, biochemical changes to eDNA stabilization processes such as organo-mineral binding90, or shifting microbial and other taphonomic pressures. As Equus sedaDNA remains relatively consistent until the rise of mesic-hydric woody shrubs during the Allerød warming (~13,500 cal BP) (Fig. 3B), this is suggestive of longer-term local declines of Mammuthus (and perhaps Bison) eDNA input rather than a shifting of biomolecular taphonomy because we would generally expect animal eDNA to breakdown or become mineral-bound88 at similar local rates. Further, Lagopus DNA spikes during major declines in megafaunal DNA. Again, implying local ecological rather than just taphonomic or methodological factors driving shifts in relative animal sedaDNA recovery. As this study used a capture enrichment approach targeting organelle genomes36 (with replicates [Supplementary Table 14]) rather than PCR metabarcoding amplicons76,112, relative changes in metagenomic signal abundance are arguably somewhat correlated with shifting eDNA inputs95,96,113,114,115,116 during periods of otherwise stable climate. As such, we suggest that major shifts in relative DNA abundance, such as Mammuthus sedaDNA nearly dropping out of the dataset after 20,000 cal BP, are ecologically informative.
The decline in local mammoth abundance that we infer from this sedaDNA record is consistent with a low frequency of 14C-dated mammoth macrofossils from the Klondike. M. primigenius macrofossils are comparatively rare in central Yukon relative to more northerly sites around Old Crow (Supplementary Fig. 1), but still relatively few of those northern macrofossils have been successfully radiocarbon dated because most are beyond radiocarbon range117,118. Much of the <30,000-year fossil record in Beringia is represented by a small number of well-preserved, and logistically accessible sites (Supplementary Fig. 1). Our faunal sedaDNA dataset somewhat conflicts with these macrofossil abundances in that the highest frequencies of DNA reads identified as Mammuthus, Equus, or Bison are distributed between 30,000–15,000 cal BP. Conversely, dated faunal remains of Mammuthus and Bison in eastern Beringia have a median concentration around 15,000 cal BP, with only Equus bones being predominant nearer to 25,000 cal BP (Supplementary Fig. 1).
Relative Bison and Equus sedaDNA signals decrease in read counts after ~15,000 cal BP. However, there is a subsequent increase around the onset of the Younger Dryas (ca. 12,900 cal BP) that may be associated with previously described Bison dispersals40,119 or other local factors (e.g. shifting mosaic vegetation patches25, herbivore land use, or taphonomy). It is worth noting that this inference is limited by there only being a single Younger Dryas core sample in this dataset. After the Younger Dryas (during the early Holocene) grazer sedaDNA nearly disappears (Fig. 3), which is correlated with the ecological turnover from forbs and graminoids to woody shrubs—predominantly Salix sp.—and a rise in avian fauna, rodents, and cervid browsers. There is a pronounced transition in the ecological signal after ~13,500 cal BP from forbs and graminoids to woody shrubs in the Lucky Lady II cores, with rises in Salix reads tightly associated with a sharp increase of Lagopus lagopus (willow ptarmigan) sedaDNA—a grouse whose habitat and subsistence patterns are based on woody shrubs120 (Fig. 3). Keesing and Young121 observed on the African savanna that when large grazing mammals were removed from an area, the rodent populations doubled, which increased the populations of predators that target small-bodied animals. Our data mirrors this observation with an increase in rodent sedaDNA after ~10,000 cal BP (Fig. 2), along with the appearance of the small forest dwelling carnivore Martes sp. (martens) who may also now be present because of trees on the landscape.
After Mammuthus primigenius and Equus sp. were functionally extirpated from the Klondike, the local ecosystem began transitioning towards boreal taxa with an associated rise in Picea (spruce) and mosses (Fig. 4). Despite significant declines in grazing megafaunal DNA, reads extend beyond their last dated macro-remains—perhaps even as late as the mid-Holocene—which has already been observed for Bison priscus39,40.
Our plant dataset is consistent with Willerslev et al.33 in which forbs were found to proportionally dominate their metabarcoded sedaDNA signal during and after the LGM. Nichols et al.112 argue that the forb dominance observed in Willerslev et al.33 was partly caused by polymerase and GC biases of their PCR metabarcoding approach favoring forbs over graminoids with the Platinum HiFi Taq polymerase targeting the short trnL (P6 loop) locus122. We have used a capture enrichment approach (indexed and reamplified with the KAPA SYBR FAST qPCR Master Mix) targeting much larger regions of the chloroplast genome (trnL [~500 bp], rbcL [~600 bp], and matK [~800 bp])36 where overall GC content is generally equivalent between the three major target families identified in Fig. 3 (see Supplementary Fig. 56). We suspect that beyond the PCR biases argued to have influenced Willerslev et al.33 (i.e. the greater relative abundance of forb sedaDNA compared to graminoids and woody plants) that this is likely the result of eDNA release and preservation characteristics of forbs with higher rates of biomass turnover. This more rapid turnover thus potentially leads to an eDNA over-representation of forbs compared with typical palynological findings. It has been argued that interpreting relative floral abundances with eDNA requires calibration. Yoccoz et al.95, for example, observed that their above-ground vegetation surveys were accurately mirrored in modern environmental soil DNA, but that functional groups (woody plants, graminoids, and forbs) varied in their proportional eDNA representation. Woody plants were most affected by this trend, being proportionally under-represented in eDNA compared to above ground biomass by 1:595, while graminoids were under-represented by 1:1.5. Conversely, forbs were over-represented by 2.5:1. GC and polymerase bias coupled with eDNA release variation, beyond simple growth form categories, complicates this further112. Nevertheless, the substantial abundance of forb DNA, even if cut by half, likely reflects an abundance of flowering herbs on the Pleistocene mammoth-steppe. This may also be under-represented palynologically due to varied pollen production between entomophilous (insect-pollinated) forbs and anemophilous (wind-pollinated) gramminoids123.
The rise in Pinaceae (notably spruce, see Fig. 4) around ~10,000 cal BP, and its growing dominance through our mid-Holocene samples, is consistent with other records from Yukon in regard to the initial development of the taiga/boreal forest124,125,126,127,128,129, and is consistent with pollen grains identified in samples younger than ca. 9200 cal BP (Supplementary Table 4). We observe similar relative sedaDNA increases in boreal flora (Fig. 4), albeit with a comparatively less abundant signal for Betula. Palynological studies frequently report an abundance of Betula with a comparatively small initial influx of Salix in the Alaskan-Yukon interior during the terminal Pleistocene shrub expansion125. While we observe a distinct rise in relative Betula sedaDNA during the Bølling–Allerød and post-Younger Dryas chronozones that persists into the Holocene, the number of Betula sedaDNA molecules are comparatively dwarfed by the immense abundances of Salicaceae (Salix [willow]) (Figs. 3–4) DNA. Betula is known to be over-represented by pollen, whereas Salix is often under-represented130,131,132,133. Our sedaDNA data suggests that Salix was more important in Beringian shrub expansion than palynological records have yet indicated.
The relative over-abundance of Salix sedaDNA compared to Betula is relevant to testing the shrub-expansion extinction model of Guthrie16, who contended that an increasing moisture regime and rise of mesic-hydric vegetation, with chemical defenses against herbivory (notably Betula nana exilis [resinous dwarf birch], but also including Salix)134,135,136, drove regional extirpations of grazing megafauna in eastern Beringia. If Salix was substantially more abundant in the Beringian shrub expansion than Betula as our sedaDNA dataset suggests, this questions whether the rise of defensive vegetation was a major driver in the extirpations as Salix is the most preferred and palatable shrub among extant subarctic browsers135,137. While Bison and Equus are considered closer towards the obligate grazer end of dietary guilds50,138, both have been observed to exhibit variable grazing and even mixed feeding139,140. Mammuthus, Equus, and Bison coprolites suggest that these taxa had a diet variably rich in forbs and graminoids, with a smaller but notable proportion of woody shrubs/trees (including alder [Alnus], birch [Betula], larch [Larix], spruce [Picea], and willow [Salix])33,141,142.
If mammalian sedaDNA abundances are rough indicators of palaeo-biomass (a correlation in need of further research), it is unclear why Mammuthus primigenius and Bison priscus (Fig. 2) relative sedaDNA signals decline prior to an expansion of woody shrubs during the Bølling-Allerød warming (Fig. 3). The abrupt increases in Cyperaceae (Carex [sedges]), Ericaceae (Arctous [bearberry], Rhododendron), Betulaceae, and Salicaceae during the Allerød are suggestive of a transition toward a moist dwarf-shrub ecosystem by ca. 13,500 cal BP. The presence of these plants suggests more continuous ground cover, better insulation, shallower permafrost, and likely boggy, wet conditions. The early Allerød rise of Fabaceae sedaDNA (particularly Astragalus and Oxytropis, Fig. 4) may also be indicative of a rise in flora with anti-herbivory defenses as these locoweeds are toxic to grazing fauna. Notably, despite otherwise rapid faunal and floral turnover, our single Younger Dryas core sample may suggest that the mammoth-steppe locally persisted through the Bølling-Allerød chronozone in the Klondike.
Mann et al.28,29 and Rabanus-Wallace et al.30 contend that a shifting moisture gradient from xeric to mesic-hydric, with the paludification of eastern Beringia, best accounts for the loss of dryland-specialists (Equus, Mammuthus), whereas mesic and mixed feeding seasonal fauna (Rangifer, Cervus, Ovibos) retained suitable habitats and hydric specialists (Alces, Homo sapiens) were able to invade new Beringian niches. While warming, an increasing moisture regime, the arrival of cervid browsers, and the rise of woody shrubs may explain much of the terminal signal decay observed for grazing specialists, this does not explain the relative declines of Mammuthus and Bison sedaDNA prior to the Bølling–Allerød chronozone (assuming that sedaDNA abundance retains a correlation with palaeo-biomass). Guthrie143 found that horses had undergone body-size declines after the LGM until their extirpation in Beringia, which likewise suggests longer-term pressures predating shrub expansion. The rise of woody plants in this dataset thus may be partially explained by both the gradual reduction in local megaherbivore ecosystem engineering over millennia and by a warming climate and shifting moisture regimes.
Unknowns in sedaDNA release, preservation, and recovery restrict what we can confidently infer from differences in relative signal abundance, but there are indications of both top-down and bottom-up contributions to the collapse of the mammoth-steppe in central Yukon. While the decline of faunal sedaDNA is likely influenced to some degree by shifting taphonomic processes (such as warming and increasing moisture causing more DNA degradation), substantial declines in megafaunal sedaDNA predates the Bølling–Allerød interstadial (14,690–12,900 cal BP)68. This could be seen as partially supporting Zimov and colleagues’5,23 keystone megaherbivore decline model that these animals were increasingly unable to maintain suitable steppe habitats due to declining populations. However, there is a substantial time lag in our dataset between a declining megafaunal sedaDNA signal after 20,000 cal BP and the relative rise of woody shrub sedaDNA ca. 13,500 cal BP. By contrast, the rise of mesic-hydric woody shrub DNA during the Allerød oscillation (13,900–12,900 cal BP) and early Holocene are clearly associated with an abrupt decline in megafaunal sedaDNA, strongly supporting the climate-induced shrub and peatland expansion model of Guthrie16 and Mann et al.28,29,30. There is potential support in this dataset for both bottom-up and top-down pressures influencing megafaunal extirpations in the Klondike. Further research is needed to determine whether indications of longer-term top-down pressures are real or an artifact of sedaDNA methodology, and to what degree each Beringian megafaunal species was differentially impacted and/or responsive to shifting ecological pressures.
The degree to which humans may have been involved in any of these transformations is hard to gauge from available evidence (see Supplementary Notes 1.2). Early (>14,000 cal BP) human presence in eastern Beringia is controversial but has been suggested based on possible anthropogenic cutmarks at Bluefish Caves144,145,146,147 and the identification of allegedly human fecal biomarkers and a coinciding rise of fire activity on the Alaskan North Slope148,149,150,151. However, these records lack unambiguous artifacts, features, or other clear indications of middle Upper Palaeolithic lifeways as seen in eastern Siberia152,153,154,155,156. At this time, there is no clear evidence for an ecologically significant human presence in eastern Beringia prior to ca. 14,000 cal BP (Supplementary Figs. 2–3). Thereafter, low fecundity megafauna72,157, who had already undergone millennia of oscillating climatological and ecological pressures, may have been vulnerable to novel anthropogenic forces25,158,159,160,161,162 that lack archaeological visibility due to the emergence of post-LGM, high mobility lifeways156,163. Currently, evidence of anthropogenic contributions to the ecological turnover in eastern Beringia remain functionally absent, being at most but one enigmatic component in a synergistic set of compounding pressures25. SedaDNA analyses of Pleistocene permafrost targeting human DNA may prove key to addressing lingering unknowns in the peopling of Beringia.
Evidence of a cryptic refugium
The persistence of Equus and Mammuthus until ~9200 cal BP and perhaps as late as ~5700 cal BP (Fig. 2), as suggested by our sedaDNA records, lies well beyond the last dated macrofossils for these taxa (Fig. 7). However, interpreting cryptic populations with sedaDNA necessitates caution. As noted previously, Arnold et al.91 found that although permafrost contains a wealth of well-preserved eDNA, the favourable characteristics of perennially frozen ground increases the likelihood for allochthonous organics to survive transport and be redeposited within younger strata. They argue that while reworking is of lesser concern when assessing first appearance dates and “abundant” sedaDNA signals, reworking of older sediments can be an inherent problem when assessing last appearance dates in high-energy fluvial contexts or in areas of thermokarst where older sediments thaw and mobilize followed by potential re-aggradation of permafrost. Arnold and colleagues highlight the careful analysis of loess sediments from the Stevens Village site in central Alaska where Haile et al.37 utilized 14C, OSL, extensive eDNA sampling on and off site, and careful sedimentological analyses to plausibly infer the late survival of Mammuthus and Equus to as late as ~10,000 cal BP. While the sediments targeted here are also loessal silts100, these materials were not recovered in the field with ancient DNA in mind, but were instead later reselected to follow-up on results presented by Murchie et al.36. Although we acknowledge that the signals for late megafaunal persistence should be interpreted with careful skepticism, and require additional supporting evidence for verification (particularly given early Holocene thaw unconformities97,98,99 in the Klondike as identified at Upper Goldbottom and Upper Quartz100), these signals are reasonable and worthy of further study for the following reasons.

Source data are provided as a Source Data file, see the following references for original data sources. A Dated fossils of select megafaunal taxa in eastern Beringia165, see Supplemental Fig. 1. B Earliest archaeological sites in northwestern North America188, see Supplemental Figs. 2–3 and Supplemental Table 2. C Central Yukon permafrost samples analyzed in this study with 2σ modelled age ranges, see Supplementary Figs. 5–7. D Greenland Ice Sheet Project 2 (GISP2) oxygen isotope profiles at 2 m intervals189. E Onset of the Younger Dryas, and alleged cosmic impact hypothesis190.
First, the ghost range signals observed for Mammuthus and Equus is observed at three different sites in 9 (Mammuthus) to 12 (Equus) separate permafrost cores and are correlated with substantial and consistent changes in vegetation. In samples younger than ~13,500 cal BP there is a complete restructuring of vegetation. To our knowledge, no plant taxa completely disappeared during the transition, which limits our ability to use the plant data to chronologically test for allochthonous sedaDNA. However, the megafaunal signal observed in the alleged ghost range cores are comparable to those observed during periods of known presence. It is reasonable to ask how many reads are sufficient to say an organism was truly present, but there cannot be any simple answer to that question. At the same time, >50 unique sedaDNA molecules identified as Elephantidae at ~9500 cal BP is significant relative to older cores considering the otherwise substantial ecological turnover.
Certainly in the case of cores with sediments younger than 13,000 cal BP, the presence of Mammuthus and Equus extending to ~9500 cal BP is highly consistent with other investigations in both northern Asia and northwestern North America37,41,42,164. Furthermore, reads for these megafaunal taxa are observed at two sites across all subsampled replicates and are associated with a completely different plant ecological signature. Here the point is that we would expect both floral and faunal sedaDNA would be reworked at roughly similar rates, which is not what is observed. Graminoids and forbs do persist, as would be expected, but there is no obviously mixed ecological signal as woody species dominate the plant metagenomics data (Fig. 4); this is despite otherwise having been observed in modern experiments to be proportionally under-represented genetically compared to their biomass by a factor of ~1:595.
The youngest signatures for Equus and Mammuthus (ca. 5700 cal BP) are of great interest because they imply local survival of these taxa long after the Pleistocene-Holocene transition. Aside from very late insular occurrences of mammoths from the Bering and Chukchi Seas41, there are no accepted radiocarbon dates on mammoth or horse fossils in mainland Beringia that fall anywhere close to the mid-Holocene (Figs. 2, 7, Supplementary Fig. 1). Although the authenticity of the identifications is not in question (Supplementary Fig. 57), the wide temporal gap between these sedaDNA molecules and dated bones is concerning. This difference needs to be evaluated in context. Palaeontological and archaeological records across much of the Arctic and Subarctic are notably sparse. Small refugial populations might have survived in remote pockets at sizes too small to be readily detected by macrofossil collections derived largely from a small set of resource extraction and development sites. The case of Mammuthus survival on St. Paul and Wrangel islands, until 5500 and 4000 cal years BP, respectively41,42, is interesting because until recent decades neither population was known to have persisted into the mid-Holocene. Bison priscus was likewise discovered to have survived throughout the Holocene in southern Yukon39,40. Conroy et al.165 observed the presence of coprophilous fungi in the Alaskan interior at Windmill and Jan Lakes until ~9000 cal BP and 4500 cal BP. This abundance of spores postdates the megafaunal turnover and is perhaps most likely related to a replacement with browsing cervids. Alternatively, it is also possible that cryptic, refugial populations of Pleistocene grazers were contributing sources. The way forward is through further testing and confirmation, which can be achieved through multiproxy sampling from a broader suite of high latitude sites where reworking is negligible, and conditions favour the widest possible metagenomic spectra.
Summary
This taxonomically rich sedaDNA dataset tracks the ecological turnover of fauna and flora in central Yukon across the Pleistocene-Holocene transition. We identify the coeval eDNA turnover of megafaunal grazers with forbs and graminoids relative to the rise of woody shrubs and boreal flora at ca. 13,500–13,000 cal BP and after 10,000 cal BP, along with the gradual decline of faunal sedaDNA after 30,000 cal BP. There is also a consistent, multi-site signal of late persistence for Equus and Mammuthus, perhaps surviving some 7000 years longer than their last dated macrofossils in eastern Beringia would indicate.
Top-down versus bottom-up perspectives on the collapse of the mammoth-steppe cannot be entirely resolved with this dataset, perhaps because both are relevant. The ancient eDNA data presented here have indications of both long term, top-down ecological pressures impacting megafaunal populations in the Klondike, arguably supporting the keystone megaherbivore model of the mammoth-steppe23,31 (albeit with unknowns regarding sedaDNA abundance and palaeo-biomass, along with a lag in vegetation turnover). There are also clear, short term, bottom-up indicators signifying climate-induced ecological transformations triggering the megafaunal extirpations between 13,500–10,000 cal BP16,28,29,30. The degree to which a decline of megafaunal DNA prior to the Bølling–Allerød interstadial can be interpreted as indicative of declines in the local populations of grazing megafauna is open for debate. But, as Equus sedaDNA remains relatively consistent until the rise of mesic-hydric woody flora during the Allerød warming, this is suggestive of longer-term local declines of woolly mammoths and steppe-bison pre-dating the ecological turnover. The abruptness of the transition thereafter is arguably indicative of both a potential lack of megafaunal engineering controlling woody expansion4,23,31,121,166 and the compounding impacts of significant climate change25. The local persistence of mammoth-steppe taxa at our single Younger Dryas point sample suggests that the mammoth-steppe locally persisted through the Bølling–Allerød warming, but that subsequent early Holocene transformations significantly shifted the ecological character of the Klondike by 10,000 cal BP.
The late Pleistocene palaeontological record of Beringian megafauna is extensive compared to many other areas of Eurasia and the Americas27. However, even with this richness, it is too sparse in well-dated and geographically diverse macrofossils to effectively tease apart the complexity of factors involved in the Pleistocene-Holocene transition and the collapse of the mammoth-steppe. The data presented in this study highlights the power of environmental DNA for the recovery of highly complex signals of ecological change from exceptionally small sediment inputs (0.3–1.35 grams), even in the absence of macro biological tissues. Using targeted enrichment for sedaDNA is also far more cost effective for high-throughput sequencing applications compared to a shotgun approach. Murchie et al.36 shotgun sequenced replicates of the same core samples studied here, and in the best sample, a shotgun approach had only 0.007% on-target sequence data (average 0.002%) of ~21 million DNA reads. This contrasts with as much as 10.8% on-target (average 4.2%) here with PalaeoChip enrichments—a 1,538x increase in on-target aDNA. If such a capture enrichment approach were paired with a robust sampling effort focused on sites with clear evidence of stratigraphic integrity across multiple regions and with tight temporal control, these genetic records could prove to be key to unravelling the ambiguities of this extinction event that has confounded Quaternary science since the eighteenth century.
Methods
Ethical approval was not required for this research as no archaeological or palaeontological materials were processed or analyzed and no human DNA was targeted or analyzed. Cores were acquired with Yukon Science and Explorer License research permits issued to DF by the Yukon Government, Department of Tourism & Culture. A description of the sampling sites, palynology, wet lab master mix recipes, qPCR results, additional bioinformatic analyses and results, and a palaeontological and archaeological background summary are included in the Supplementary Information.
Summary
Twenty-one core samples of loessal permafrost silts recovered from the Klondike region of Yukon, Canada (Fig. 1, Table 1)—dating between 30,000–4,000 calibrated years before present (cal BP)—were processed for sedimentary ancient DNA (sedaDNA) to evaluate changing biomolecular signals of plants and animals during the late Pleistocene-Holocene transition in eastern Beringia. We used Bayseian age modelling in conjunction with stratigraphic and cryostratigraphic observations to estimate core dates, and a subset of the core samples were processed in parallel for palynology. Despite the exceptional preservation and richness of the sedaDNA, very few pollen grains could be found in the samples (Supplementary Table 4).
For ancient DNA processing, we utilized a sedaDNA modified Dabney et al.167 extraction procedure with the long cold spin inhibitor removal technique as described in Murchie et al.36, and prepared double stranded, dual-indexed libraries168,169 for targeted enrichment. We used the PalaeoChip Arctic-1.0 plant and animal baits36 to capture enrich these libraries for chloroplast barcoding loci (trnL, matK, rbcL) of Arctic/Subarctic plants and for whole mitochondrial genomes (or singular loci where mitogenomes were unavailable at the time of bait-design in 2017) of extinct and extant northern animals (focused on megafauna). Libraries were sequenced on an Illumina HiSeq 1500 with 2 × 90 paired-end read chemistry.
After trimming, merging, and filtering the sequenced reads, BLASTn170 was used to taxonomically identify the reads to the top 600 hits against a July 2019 local copy of the GenBank database171,172, which was used as the input for MEGAN Community Edition173,174 (v6.19.7, https://github.com/husonlab/megan-ce) and PIA (Phylogenetic Intersection Analysis, v 5.3, https://github.com/Allaby-lab/PIA)94. The outputs from MEGAN are plotted in the main text, while plots of individual extraction replicates from both MEGAN and PIA are included in the supplement (Supplementary Figs. 34–55). MapDamage175 (v2.0.3, https://ginolhac.github.io/mapDamage/) was used to assess the aDNA damage signals of taxonomically identified taxa (Supplementary Figs. 13–28).
Field sampling
The cores used in this analysis were previously studied by D’Costa et al.106, Mahony100, and Sadoway109 and have since been kept in cold storage at the McMaster Ancient DNA Centre and the Permafrost Archives Laboratory at the University of Alberta. Permafrost cores were collected between June and August of 2010, 2012, and 2013 with research permits issued to DF from the Yukon Heritage Branch. These cores were sampled at placer gold mining exposures chosen for the quality of the exposure and expected age of the sediments. Prior to sample collection by all three original research teams, the sampling area was cleared of eroded materials back to frozen sediments to create a fresh coring surface for a ~10 cm diameter coring tube ~30 cm in length. Horizontal core samples were drilled with a small portable gas-powered drill (Echo), recovered frozen, stored individually in plastic bags, immediately placed in a −20 °C chest freezer, and transported in the freezer to the University of Alberta or McMaster University for subsampling. Core locations were recorded with a GPS in the field and these locations along with stratigraphic information were recorded in a field notebook at the time of sampling. Horizontal permafrost cores were collected from Bear Creek, Upper Quartz, and Upper Goldbottom. Vertical cores were taken from Lucky Lady II (Fig. 1, Supplementary Methods 2.1).
Radiocarbon dating and Bayesian age-depth modelling
Plant macrofossils were picked from thawed samples using a dissecting microscope, dried and pre-treated for AMS dating at the University of Alberta along with known-age wood standards (c.f. Mahony100). Pre-treatment of all samples followed standard acid-base-acid procedures. Solutions heated to 70 °C and placed in 1 M HCl for 30 min, followed by 60-min washes in 1 M NaOH until the solution became clear. Finally, samples were placed in 1 M HCl for 30 min and rinsed with ultrapure water until they became neutral. Measurements of CO2 production, graphitization and radiocarbon abundance in all samples were completed at the Keck-Carbon Cycle AMS facility (UCIAMS).
Age-depth models for Lucky Lady II, Upper Goldbottom and Upper Quartz were first presented by Sadoway109 and Mahony100. To refine the chronologies of these records we developed new Bayesian age-depth models for each study site using Oxcal v.4.4.2110 and the IntCal20 calibration curve111. In each case a P_Sequence depositional model was developed along sampling transects or vertical cores using a variable K parameter (increments per unit length)176 and a general Outlier_Model, with a 5% prior probability of any 14C date being a statistical outlier177. Boundaries were placed at the contacts between sediment units where changes in accumulation rates are likely to have taken place.
The Upper Quartz age-depth model (Supplementary Fig. 5) is developed from two P_Sequence depositional models run either side sedimentological boundary at 3.8 m which represents an unconformity of several thousand years. The lower P_Sequence includes two 14C dates (UCIAMS-114733 and UCIAMS-114710) and provides chronology for SedaDNA samples; PHP-22, PHP-23, PHP-24, and PHP-25. The upper P_Sequence includes three 14C dates (UCIAMS-114899, UCIAMS-114733, and UCIAMS-114710) and provides chronology for SedaDNA samples; PHP-19, PHP-20, and PHP-21.
Lucky Lady II includes three vertical cores which were sampled for SedaDNA (LLII-12, LL2C, and LL2S) (Supplementary Fig. 6). A prominent palaeo-soil that can be traced laterally for hundreds of metres around the exposure is present in the LLII-12 and LL2C cores, and 14C dates associated with this horizon were used to combine these cores with a single P_Sequence model (UCIAMS-56390, UCIAMS-114725, UCIAMS-143307, and UCIAMS-143308). Core LLII-12 includes three 14C dates (UCIAMS-240139, UCIAMS-122284, and UCIAMS-122273), as well as a palaeo-soil isochron, and provides chronology for SedaDNA samples PHP-9, PHP-8, and PHP-6. Core LL2C includes three 14C dates (UCIAMS-142198, UCIAMS-142197, and UCIAMS-143306) as well the palaeo-soil isochron, and provides chronology for SedaDNA samples PHP-4, PHP-5, and PHP-7. A boundary was placed at 3.5 m (at the contact between units 1 and 2) where grey silts including graminoid vegetation are replaced by organic-rich grey and black silts with in situ shrub vegetation. Core LL2-S includes three 14C dates (UCIAMS-143296, UCIAMS-142212, and UCIAMS-142211) and provides chronology for SedaDNA samples PHP-3 and PHP-2.
The Upper Goldbottom age-depth model (Supplementary Fig. 7) is developed from two P_Sequence depositional models run either side of a sedimentological boundary at 22.5 m which represents an unconformity of several thousand years. The lower P_Sequence provides chronology for sedaDNA samples PHP-14, PHP-13, and PHP-15, and includes six 14C dates (UCIAMS-122282, UCIAMS-114712, UCIAMS-114714, UCIAMS-142208, UCIAMS-114716, and UCIAMS-122274) as well as the Dawson tephra which has been dated to 29,055–29,470 cal BP178. The depths of three 14C dates obtained from Arctic ground squirrel middens (UCIAMS-122282, UCIAMS-114712 and UCIAMS-114714) were adjusted by 0.8 m to account for burrowing depth. This is likely to be a conservative estimate of active-layer depths during cold stages in the Yukon which were deeper than present. One 14C date caused a significant age-reversal and so was excluded from the age-depth model (UCIAMS-240141). Boundaries were placed at 12.6 m and 18 m (at the contacts between units 1, 2, and 3). The upper P_Sequence provides chronology for sedaDNA samples PHP-11 and PHP-12 and includes two 14C dates (UCIAMS-114898 and UCIAMS-240143). A further two 14C dates were excluded from the model as they caused significant age-reversals (UCIAMS-114910 and UCIAMS-114906).
The single Bear Creek sample (PHP-1) used in this analysis was not re-dated. This core was recovered from immediately beneath the Dawson tephra179,180,181,182, which is dated to 29,055–29,470 cal BP178. As such, this sample can be estimated as being ≳ 30,000 cal BP106. See Supplementary Methods 2.1 for a site description.
Ancient DNA wet lab
Laboratory work was conducted in clean rooms at the McMaster Ancient DNA Centre, which are subdivided into dedicated facilities for sample preparation (separate facilities for subsampling eDNA and discrete materials), stock solution setup, and DNA extraction through library preparation (collectively “pre-amplification rooms”). The post-indexing amplification clean room (enrichment) is in a physically isolated facility from the Centre’s standard aDNA labs, while the subsequent high-copy PCR workspace is in a separate building; the centre has a unidirectional workflow progressing from low-copy to high-copy facilities to reduce the chance of cross-contamination. Each dedicated workspace is physically separated with air pressure gradients between rooms to reduce exogenous airborne contamination. Prior to all phases of laboratory work, dead air hoods and workspaces were cleaned using a 6% solution of sodium hypochlorite (commercial bleach) followed by a wash with Nanopure purified water (Barnstead) and 30 min of UV irradiation at >100 mJ/cm2.
Subsampling
Permafrost sedaDNA subsamples were only taken from core interiors and care was taken to ensure that none of the sampling tools or interior surfaces were exposed any materials that had come in physical contact with the core exteriors (Supplementary Fig. 4). Permafrost samples from Lucky Lady II had been previously subsampled by Sadoway109 at the University of Alberta. These subsamples were homogenized by core and transported to McMaster University by Sadoway where they have remained in cold storage. All other subsamples from Upper Quartz and Upper Goldbottom were processed as follows.
Metal sampling tools were cleaned with commercial bleach, rinsed with Nanopure water immediately thereafter, UV irritated on both sides for >30 min, then heated overnight in an oven at ~130 °C. Once the tools had cooled the next day, work surfaces were cleaned with bleach and Nanopure water and covered with sterile lab-grade tin foil. Sediment cores previously split into disks106,109 and stored at −20 °C had the upper ~1 mm of external sediment chiselled off to create a fresh sampling area free of exogenous contaminants. For those cores that had not yet been split, a bleach and UV decontaminated handsaw was used to create a groove ~1–2 cm deep around the circumference of the core. A ~1 inch chisel was placed into the groove and a hammer was used to gradually split the core through percussion around the circumference. Once split, this opened a fresh interior surface previously unexposed to sampling equipment. For both the previously split and newly split cores, a small (~1/4 inch) decontaminated chisel was then used to carefully remove interior sediment from the core, which was collected in a weigh boat. After enough material was acquired for multiple extractions (~2–5 g), the core was covered in sterile tin foil and re-frozen. The subsampled material in the weigh boat was homogenized by manually stirring using a small metal chisel as the sediment thawed. This sediment was transferred to a 50 mL falcon tube and refrozen. Thereafter, the work area was thoroughly cleaned with bleach and Nanopure water, all plastic-ware was discarded, and metal tools were placed across the room for decontamination. The now decontaminated workspace was prepared again with sterile tin foil and another core sample. Gloves were changed frequently throughout subsampling (multiple times per core) to minimize cross and exogenous contamination. New metal tools that had been bleach, UV, and heat decontaminated from the previous day were used for each new core and all sterile tools remained isolated in the oven during subsampling. The homogenized sediments for each core were later subsampled for subsequent DNA extractions.
Lysis and purification
We followed the lysing and sedaDNA extraction procedure described in Murchie et al.36. The first round of sediments (PHP) were lysed with an input of 0.3 g. Subsequent experiments determined that this resulted in a higher inhibitor load for certain samples leading to ~10–20% failed or suboptimal adapter ligation efficiencies during library preparations (Supplementary Figs. 8–9). For the second round of extractions (PHPii), we reduced the input to 0.15 g, but used two PowerBead lysing tubes per sample that were pooled on the same Roche column following the long cold spin.
Subsamples were lysed with a digest solution (Supplementary Table 5) preloaded into Dneasy PowerBead tubes, then vortexed for 20 min using a TissueLyser II. Thereafter, the tubes were briefly centrifuged to remove liquid from the lids, and proteinase K was pipetted into each tube individually. The tubes were then briefly finger vortexed to disturb the sediment-bead pellets that had formed at the base and the tubes and were loaded in an incubator to oscillate overnight at 35 °C. The next day, the PowerBead tubes were centrifuged at 10,000 × g for 5 min and the supernatant was transferred to a 2 mL MAXYMum Recovery tube and stored at −20 °C for later purifications.
For sedaDNA purification, the digestion supernatant (≈1.25 mL) was thawed, briefly centrifuged, and added to ≳16.25 mL (13 volumes) of high-volume guanidinium binding buffer (Supplementary Table 6) in a 50 mL falcon tube and mixed by repeated inversion. The 50 mL tubes were loaded into a refrigerated centrifuge for the Murchie et al.36 long cold spin, where they were centrifuged at 2500 × g at 4 °C for ~20 h overnight. Thereafter, the falcon tubes were carefully removed from the centrifuge buckets, and the supernatant was decanted, taking care to not disturb the darkly coloured pellet that had formed during the cold spin. The binding buffer was passed through a high-volume silica-column (High Pure Extender Assembly, Roche Diagnostics) over multiple rounds of centrifugation and extraction proceeded as per Dabney et al.167 with binding and wash centrifugation at 3300 × g, two rounds of PE wash, followed by two 30 s dry spins at 16,000 × g with the tubes rotated 180° between spins to minimize the chance of ethanol retention. Purified DNA was eluted off the silica columns with two volumes of 25 µL EBT (each while waiting 5 min after EBT loading to maximize elution, then centrifuging at 16,000 × g for 1 min). Prior to all subsequent experiments the extracts were centrifuged at 16,000 × g for ≳5 min to pellet any remaining co-eluted inhibitors. Care was taken when subsampling these extracts to avoid disturbing any pellet precipitates.
Library preparation, quantitative PCR, and indexing
Doubled stranded libraries were prepared for each extract as described in Meyer and Kircher168 with modifications from Kircher et al.169 and a modified end-repair reaction to account for the lack of uracil excision (Supplementary Table 7). Samples were purified after blunt-end repair with a QIAquick Nucleotide Removal Kit (QIAGEN) (to maximally retain small fragments) and after adapter ligation (Supplementary Tables 8–9) and indexing (Supplementary Table 10) with a MinElute PCR Purification Kit (QIAGEN). Pre-indexing total library adapted DNA concentrations were estimated as a filtering step to determine whether a sample was successfully convereted into libraries with the short amplification qPCR assay (Supplementary Table 11).
Targeted capture with PalaeoChip
In-solution enrichments were carried out using the previously designed PalaeoChip Arctic v1.0 bait set36. This bait set targets whole mitochondrial genomes from approximately 180 extinct and extant Holarctic fauna, and the chloroplast barcoding loci (trnL, rbcL, and matK) from approximately 2100 species of plants. See Murchie et al.36 for further details on the design of PalaeoChip Arctic-1.0.
Enrichments were performed using a modified version of the myBaits v4.1 protocol (Daicel Arbor Biosciences). In summary, hybridization and bait mixes were prepared to the concentrations in Supplementary Table 12. For each library, 7 µL of template was combined with 5 µL of the library block master mix (using xGens, Human Cot-1 DNA, and Salmon Sperm). Hybridization and bait mixes were combined and pre-warmed to 60 °C before being combined with the library-block mixture. The final reaction for batch 1 (PHP) was incubated for 48 h at 55 °C for bait-library hybridization. The second round of libraries (PHPii) were enriched with a hybridization temperature of 60 °C over ~72 h to improve off-target exclusion.
After the hybridization, beads were dispensed (20 µL per reaction [rxn]), washed with 200 µL/rxn of binding buffer, then resuspended in 20 µL/rxn binding buffer and aliquoted into PCR strips. Baits were captured using 20 µL/rxn of the bead binding buffer, incubated at 55 °C for 2.5 min (60 °C for the second round), finger vortexed and spun down, then incubated for another 2.5 min. Beads were pelleted and the supernatant (the non-captured library fraction) was removed and stored at −20 °C as per Klunk et al.183. The beads were resuspended in 180 µL of 60 °C Wash Buffer X per tube and washed four times following the Mybaits v4.1 protocol. Beads were resuspended in 18.8 µL EBT, PCR reamplified for 12 cycles (Supplementary Table 10), then purified with MinElute columns following manufacturer’s protocols and eluted in 15 µL EBT.
Total quantification, pooling, size selection, and sequencing
Libraries were quantified using the long-amplification total library qPCR assay (Supplementary Table 13) and pooled to equimolar concentrations. Pools were size-selected with gel excision following electrophoresis for molecules ranging between 150–500 bp. Gel plugs were purified using the QIAquick Gel Extraction Kit (QIAGEN), according to manufacturer’s protocol, then sequenced on an Illumina HiSeq 1500 with a 2 × 90 bp paired-end protocol at the Farncombe Metagenomics Facility (McMaster University, ON).
Bioinformatics
Reads were demultiplexed with bcl2fastq (v1.8.4), converted to bam files with fastq2bam (https://github.com/grenaud/BCL2BAM2FASTQ), then trimmed and merged with leeHom184 using ancient DNA specific parameters (–ancientdna). Reads were mapped to a concatenation of the PalaeoChip Arctic-1.0 plant and animal probe references with network-aware-BWA185 (https://github.com/mpieva/network-aware-bwa) with a maximum edit distance of 0.01 (-n 0.01), allowing for a maximum two gap openings (-o 2), and with seeding effectively disabled (-l 16500). Mapped reads that were merged or unmerged but properly paired were extracted with libbam (https://github.com/grenaud/libbam), collapsed based on unique 5′ and 3′ positions with biohazard (https://bitbucket.org/ustenzel/biohazard) (for PCR deduplication), and converted to fasta files and restricted to a minimum length of 24 bp. Fasta files were additionally filtered to remove any reads with lingering sequence similarity to the Illumina adapter sequences (…/fasta2oneline.pl input.fasta | agrep -v -1 AGATCGGAA | agrep -v -1 TTCCGATCT | tr “\t” “\n” | tail -n + 2 > output.fasta) and were string deduplicated using the NGSXRemoveDuplicates module of NGSeXplore (https://github.com/ktmeaton/NGSeXplore).
These filtered fastas were used as the input for BLASTn170, which were aligned against a July 2019 local copy of the GenBank NCBI (National Center for Biotechnology Information;171,172) nucleotide database set to return the top 600 alignments (unique accession hits) per read with e-values less than 1.0E−5 (flags: -num_alignments 600 -max_hsps 1 -evalue 0.00001). The BLASTn outputs were then passed to MEGAN (Community Edition, v.6.19.7)173,174 where the BLASTn results were filtered through a lowest common ancestor (LCA) algorithm using the following parameters:
-
Min-score = 50 (default)
-
Max expected (e-value) = 1.0E−5
-
Minimum percent identity = 95%
-
Allows 1 base mismatch at 24 bp, 2 at 50 bp, and 3 at 60 bp to account for cytosine deamination and other aDNA characteristic damage or sequencing errors.
-
-
Top percent consideration of hits based on bit-score = 20%.
-
More conservative taxonomic assignments than the 10% default by taking more of the top hits into consideration for LCA assignment.
-
-
Minimum read support = 3
-
Number of unique reads aligning to an NCBI accession sequence for that taxon to be considered for LCA binning.
-
-
Minimum complexity = 0.3
-
Default minimum complexity filter.
-
-
LCA weighted algorithm at 80%.
-
Two rounds of analysis that increases LCA specificity by taking all taxonomic assignments of a library into consideration.
-
A second set of BLASTn files, identical to those utilized in MEGAN except for being in a different output format (-outfmt “6 std staxids”) were passed to PIA (Phylogenetic Intersection Analysis)94 using default inputs. The PIA output was converted to a MEGAN readable format using scripts available on the PIA-accessories GitHub (https://github.com/Allaby-lab/PIA-accessories). All libraries from both the MEGAN-LCA and PIA taxonomic binning approaches were then compiled using MEGAN’s compare feature with absolute read counts. Plant and animal sequences were inspected separately; taxonomic nodes deemed to be ecologically informative (through data exploration prioritizing taxa with high read counts, distinct changes in read abundance through time, or are otherwise known to be ecologically significant organisms) were selected and plotted using MEGAN’s built-in bubble chart feature. In these inter-sample comparative charts, absolute assigned read counts are retained, but the bubble sizes are scaled (either square-root or logarithmic) to visually normalize between samples of differing sequence depths. As these logarithmically scaled bubbles make it difficult to see change through time (whereas linearly scaled bubbles obscure all but the most abundant taxon nodes), a normalized set of MEGAN libraries was used to create stacked proportional charts to more clearly observe changing sedaDNA taxon assignments through time by taking sequencing depth into account. A PCoA was also created through MEGAN with these normalized libraries. All MEGAN charts were exported as EPS files and were visually fine-tuned in Adobe Illustrator186.
Thereafter, the closest available genetic references were obtained for notable taxa identified with the MEGAN-LCA and PIA taxonomic binning approaches. All samples were mapped to these references using the same aforementioned procedures but with an additional map-quality filter set to ≥30 with samtools (https://github.com/samtools/samtools), then assessed for ancient DNA typical damage signals using mapDamage175 (v 2.0.3).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The sedaDNA sequence data generated in this study that can be mapped to the PalaeoChip bait reference sequences have been deposited as bam files in the NCBI SRA database under BioProject: PRJNA722670, Accessions: SRR14265632– SRR14265692. Metagenomic data derived from these mapped reads are provided in the supplementary information and source data files. PalaeoChip reference and bait sequences are available at https://doi.org/10.5281/zenodo.5643845. Source data are provided with this paper.
Code availability
All software utilized in this analysis are available online and are referenced and linked to here or in the supplementary information.
References
Dirzo, R. et al. Defaunation in the Anthropocene. Science 345, 401–406 (2014).
Pimm, S. L. et al. The biodiversity of species and their rates of extinction, distribution, and protection. Science 344, 1246752 (2014).
Boivin, N. L. et al. Ecological consequences of human niche construction: examining long-term anthropogenic shaping of global species distributions. Proc. Natl Acad. Sci. USA 113, 6388–6396 (2016).
Asner, G. P., Vaughn, N., Smit, I. P. J. & Levick, S. Ecosystem-scale effects of megafauna in African savannas. Ecography (Cop.). 39, 240–252 (2016).
Bakker, E. S. et al. Combining paleo-data and modern exclosure experiments to assess the impact of megafauna extinctions on woody vegetation. Proc. Natl Acad. Sci. USA 113, 847–855 (2016).
Bakker, E. S., Pagès, J. F., Arthur, R. & Alcoverro, T. Assessing the role of large herbivores in the structuring and functioning of freshwater and marine angiosperm ecosystems. Ecography (Cop.). 39, 162–179 (2016).
Brault, M. O., Mysak, L. A., Matthews, H. D. & Simmons, C. T. Assessing the impact of late Pleistocene megafaunal extinctions on global vegetation and climate. Clim 9, 1761–1771 (2013).
Doughty, C. E., Faurby, S. & Svenning, J. C. The impact of the megafauna extinctions on savanna woody cover in South America. Ecography (Cop.). 39, 213–222 (2016).
Doughty, C. E., Wolf, A. & Malhi, Y. The legacy of the Pleistocene megafauna extinctions on nutrient availability in Amazonia. Nat. Geosci. 6, 761–764 (2013).
Doughty, C. E. et al. Global nutrient transport in a world of giants. Proc. Natl Acad. Sci. USA 113, 1–6 (2015).
Malhi, Y. et al. Megafauna and ecosystem function from the Pleistocene to the Anthropocene. Proc. Natl Acad. Sci. USA 113, 838–846 (2016).
Smith, F. A. et al. Exploring the influence of ancient and historic megaherbivore extirpations on the global methane budget. Proc. Natl Acad. Sci. USA 113, 201502547 (2015).
le Roux, E., Kerley, G. I. H. & Cromsigt, J. P. G. M. Megaherbivores modify trophic cascades triggered by fear of predation in an African Savanna Ecosystem. Curr. Biol. 28, 2493–2499.e3 (2018).
Boulanger, M. T. & Lyman, R. L. Northeastern North American Pleistocene megafauna chronologically overlapped minimally with Paleoindians. Quat. Sci. Rev. 85, 35–46 (2013).
Rozas-Dávila, A., Valencia, B. G. & Bush, M. B. The functional extinction of Andean megafauna. Ecology 97, 2533–2539 (2016).
Guthrie, R. D. New Carbon Dates Link Climatic Change with Human Colonization and Pleistocene Extinctions. Nature 441, 207–209 (2006).
Meltzer, D. J. Overkill, glacial history, and the extinction of North America’s Ice Age megafauna. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2015032117 (2020).
Sandom, C., Faurby, S., Sandel, B. & Svenning, J.-C. Global late Quaternary megafauna extinctions linked to humans, not climate change. Proc. R. Soc. Lond. B Biol. Sci. 281, 20133254 (2014).
Martin, P. S. in Quaternary Extinctions: A Prehistoric Revolution (eds. Martin, P. S. & Klein, R. G.) 354–403 (University of Arizona Press, 1984).
Braje, T. J. & Erlandson, J. M. Human acceleration of animal and plant extinctions: a late Pleistocene, Holocene, and Anthropocene continuum. Anthropocene 4, 14–23 (2013).
Smith, F. A., Smith, R. E. E. E., Lyons, S. K. & Payne, J. L. Body size downgrading of mammals over the late Quaternary. Science. 360, 310–313 (2018).
Barnosky, A. D., Koch, P. L., Feranec, R. S., Wing, S. L. & Shabel, A. B. Assessing the causes of late pleistocene extinctions on the continents. Science 306, 70–75 (2004).
Zimov, S. A. et al. Steppe-Tundra Transition: A Herbivore-Driven Biome Shift at the End of the Pleistocene. Am. Nat. 146, 765–794 (1995).
Lorenzen, E. D. et al. Species-specific responses of Late Quaternary megafauna to climate and humans. Nature 479, 359–364 (2011).
Mann, D. H., Groves, P., Gaglioti, B. V. & Shapiro, B. A. Climate-driven ecological stability as a globally shared cause of Late Quaternary megafaunal extinctions: the Plaids and Stripes Hypothesis. Biol. Rev. 94, 328–352 (2019).
Zazula, G. D. et al. American mastodon extirpation in the Arctic and Subarctic predates human colonization and terminal Pleistocene climate change. Proc. Natl Acad. Sci. USA 111, 18460–18465 (2014).
Stuart, A. J. Late Quaternary megafaunal extinctions on the continents: a short review. Geol. J. 50, 414–433 (2015).
Mann, D. H., Groves, P., Kunz, M. L., Reanier, R. E. & Gaglioti, B. V. Ice-age megafauna in Arctic Alaska: extinction, invasion, survival. Quat. Sci. Rev. 70, 91–108 (2013).
Mann, D. H. et al. Life and extinction of megafauna in the ice-age Arctic. Proc. Natl Acad. Sci. USA 112, 14301–14306 (2015).
Rabanus-Wallace, M. T. et al. Megafaunal isotopes reveal role of increased moisture on rangeland during late Pleistocene extinctions. Nat. Ecol. Evol. 1, 1–5 (2017).
Zimov, S. A., Zimov, N. S., Tikhonov, A. N. & Chapin, I. S. Mammoth steppe: a high-productivity phenomenon. Quat. Sci. Rev. 57, 26–45 (2012).
Owen-Smith, N. Pleistocene extinctions: the pivotal role of megaherbivores. Paleobiology 13, 351–362 (1987).
Willerslev, E. et al. Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51 (2014).
Jackson, S. T. Representation of flora and vegetation in Quaternary fossil assemblages: known and unknown knowns and unknowns. Quat. Sci. Rev. 49, 1–15 (2012).
Froese, D. G. et al. The Klondike goldfields and Pleistocene environments of Beringia. GSA Today 19, 4–10 (2009).
Murchie, T. J. et al. Optimizing extraction and targeted capture of ancient environmental DNA for reconstructing past environments using the PalaeoChip Arctic-1.0 bait-set. Quat. Res. 99, 305–328 (2021).
Haile, J. et al. Ancient DNA reveals late survival of mammoth and horse in interior Alaska. Proc. Natl Acad. Sci. USA 106, 22352–22357 (2009).
Clark, P. U. The last glacial maximum. Science 325, 710–714 (2009).
Zazula, G. D. et al. A middle Holocene steppe bison and paleoenvironments from the versleuce meadows, Whitehorse, Yukon, Canada. Can. J. Earth Sci. 54, 1138–1152 (2017).
Heintzman, P. D. et al. Bison phylogeography constrains dispersal and viability of the Ice Free Corridor in western Canada. Proc. Natl Acad. Sci. USA 113, 8057–8063 (2016).
Graham, R. W. et al. Timing and causes of mid-Holocene mammoth extinction on St. Paul Island, Alaska. Proc. Natl Acad. Sci. USA 113, 9310–9314 (2016).
Vartanyan, S. L., Arslanov, K. A., Karhu, J. A., Possnert, G. & Sulerzhitsky, L. D. Collection of radiocarbon dates on the mammoths (Mammuthus primigenius) and other genera of Wrangel Island, northeast Siberia, Russia. Quat. Res. 70, 51–59 (2008).
Faith, J. T. & Surovell, T. A. Synchronous extinction of North America’s Pleistocene mammals. Proc. Natl Acad. Sci. USA 106, 20641–20645 (2009).
Signor, P. W. & Lipps, J. H. Sampling bias, gradual extinction patterns and catastrophes in the fossil record. GSA Spec. Pap. 190, 291–296 (1982).
Fiedel, S. in American Megafaunal Extinctions at the End of the Pleistocene (ed. Haynes, G.) 21–37 (Springer Netherlands, 2009).
Graf, K. E. Uncharted Territory: Late Pleistocene Hunter-Gatherer Dispersals in the Siberian Mammoth-Steppe (University of Nevada, 2008).
Kuzmina, S. A. et al. The late Pleistocene environment of the Eastern West Beringia based on the principal section at the Main River, Chukotka. Quat. Sci. Rev. 30, 2091–2106 (2011).
Hoffecker, J. F., Elias, S. A. & Rourke, D. H. O. Out of Beringia? Science 343, 979–980 (2014).
Zazula, G. D. et al. Ice-age steppe vegetation in East Beringia. Nature 423, 603 (2003).
Guthrie, R. D. Origin and causes of the mammoth steppe: a story of cloud cover, woolly mammal tooth pits, buckles, and inside-out Beringia. Quat. Sci. Rev. 20, 549–574 (2001).
Pavelková Řičánková, V., Robovský, J. & Riegert, J. Ecological structure of recent and last glacial mammalian faunas in northern Eurasia: the case of Altai-Sayan refugium. PLoS ONE 9, e85056 (2014).
Bocherens, H. Isotopic tracking of large carnivore palaeoecology in the mammoth steppe. Quat. Sci. Rev. 117, 42–71 (2015).
Ritchie, J. C. & Cwynar, L. C. in Paleoecology of Beringia (eds. Hopkins, D. M. et al.) 113–126 (Academic Press, 1982).
Zhu, D. et al. The large mean body size of mammalian herbivores explains the productivity paradox during the Last Glacial Maximum. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-018-0481-y (2018).
Hopkins, D. M., Matthews, J. V., and Schweger, C. E. eds. Paleoecology of Beringia. (Academic Press, 1982).
Stivrins, N. et al. Biotic turnover rates during the Pleistocene-Holocene transition. Quat. Sci. Rev. 151, 100–110 (2016).
Bakker, E. S., Ritchie, M. E., Olff, H., Milchunas, D. G. & Knops, J. M. H. Herbivore impact on grassland plant diversity depends on habitat productivity and herbivore size. Ecol. Lett. 9, 780–788 (2006).
Bradshaw, R. H. W., Hannon, G. E. & Lister, A. M. A long-term perspective on ungulate-vegetation interactions. Ecol. Manag. 181, 267–280 (2003).
Gill, J. L. Ecological impacts of the late Quaternary megaherbivore extinctions. N. Phytologist 201, 1163–1169 (2014).
Gill, J. L., Williams, J. W., Jackson, S. T., Donnelly, J. P. & Schellinger, G. C. Climatic and megaherbivory controls on late-glacial vegetation dynamics: a new, high-resolution, multi-proxy record from Silver Lake, Ohio. Quat. Sci. Rev. 34, 66–80 (2012).
Gill, J. L., Williams, J. W., Jackson, S. T., Lininger, K. B. & Robinson, G. S. Pleistocene megafaunal collapse, novel plant communities, and enhanced fire regimes in North America. Science 326, 1100–1103 (2009).
Johnson, C. N. Ecological consequences of Late Quaternary extinctions of megafauna. Proc. Biol. Sci. 276, 2509–2519 (2009).
Owen-Smith, N. Megaherbivores: The Influence of Very Large Body Size on Ecology (Cambridge University Press, 1992).
Wright, J. P. & Jones, C. G. The concept of organisms as ecosystem engineers ten years on: progress, limitations, and challenges. Bioscience 56, 203 (2006).
Gutierrez, J. L. & Jones, C. G. Physical ecosystem engineers as agents of biogeochemical heterogeneity. Bioscience 56, 227 (2006).
Berke, S. K. Functional groups of ecosystem engineers: a proposed classification with comments on current issues. Integr. Comp. Biol. 50, 147–157 (2010).
Ries, L., Fletcher, R. J. J., Battin, J. & Sisk, T. D. Ecological responses to habitat edges: Mechanisms, models, and variability explained. Annu. Rev. Ecol., Evolution, Syst. 35, 491–522 (2004).
Rasmussen, S. O. et al. A new Greenland ice core chronology for the last glacial termination. J. Geophys. Res. Atmos. 111, 1–16 (2006).
Swift, J. A. et al. Micro methods for Megafauna: novel approaches to late quaternary extinctions and their contributions to faunal conservation in the Anthropocene. Bioscience 69, 877–887 (2019).
Andersen, K. et al. Meta-barcoding of ‘dirt’ DNA from soil reflects vertebrate biodiversity. Mol. Ecol. 21, 1966–1979 (2012).
Comandini, O. & Rinaldi, A. C. Tracing megafaunal extinctions with dung fungal spores. Mycologist 18, 140–142 (2004).
Säterberg, T., Sellman, S. & Ebenman, B. High frequency of functional extinctions in ecological networks. Nature 499, 468–470 (2013).
Courchamp, F., Berec, L. & Gascoigne, J. Allee Effects in Ecology and Conservation. Allee Effects in Ecology and Conservation (Oxford University Press, 2008).
Allee, W. C. Animal aggregations. Q. Rev. Biol. 2, 367–398 (1927).
Allee, W. C. & Bowen, E. S. Studies in animal aggregations: mass protection against colloidal silver among goldfishes. J. Exp. Zool. 61, 185–207 (1932).
Taberlet, P., Bonin, A., Zinger, L. & Coissac, E. Environmental DNA: For Biodiversity Research and Monitoring. (Oxford University Press, 2018).
Edwards, M. E. et al. Metabarcoding of modern soil DNA gives a highly local vegetation signal in Svalbard tundra. Holocene 28, 2006–2016 (2018).
Slon, V. et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science 356, 605–608 (2017).
Anderson-Carpenter, L. L. et al. Ancient DNA from lake sediments: bridging the gap between paleoecology and genetics. BMC Evol. Biol. 11, 1–15 (2011).
Bellemain, E. et al. Fungal palaeodiversity revealed using high-throughput metabarcoding of ancient DNA from arctic permafrost. Environ. Microbiol. 15, 1176–1189 (2013).
Ahmed, E. et al. Archaeal community changes in Lateglacial lake sediments: evidence from ancient DNA. Quat. Sci. Rev. 181, 19–29 (2018).
Niemeyer, B., Epp, L. S., Stoof-Leichsenring, K. R., Pestryakova, L. A. & Herzschuh, U. A comparison of sedimentary DNA and pollen from lake sediments in recording vegetation composition at the Siberian treeline. Mol. Ecol. Resour. 17, e46–e62 (2017).
Rawlence, N. J. et al. Using palaeoenvironmental DNA to reconstruct past environments: progress and prospects. J. Quat. Sci. 29, 610–626 (2014).
Blum, S. A. E., Lorenz, M. G. & Wackernagel, W. Mechanism of retarded DNA degradation and prokaryotic origin of DNases in nonsterile soils. Syst. Appl. Microbiol. 20, 513–521 (1997).
Greaves, M. P. & Wilson, M. J. The degradation of nucleic acids and montmorillonite-nucleic-acid complexes by soil microorganisms. Soil Biol. Biochem. 2, 257–268 (1970).
Gardner, C. M. & Gunsch, C. K. Adsorption capacity of multiple DNA sources to clay minerals and environmental soil matrices less than previously estimated. Chemosphere 175, 45–51 (2017).
Lorenz, M. G. & Wackernagel, W. Adsorption of DNA to sand and variable degradation rates of adsorbed DNA. Appl. Environ. Microbiol. 53, 2948–2952 (1987).
Ogram, A., Sayler, G., Gustin, D. & Lewis, R. DNA adsorption to soils and sediments. Environ. Sci. Technol. 22, 982–984 (1988).
Lorenz, M. G. & Wackernagel, W. Adsorption of DNA to sand and variable degradation of adsorbed DNA. Appl. Environ. Microbiol. 53, 2948–2952 (1987).
Morrissey, E. M. et al. Dynamics of extracellular DNA decomposition and bacterial community composition in soil. Soil Biol. Biochem. 86, 42–49 (2015).
Arnold, L. J. et al. Paper II - Dirt, dates and DNA: OSL and radiocarbon chronologies of perennially frozen sediments in Siberia, and their implications for sedimentary ancient DNA studies. Boreas 40, 417–445 (2011).
Allentoft, M. E. et al. The half-life of DNA in bone: measuring decay kinetics in 158 dated fossils. Proc. R. Soc. B Biol. Sci. https://doi.org/10.1098/rspb.2012.1745 (2012).
Kistler, L., Ware, R., Smith, O., Collins, M. & Allaby, R. G. A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res. 45, 6310–6320 (2017).
Cribdon, B., Ware, R., Smith, O., Gaffney, V. & Allaby, R. G. PIA: more accurate taxonomic assignment of metagenomic data demonstrated on sedaDNA from the North Sea. Front. Ecol. Evol. 8, 1–12 (2020).
Yoccoz, N. G. et al. DNA from soil mirrors plant taxonomic and growth form diversity. Mol. Ecol. 21, 3647–3655 (2012).
Doi, H. et al. Environmental DNA analysis for estimating the abundance and biomass of stream fish. Freshw. Biol. 62, 30–39 (2017).
Burn, C. R., Michel, F. A. & Smith, M. W. Stratigraphic, isotopic, and mineralogical evidence for an early Holocene thaw unconformity at Mayo, Yukon Territory. Can. J. Earth Sci. 23, 794–803 (1986).
Kotler, E. & Burn, C. R. Cryostratigraphy of the Klondike ‘muck’ deposits, west-central Yukon Territory. Can. J. Earth Sci. 37, 849–861 (2000).
Fraser, T. A. & Burn, C. R. On the nature and origin of ‘muck’ deposits in the Klondike area, Yukon Territory. Can. J. Earth Sci. 34, 1333–1344 (1997).
Mahony, M. E. 50,000 years of paleoenvironmental change recorded in meteoric waters and coeval paleoecological and cryostratigraphic indicators from the Klondike goldfields, Yukon, Canada. (University of Alberta, 2015). https://doi.org/10.7939/R34T6FF58.
Lydolph, M. C. et al. Beringian paleoecology inferred from permafrost-preserved fungal DNA. Appl. Environ. Microbiol. 71, 1012–1017 (2005).
Willerslev, E. et al. Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 300, 791–795 (2003).
Haile, J. et al. Ancient DNA chronology within sediment deposits: are paleobiological reconstructions possible and is DNA leaching a factor? Mol. Biol. Evol. 24, 982–989 (2007).
Willerslev, E., Hansen, A. J. & Poinar, H. N. Isolation of nucleic acids and cultures from fossil ice and permafrost. Trends Ecol. Evol. 19, 141–147 (2004).
Hansen, A. J. et al. Crosslinks rather than strand breaks determine access to ancient DNA sequences from frozen sediments. Genetics 173, 1175–1179 (2006).
D’Costa, V. M. et al. Antibiotic resistance is ancient. Nature 477, 457–461 (2011).
Johnson, S. S. et al. Ancient bacteria show evidence of DNA repair. Proc. Natl Acad. Sci. USA 104, 14401–14405 (2007).
Hebsgaard, M. B. et al. ‘The Farm Beneath the Sand’- an archaeological case study on ancient ‘dirt’ DNA. Antiquity 83, 430–444 (2009).
Sadoway, T. R. A Metagenomic Analysis of Ancient Sedimentary DNA Across the Pleistocene-Holocene Transition (McMaster University, 2014).
Bronk Ramsey, C. Deposition models for chronological records. Quat. Sci. Rev. 27, 42–60 (2008).
Reimer, P. J. et al. The IntCal20 Northern Hemisphere Radiocarbon Age Calibration Curve (0-55 cal kBP). Radiocarbon 62, 725–757 (2020).
Nichols, R. V. et al. Minimizing polymerase biases in metabarcoding. Mol. Ecol. Resour. 18, 927–939 (2018).
Wei, N., Nakajima, F. & Tobino, T. A microcosm study of surface sediment environmental DNA: decay observation, abundance estimation, and fragment length comparison. Environ. Sci. Technol. 52, 12428–12435 (2018).
Matesanz, S. et al. Estimating belowground plant abundance with DNA metabarcoding. Mol. Ecol. Resour. 19, 1265–1277 (2019).
Takahara, T., Minamoto, T., Yamanaka, H., Doi, H. & Kawabata, Z. Estimation of fish biomass using environmental DNA. PLoS ONE 7, 3–10 (2012).
Doi, H. et al. Use of droplet digital PCR for estimation of fish abundance and biomass in environmental DNA surveys. PLoS ONE 10, 1–11 (2015).
Debruyne, R. et al. Out of America: ancient DNA evidence for a new world origin of late Quaternary Woolly Mammoths. Curr. Biol. 18, 1320–1326 (2008).
Metcalfe, J. Z., Longstaffe, F. J. & Zazula, G. D. Nursing, weaning, and tooth development in woolly mammoths from Old Crow, Yukon, Canada: Implications for Pleistocene extinctions. Palaeogeogr. Palaeoclimatol. Palaeoecol. 298, 257–270 (2010).
Shapiro, B. et al. Rise and fall of the Beringian steppe bison. Science 306, 1561–1565 (2004).
Sinclair, P. H., Nixon, W. A., Eckert C. D. & Hughes, N. L.Hughes, eds. Birds of the Yukon Territory. (UBC Press, 2003).
Keesing, F. & Young, T. P. Cascading consequences of the loss of large mammals in an African Savanna. Bioscience 64, 487–495 (2014).
Taberlet, P. et al. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 35, e14 (2007).
Chevalier, M. et al. Pollen-based climate reconstruction techniques for late Quaternary studies. Earth-Sci. Rev. 210, 103384 (2020).
Wang, X.-C. & Geurts, M.-A. Post-glacial vegetation history of the Ittlemit Lake basin, southwest Yukon Territory. Arctic 44, 23–30 (1991).
Wang, X.-C. & Geurts, M.-A. Late Quaternary pollen records and vegetation history of the southwest Yukon Territory: a review. Geogr. Phys. Quat. 45, 175–193 (1991).
Rainville, R. A. & Gajewski, K. Holocene environmental history of the Aishihik region, Yukon, Canada. Can. J. Earth Sci. 50, 397–405 (2013).
Lacourse, T. & Gajewski, K. Late Quaternary vegetation history of Sulphur Lake, southwest Yukon Territory, Canada. Arctic 53, 27–35 (2000).
Bunbury, J. & Gajewski, K. Postglacial climates inferred from a lake at treeline, southwest Yukon Territory, Canada. Quat. Sci. Rev. 28, 354–369 (2009).
Gajewski, K., Bunbury, J., Vetter, M., Kroeker, N. & Khan, A. H. Paleoenvironmental studies in Southwestern Yukon. Arctic 67, 58–70 (2014).
Schofield, E. J., Edwards, K. J. & McMullen, A. J. Modern Pollen-Vegetation Relationships in Subarctic Southern Greenland and the Interpretation of Fossil Pollen Data from the Norse landnám. J. Biogeogr. 34, 473–488 (2007).
Pennington, W. & Tutin, T. G. Modern pollen samples from west greenland and the interpretation of pollen data from the british late-glacial (late Devesian). N. Phytol. 84, 171–201 (1980).
Bradshaw, R. H. W. Modern pollen-representation factors for Woods in South-East England. J. Ecol. 69, 45 (1981).
Roy, I. et al. Over-representation of some taxa in surface pollen analysis misleads the interpretation of fossil pollen spectra in terms of extant vegetation. Trop. Ecol. 59, 339–350 (2018).
Bryant, J. P. et al. Biogeographic evidence for the evolution of chemical defense by boreal birch and willow against mammalian browsing. Am. Nat. 134, 20–34 (1979).
Christie, K. S. et al. The role of vertebrate herbivores in regulating shrub expansion in the Arctic: a synthesis. Bioscience 65, 1123 (2015).
Bryant, J. P. et al. Can antibrowsing defense regulate the spread of woody vegetation in arctic tundra? Ecography (Cop.). 37, 204–211 (2014).
Bryant, J. P. & Kuropat, P. J. Selection of winter forage by subarctic browsing vertebrates: the role of plant chemistry. Annu. Rev. Ecol. Syst. 11, 261–285 (1980).
Fox-Dobbs, K., Leonard, J. A. & Koch, P. L. Pleistocene megafauna from eastern Beringia: Paleoecological and paleoenvironmental interpretations of stable carbon and nitrogen isotope and radiocarbon records. Palaeogeogr. Palaeoclimatol. Palaeoecol. 261, 30–46 (2008).
Gardner, C., Berger, M. & Taras, M. Habitat assessment of potential wood bison relocation sites in Alaska. Arctic 1–30 (2007).
Jiménez-Hidalgo, E. et al. Species diversity and paleoecology of late pleistocene horses from Southern Mexico. Front. Ecol. Evol. 7, 1–18 (2019).
van Geel, B. et al. The ecological implications of a Yakutian mammoth’s last meal. Quat. Res. 69, 361–376 (2008).
van Geel, B. et al. Palaeo-environmental and dietary analysis of intestinal contents of a mammoth calf (Yamal Peninsula, northwest Siberia). Quat. Sci. Rev. 30, 3935–3946 (2011).
Guthrie, R. D. Rapid body size decline in Alaskan Pleistocene horses before extinction. Nature 426, 169–171 (2003).
Bourgeon, L. Bluefish Cave II (Yukon Territory, Canada): Taphonomic Study of a Bone Assemblage. PaleoAmerica 1, 105–108 (2015).
Bourgeon, L., Burke, A. & Higham, T. Earliest human presence in North America dated to the last glacial maximum: new radiocarbon dates from Bluefish Caves, Canada. PLoS ONE 12, e0169486 (2017).
Bourgeon, L. Revisiting the mammoth bone modifications from Bluefish Caves (YT, Canada). J. Archaeol. Sci. Rep. 37, 102969 (2021).
Bourgeon, L. & Burke, A. Horse exploitation by Beringian hunters during the Last Glacial Maximum. Quat. Sci. Rev. 261, (2021).
Vachula, R. S., Sae-Lim, J. & Russell, J. M. Sedimentary charcoal proxy records of fire in Alaskan tundra ecosystems. Palaeogeogr. Palaeoclimatol. Palaeoecol. 541, 109564 (2020).
Vachula, R. S. Alaskan lake sediment records and their implications for the Beringian standstill hypothesis. PaleoAmerica 6, 303–307 (2020).
Vachula, R. S. et al. Evidence of Ice Age humans in eastern Beringia suggests early migration to North America. Quat. Sci. Rev. 205, 35–44 (2019).
Vachula, R. S. et al. Sedimentary biomarkers reaffirm human impacts on northern Beringian ecosystems during the Last Glacial period. Boreas 49, 514–525 (2020).
Abramova, Z. A. in Paleolit Kavkaza i Severnoi Azii (ed. Boriskovskii, P. I.) 145–243 (Nauka, 1989).
Abramova, Z. A., Astakhov, S. N., Vasil’ev, S. A., Ermolva, N. M. & Lisitsyn, N. F. Paleolit Eniseya. (Nauka, 1991).
Goebel, T. in Encyclopedia of prehistory. Vol 2: Arctic and Subarctic (eds. Peregrine, P. N. & Ember, M.) 192–196 (Kluwer Academic Publishers, 2002).
Ermolova, N. M. Teriofauna doliny Angary v pozdem antropogene. (Nauka, 1978).
Hoffecker, J. F. & Elias, S. A. Human Ecology of Beringia. (Columbia University Press, 2007).
Johnson, C. N. Determinants of loss of mammal species during the Late Quaternary ‘megafauna’ extinctions: life history and ecology, but not body size. Proc. Biol. Sci. 269, 2221–2227 (2002).
Laland, K. N. & O’Brien, M. J. Niche Construction Theory and Archaeology. J. Archaeol. Method Theory 17, 303–322 (2010).
Riede, F. Adaptation and niche construction in human prehistory: a case study from the southern Scandinavian Late Glacial. Philos. Trans. R. Soc. Lond. 366, 793–808 (2011).
Roos, C. I., Zedeño, M. N., Hollenback, K. L. & Erlick, M. M. H. Indigenous impacts on North American Great Plains fire regimes of the past millennium. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.1805259115 (2018).
Pinter, N., Fiedel, S. & Keeley, J. E. Fire and vegetation shifts in the Americas at the vanguard of Paleoindian migration. Quat. Sci. Rev. 30, 269–272 (2011).
Haynes, G. Extinctions in North America’s Late Glacial landscapes. Quat. Int. 285, 89–98 (2013).
Graf, K. E. in Paleoamerican Odyssey (eds. Graf, K. E., Ketron, C. V. & Waters, M. R.) 65–80 (Texas A&M University Press, 2014).
Pečnerová, P. et al. Mitogenome evolution in the last surviving woolly mammoth population reveals neutral and functional consequences of small population size. Evol. Lett. 1, 292–303 (2017).
Conroy, K. J. et al. Tracking late-Quaternary extinctions in interior Alaska using megaherbivore bone remains and dung fungal spores. Quat. Res. https://doi.org/10.1017/qua.2020.19 (2020).
Bakker, E. S. et al. Combining paleo-data and modern exclosure experiments to assess the impact of megafauna extinctions on woody vegetation. Proc. Natl Acad. Sci. USA 113, 847–855 (2016).
Dabney, J. et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl Acad. Sci. USA 110, 15758–15763 (2013).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 5, pdb.prot5448 (2010).
Kircher, M., Sawyer, S. & Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, 1–8 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Agarwala, R. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 44, D7–D19 (2016).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 41, D36–D42 (2013).
Huson, D. H. et al. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 12, e1004957 (2016).
Huson, D. H., Auch, A. F., Qi, J. & Schuster, S. C. MEGAN analysis of metagenomic data. Genome Res. 17, 377–386 (2007).
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. & Orlando, L. MapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Bronk Ramsey, C. & Lee, S. Recent and planned developments of the program OxCal. Radiocarbon 55, 720–730 (2013).
Bronk Ramsey, C. Dealing with outliers and offsets in radiocarbon dating. Radiocarbon 51, 1023–1045 (2009).
Davies, L. J., Jensen, B. J. L., Froese, D. G. & Wallace, K. L. Late Pleistocene and Holocene tephrostratigraphy of interior Alaska and Yukon: key beds and chronologies over the past 30,000 years. Quat. Sci. Rev. 146, 28–53 (2016).
Westgate, J. A., Preece, S. J., Kotler, E. & Hall, S. Dawson tephra: a prominent stratigraphic marker of Late Wisconsinan age in west-central Yukon, Canada. Can. J. Earth Sci. 37, 621–627 (2000).
Froese, D., Westgate, J., Preece, S. & Storer, J. Age and significance of the Late Pleistocene Dawson tephra in eastern Beringia. Quat. Sci. Rev. 21, 2137–2142 (2002).
Zazula, G. D. et al. Vegetation buried under Dawson tephra (25,300 14C years BP) and locally diverse late Pleistocene paleoenvironments of Goldbottom Creek, Yukon, Canada. Palaeogeogr. Palaeoclimatol. Palaeoecol. 242, 253–286 (2006).
Froese, D. G., Zazula, G. D. & Reyes, A. V. Seasonality of the late Pleistocene Dawson tephra and exceptional preservation of a buried riparian surface in central Yukon Territory, Canada. Quat. Sci. Rev. 25, 1542–1551 (2006).
Klunk, J. et al. Genetic resiliency and the Black Death: no apparent loss of mitogenomic diversity due to the Black Death in medieval London and Denmark. Am. J. Phys. Anthropol. 169, 240–252 (2019).
Renaud, G., Stenzel, U. & Kelso, J. LeeHom: Adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res 42, e141 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Adobe Inc. Adobe Illustrator. (2020). https://adobe.com/products/illustrator.
Lebart, L., Morineau, A. & Tabard, N. Techniques De La Description Statistique Méthodes Et Logiciels Pour L’analyse Des Grands Tableaux. (Dunod, 1977).
Potter, B. A. et al. Current evidence allows multiple models for the peopling of the Americas. Sci. Adv. 4, 1–9 (2018).
Grootes, P. M. & Stuiver, M. Oxygen 18/16 variability in Greenland snow and ice with 10-3- to 105-year time resolution. J. Geophys. Res. Ocean. 102, 26455–26470 (1997).
Wolbach, W. S. et al. Extraordinary Biomass-Burning Episode and Impact Winter Triggered by the Younger Dryas Cosmic Impact ∼12,800 Years Ago. 2. Lake, Marine, and Terrestrial Sediments. J. Geol. 126, 185–205 (2018).
Acknowledgements
Many thanks to the CANA Foundation for their support of a PDF and operating costs to T.J.M. and H.N.P. This work was partially funded by the Belmont Forum and BiodivERsA grants (to D.F. and H.N.P.) for the Future ArcTic Ecosystems (FATE) research consortium, as well as NSERC Discovery grants to D.F. and H.N.P. We wish to also thank the Garfield Weston Foundation for their support of T.J.M., as well as McMaster University, the Arctic Institute of North America, Polar Knowledge Canada, and the Social Sciences and Humanities Research Council of Canada for each funding various components of this research. Thank you to the placer gold mining community of the Klondike and the Tr’ondëk Hwëch’in for their continued support of our research and access to study sites in central Yukon. Additional thanks to Ana T. Duggan for the development and continued maintenance of her ancient DNA pipeline at the McMaster Ancient DNA centre. Further thanks to Brian Golding for providing access to his computational resources. Thanks to Shanti Morell-Hart, Brian Golding, and Peter Heintzman for their guidance and support during T.J.M.’s doctoral research. Thanks to all members and affiliates of the McMaster Ancient DNA Centre for their ongoing support of this work, as well as the admin and faculty of the Anthropology department at McMaster University. Special thanks to Debi Poinar, Melanie Kuch, Jennifer Klunk, Marissa L. Ledger, and Kévin Roche for their wet-lab assistance during the Quaternary Research PalaeoChip paper that laid the groundwork for this study.
Author information
Authors and Affiliations
Contributions
T.J.M. Project conception and design, aDNA wet lab, primary computational analyses, writing, figure design, coordination. A.J.M. Dating, age-modelling, taxonomic and shrub expansion consultation, editing. M.E.M. Original field coring, radiometric dating, and stratigraphic analysis. G.S.L. Deamination/depurination analyses, editing. S.C. Subsampling for radiocarbon dating, palynology, editing. T.S. Original field coring, Lucky Lady II subsampling, and preliminary sedaDNA investigations. E.K. DNA wet lab, design consultation, editing, G.Z. Project design, taxonomic consultation, editing, R.D.E.M. Project design, taxonomic consultation, editing, D.F. Field work and core collection, stratigraphic analyses, dating, project design, consultation, editing, H.N.P. Project design and coordination, consultation, editing
Corresponding authors
Ethics declarations
Competing interests
T.J.M., H.N.P., and R.D.E.M. are members of the CANA Foundation science advisory board, a non-profit organization with horse re-wilding initiatives. T.J.M. and H.N.P. are currently supported by CANA through a PDF to T.J.M. and consumable costs. The research presented in this paper was completed long prior to funding by CANA. The authors declare no other competing interests.
Peer review information
Nature Communications thanks Eric Capo, Daniel Mann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Murchie, T.J., Monteath, A.J., Mahony, M.E. et al. Collapse of the mammoth-steppe in central Yukon as revealed by ancient environmental DNA. Nat Commun 12, 7120 (2021). https://doi.org/10.1038/s41467-021-27439-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-27439-6
This article is cited by
-
Environmental DNA identifies coastal plant community shift 1,000 years ago in Torrens Island, South Australia
Communications Earth & Environment (2024)
-
Paleoecology of extinct species
BMC Ecology and Evolution (2023)
-
Rewilding Risks for Peatland Permafrost
Ecosystems (2023)
-
Sedimentary DNA for tracking the long-term changes in biodiversity
Environmental Science and Pollution Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
