
Abstract
Securinega alkaloids have fascinated the synthetic chemical community for over six decades. Historically, major research foci in securinega alkaloid synthesis have been on the efficient construction of the fused tetracyclic framework that bears a butenolide moiety and tertiary amine-based heterocycles. These “basic” securinega alkaloids have evolved to undergo biosynthetic oxidative diversifications, especially on the piperidine core. However, a general synthetic solution to access these high-oxidation state securinega alkaloids is lacking. In this study, we have completed the total synthesis of various C4-oxygenated securinine-type alkaloids including securingines A, C, D, securitinine, secu’amamine D, phyllanthine, and 4-epi-phyllanthine. Our synthetic strategy features stereocontrolled oxidation, rearrangement, and epimerization at N1 and C2–C4 positions of the piperidine core within (neo)securinane scaffolds. Our discoveries provide a fundamental synthetic solution to all known securinine-type natural products with various oxidative and stereochemical variations around the central piperidine ring.
Similar content being viewed by others
Introduction
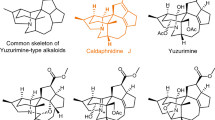
Securinega alkaloids have fascinated chemists for over 60 years since the first isolation of securinine1,2. Plants that contain these alkaloids have been applied in traditional medicines for the treatment of malaria and other diseases3,4,5. The basic securinine framework can undergo biosynthetic oxidations to yield various natural products with oxidative decorations around the piperidine moiety. In that regard, recent isolation campaigns from Securinega suffruticosa resulted in an outburst of discoveries of novel C4-oxygenated securinine-type alkaloids that show oxidative and (or) stereochemical variations around the piperidine ring A (Fig. 1)6,7,8,9,10,11. With respect to the structure determination of natural products, the continued development of computational chemistry has enabled the estimation of spectroscopic data of complex molecules12. Recently, we proposed an alternative structure of securingine A (7) based on its calculated ground-state structure and DP4 + probability analysis13. Kutateladze and coworkers suggested structure revisions of securingines C (8) and D (9) based on their machine learning (ML)-based hybrid density functional theory/parametric computations of nuclear magnetic resonance spectra14. However, these computationally proposed structures have yet to be experimentally confirmed via total synthesis.

Structures of securinine, allosecurinine, and representative C4-oxygenated securinine-type natural products are depicted. Both the originally proposed structure and the computationally predicted structure are presented for securingines A, C, and D.
The tetracyclic structure of securinega alkaloids that features butenolide and tertiary amine moieties has served as an excellent platform for the development of new synthetic strategies and tactics15,16,17. Over 25 reports have depicted the synthesis of securinine-type alkaloids with piperidine-based A-ring. Among those works, Busqué and de March’s total synthesis of allosecurinine (2) that featured a vinylogous Mannich reaction18 between hemiaminal 10 and silyl dienol ether derivative 11 and an intramolecular N-alkylation greatly inspired us at the onset of our synthetic campaign by providing us with guiding footprints on how to efficiently assemble the eastern menisdaurilide-based and the western piperidine-based fragments (Fig. 2a)19. Furthermore, Gademann’s elegant total synthesis of allosecurinine (2), which was enabled by an intramolecular aza-Michael addition to access secu’amamine E (13) and its subsequent mesylation-based 1,2-amine shift (Fig. 2b)20,21 has enabled us to explore new chemistries within the neosecurinane framework22. For the synthesis of tetrahydro-1,2-oxazine-based securinine-type natural products, Horii, Parello and coworkers reported the biomimetic oxidation of allosecurinine (2) to phyllantidine (14, Fig. 2c) in 197223. Importantly, the tetrahydro-1,2-oxazine core could also be established by the Kerr group via a Lewis acid-catalyzed three-component homo [3 + 2] dipolar cycloaddition24 and by the Wood group via an acyloxy nitroso ring expansion strategy25 en route to their total synthesis of phyllantidine (14), respectively. With respect to the C4-oxygenated high-oxidation state securinega alkaloid, the Weinreb group’s total synthesis of phyllanthine (5) via SmI2-mediated intramolecular ketonitrile coupling and stereoselective imino Diels–Alder reaction has remained as the only synthesis of securinega alkaloid with a C4-oxygenation26,27.

a Busqué and de March’s biosynthetically inspired synthesis of allosecurinine (ref. 19); b Gademann’s biosynthetically inspired synthesis of secu’amamine E and allosecurinine (ref. 22); c Horii and Parello’s biomimetic conversion of allosecurinine to phyllantidine (ref. 23); d Pre- and post-modification of the securinega framework (this work). Boc tbutoxycarbonyl, TIPS triisopropylsilyl, TBDPS tbutyldiphenylsilyl.
Building on these seminal prior synthetic studies, we envisioned establishing a general synthetic solution for various C4-oxygenated securinine-type alkaloids. Structural examination of C4-oxygenated securinine-type alkaloids revealed key challenges en route to our grand goal. First, the isolation of both phyllanthine (5) and 4-epi-phyllanthine (4) with R- and S-configuration at C4, respectively, hinted at the necessity to develop a synthetic strategy that can introduce the methoxy group at C4 with complete stereocontrol (Fig. 2d). Next, structural comparison of 4-epi-phyllanthine (4) and securitinine (3) unveiled the importance of stereochemical flexibility and control at C2 methine in our synthetic approach. Then, the isolations of both securingine A (7b) and secu’amamine D (6) highlighted the importance of control over the atom connectivity after the oxidation of the tertiary amine moiety. After that, the isolation of securingine C (8b) necessitated a method to stereoselectively epoxidize the C3–C4 site. Finally, structural comparison of securingines C (8b) and D (9b) asked for a method that can regioselectively oxygenate the C2 position. Herein, we introduce a unified synthetic route to high-oxidation state securinine-type alkaloids that addresses all these key challenges.
Results
Decagram-scale synthesis of O-silylated menisdaurilide
Our study commenced with the synthesis of the right-hand fragment of the securinine framework. Despite previous reports on enantioselective total synthesis of menisdaurilide28,29,30,31, a practical decagram-scale synthetic process to enantioenriched menisdaurilide or its derivative had not been described when we started our studies. In 2019, Peixoto and coworkers reported an elegant 5-step synthesis of (±)-O-tbutyldimethylsilylmenisdaurilide that delivered 2.5 g of material in a single pass. However, enantiomerically enriched menisdaurilide derivative could be obtained only after semi-preparative chiral HPLC separation31. Resolution of (±)-O-tbutyldimethylsilylmenisdaurilide via its derivatization with the enantiomerically enriched carboxylic acid was possible but required multiple flash chromatographic separations32. Hence, we first aimed to develop a decagram-scale synthesis of enantiomerically enriched menisdaurilide derivative (Fig. 3). Our synthesis commenced with enantioselective catalytic desymmetrization of 15 previously reported by Snapper, Hoveyda, and coworkers33,34. Enantioenriched (95% ee) alcohol 18 was subjected to hydroxyl-directed epoxidation reaction conditions to afford epoxide 19 in 98% yield. Alcohol 19 was then allowed to react with Dess–Martin periodinane and subsequently with silica gel to yield γ-hydroxyenone 20 in 94% yield. TBDPS protection of alcohol 20 (95% yield) followed by selective TBS deprotection in the presence of zinc bromide (83% yield) yielded α-hydroxyketone 21. Finally, after ester formation between 21 and diethylphosphonoacetic acid (96% yield), the resulting phosphonate 22 was treated with potassium carbonate and 18-crown-6 to afford the HWE reaction product, O-silylated menisdaurilide derivative 23 (61% yield, 95% ee). It is important to note that all synthetic steps from precursor 15 were conducted on a >10 g scale which enabled the decagram access to O-tbutyldiphenylsilylmenisdaurilide (23) in a single pass (Fig. 3).

Reagents and conditions: (a) 16 (0.2 equiv), 17 (0.1 equiv), TBSCl (2.0 equiv), iPr2EtN (1.2 equiv), THF, –40 °C, 99% (95% ee); (b) VO(OEt)3 (0.05 equiv), TBHP (4.0 equiv), CH2Cl2, 0 °C to 23 °C, 98%; (c) DMP (1.25 equiv), CH2Cl2, 23 °C; SiO2, 94%; (d) TBDPSCl (1.2 equiv), imidazole (1.2 equiv), DMAP (0.1 equiv), CH2Cl2, 0 °C to 23 °C, 95%; (e) ZnBr2 (5.0 equiv), H2O (5.0 equiv), CH2Cl2, 50 °C, 83%; (f) diethylphosphonoacetic acid (2.0 equiv), EDCI (2.0 equiv), CH2Cl2, 23 °C, 96%; (g) K2CO3 (5.0 equiv), 18-crown-6 ether (5.0 equiv), THF, 0 °C, 61%. TBS tbutyldimethylsilyl, TBHP tbutyl hydroperoxide, DMP Dess–Martin periodinane, DMAP 4-dimethylaminopyridine, EDCI 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide.
Total synthesis of securingine A and secu’amamine D
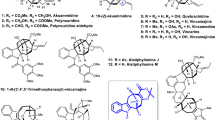
With a robust and scalable synthetic access to 23, we embarked on a divergent synthesis of N–O bond containing securingine A (7b) and secu’amamine D (6). Our first challenge was the enantioselective introduction of the methoxy group at the C4 position of lactam 24. In 2009, the Yun group reported an enantioselective conjugate borylation of cyclic enones and lactones catalyzed by copper–Taniaphos complex35. Inspired by this report, we conducted asymmetric conjugate borylation of lactam 24 in the presence of B2pin2, CuCl, (S)-Taniaphos (25), and NaOtBu. The resulting boronic ester was oxidized with sodium perborate to yield alcohol 26 in 54% over 2 steps and was subsequently methylated to yield C4-methoxy adduct 27 (72% yield). To our pleasure, chiral 1H NMR analysis of 27 that lacks a chromophore using cationic cobalt complex developed by the Kim group36 revealed that the conjugate borylation occurred in 96% ee (Fig. 4). Lactam 27 was then reduced to hemiaminal 28 and allowed to react with silyl dienol ether 11 accessed from 23 under the vinylogous Mannich reaction conditions developed by Busqué, de March, and coworkers to forge the tricyclic structure19. Subsequent TBAF-mediated silyl deprotection of the resulting tricycle afforded alcohol 29 in 61% yield over 2 steps from 23. Carbamate 29 was subsequently subjected to a reaction sequence that involved TFA-mediated Boc deprotection and base-promoted intramolecular aza-Michael reaction to produce tetracycle 3022.

Reagents and conditions: (a) B2pin2 (1.1 equiv), CuCl (0.02 equiv), NaOtBu (0.03 equiv), 25 (0.04 equiv), MeOH (2.0 equiv), THF, 23 °C; (b) NaBO3·H2O (5.0 equiv), THF:H2O (1:1 v/v), 23 °C, 54% (2 steps); (c) Me3OBF4 (3.0 equiv), proton-sponge (4.0 equiv), CH2Cl2, 0 °C to 23 °C, 72% (96% ee); (d) LiEt3BH (1.2 equiv), THF, –78 °C; (e) TIPSOTf (1.2 equiv), Et3N (2.0 equiv), Et2O, 0 °C to 23 °C; 28 (1.5 equiv), Bu2BOTf (1.2 equiv), Et2O, –78 °C; (f) TBAF (2.2 equiv), THF, 23 °C, 61% (2 steps); (g) TFA:CH2Cl2 (1:1 v/v), 23 °C; (h) Et3N:MeOH (1:2 v/v), 50 °C, 90% (2 steps); (i) m-CPBA (1.1 equiv), K2CO3 (3.0 equiv), CH2Cl2, 0 °C to 23 °C, 83%; (j) MsCl (3.0 equiv), Et3N (6.0 equiv), CH2Cl2, 0 °C, 87%; (k) m-CPBA (1.1 equiv), K2CO3 (3.0 equiv), CH2Cl2, 0 °C to 23 °C, 75%. B2pin2 bis(pinacolato)diboron, TBAF tetra-n-butylammonium fluoride, TFA trifluoroacetic acid, m-CPBA 3-chloroperbenzoic acid.
To our delight, when amine 30 was treated with m-CPBA and potassium carbonate, the first synthetic sample of securingine A (7b) was obtained in 85% yield. Spectroscopic data of our synthetic sample of 7b were in complete agreement with those from the isolation report9. Hence, our first total synthesis of securingine A (7b) unambiguously corroborated its computationally proposed structural revision13. The presumed mechanism of the transformation of 30 to securingine A (7b) involves initial N-oxidation of 30 to 31 followed by a Cope elimination to result in intermediate 32. The hydroxyl amine moiety of 32 would then undergo intramolecular 1,4-conjugate addition to give securingine A (7b). On the other hand, when securitinine (3), accessed by treating 30 with MsCl and Et3N20,21,22,31, was allowed to react with m-CPBA and potassium carbonate, secu’amamine D (6) was obtained in 75% yield via a 1,2-Meisenheimer rearrangement23,37. Mechanistically, it is notable that the formation of relatively more stable allylic radical intermediate enabled homolytic cleavage of C7–N1 in N-oxide intermediate 3338 (contrary to the heterolytic C–N cleavage of intermediate 31) and subsequent intramolecular radical recombination to produce secu’amamine D (6).
Total synthesis of securingines C and D
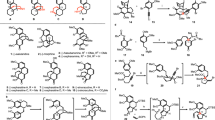
We then turned our attention to the total synthesis of epoxide-containing securinine-type alkaloids securingines C (8b) and D (9b). After numerous experimentations, we discovered that fructose-derived Shi’s ketone 35 could effectively catalyze the epoxidation of α,β-unsaturated lactam 24 in 71% yield (2 cycles) and 3:1 er (Fig. 5)39. The resulting epoxide was reduced to hemiaminal intermediate 37 and allowed to react with silyl dienol ether 11 in the presence of Bu2BOTf to forge the tricyclic intermediate19, which after TBAF-mediated deprotection of the TBDPS group yielded alcohol 38 (46% isolated yield of the desired diastereomer). Markedly, the diastereomeric byproduct that originated from the enantiomer of 36 could be readily separated from 38 by silica gel column chromatography which practically solved the issue related with the moderate enantioselectivity during the epoxidation of 24. The subjection of the resulting alcohol 38 to a three-step sequence involving mesylation of the alcohol moiety, Boc deprotection, and intramolecular N-alkylation afforded the first synthetic sample of securingine C (8b). Spectroscopic data of the synthetic sample were in complete accordance with the isolation report9. Since the relative configuration of the epoxide moiety was unambiguously confirmed by a single-crystal X-ray diffraction analysis of the downstream derivative of 8b (vide infra), we experimentally confirmed the computationally proposed structure revision of securingine C (8b) by the Kutateladze group14.

Reagents and conditions: (a) Oxone (10.0 equiv), NaHCO3 (30.0 equiv), 35 (0.5 equiv), Na2EDTA (0.0005 equiv), Bu4NHSO4 (0.1 equiv), MeCN, 0 °C to 23 °C, 71% (3:1 er) after 2 cycles; (b) LiEt3BH (1.2 equiv), THF, –78 °C; (c) Bu2BOTf (1.2 equiv), Et2O, –78 °C; (d) TBAF (2.2 equiv), THF, 23 °C, 46% (2 steps); (e) MsCl (3.0 equiv), Et3N (6.0 equiv), CH2Cl2, 0 °C; (f) TFA: CH2Cl2 (1:1 v/v), 23 °C, 63% (2 steps); (g) m-CPBA (1.1 equiv), VO(acac)2 (1.0 equiv), CH2Cl2, 0 °C, 54%. EDTA ethylenediaminetetraacetic acid, VO(acac)2 vanadyl acetylacetonate.
With efficient access to securingine C (8b), its regio- and stereoselective C–H hydroxylation at C2 position was explored. We previously reported a C2-selective enamine formation of (viro)allosecurinine via VO(acac)2-mediated Polonovski reaction32. Based on these results, we treated securingine C (8b) with m-CPBA and subsequently with VO(acac)2 to access intermediate 39. Pleasantly, 39 underwent intramolecular syn-elimination to forge C2-iminium ion 40, the regioisomer that could not be accessed under conventional E2(anti-elimination)-based Polonovski reaction conditions32. Hydroxyvanadium intermediate consequently trapped iminium ion 40 from the sterically more accessible re face to yield the first synthetic sample of securingine D (9b) upon aqueous work-up in 54% yield. Spectroscopic data of the synthetic sample were in complete agreement with those of the natural sample9. Importantly, single-crystal X-ray diffraction analysis of securingine D (9b, CCDC 2170237) unequivocally established the absolute and relative stereochemistry of the synthetic securingine D (9b). Hence, the machine learning-based structural revision of securingine D (9b)14 was confirmed via its total synthesis.
C2-epimerization of securinine-type alkaloids
Next, we sought to find a solution for the C2-epimerization of securinine-type alkaloids. In that regard, we turned our attention to hydrogen atom transfer (HAT)-mediated epimerization reaction40. Recently, Ellman, Houk, Mayer, and coworkers reported a combined photocatalytic and HAT-based strategy for the epimerization of C2-substituted piperidine41. They showed that the thiophenyl radical generated by photoredox catalyst can undergo reversible polarity matched HAT with C2-substituted piperidines to eventually yield the energetically more stable piperidine products (Fig. 6a). Encouraged by this report, we allowed 30 to react with [Ir{dF(CF3)ppy}2(dtbpy)]PF6 (1 mol%) and PhSH (1 equiv) in methanol under blue LED irradiation. To our delight, C2-epimerized product 42 was obtained in 63% yield along with 18% of 30 via radical intermediate 41 (Fig. 6b). DFT-calculated difference of the solution-phase free energy between 42 and 30 supported the favorable formation of 42 over 30. The calculated ground-state conformation of compound 30 showed that the C4-methoxy moiety resides in the axial position while that of compound 42 is positioned in the equatorial position (See the supporting information for detail). Indeed, the observed formation ratio between 42 and 30 (42:30 = 3.5:1) was in line with the predicted ratio based on the A-value of the methoxy group (0.6 kcal/mol, corresponding to 2.8:1 ratio at 23 °C). Furthermore, resubjection of 42 to the aforementioned reaction conditions resulted in the formation of 42 and 30 in approximately 3:1 ratio, corroborating the thermodynamic equilibrium between these two species. Subsequent 1,2-amine shift via a mesylation of the resulting hydroxyl moiety within compound 42 afforded the first synthetic sample of 4-epi-phyllanthine (4).

a Ellman’s light-mediated piperidine epimerization (ref. 41); b Light-mediated C2-epimerization of 30 enables the synthesis of 4-epi-phyllanthine (4). ET electron transfer, HAT hydrogen atom transfer.
The prevalence of C2-epimeric pairs in securinega alkaloids prompted us to test the generality of light-mediated HAT-based epimerization strategy by applying it to (neo)securinane alkaloids without the C4-methoxy moiety. Hence, we investigated the potential interconversion between allosecurinine and securinine. Firstly, allosecurinine (2)32 was transformed to secu’amamine E (13) following our previously reported deconstructive functionalization method as a means to mask the Michael acceptor moiety (Fig. 7)42. Secu’amamine E (13) was then subjected to the photoredox-catalyzed HAT-mediated C2-epimerization reaction conditions and the C2-epimer ent-virosine B (44) was obtained in 44% yield along with secu’amamine E (13, 38% yield). Importantly, resubjection of ent-virosine B (44) to the same reaction conditions resulted in the formation of both 13 and 44 in 38% and 31% yield, consistent with the thermodynamic equilibrium between these two compounds (See the supporting information for DFT-calculation-based analysis). ent-Virosine B (44) was then transformed to securinine (1) upon mesylation of the alcohol moiety and consequent 1,2-amine shift. Finally, direct C2-epimerization of the securinine framework (azabicyclo[3.2.1]octane core) without the intermediacy of the neosecurinine-type (azabicyclo[2.2.2]octane core) intermediate was explored. Delightfully, when securinine (1) was allowed to react with [Ir{dF(CF3)ppy}2(dtbpy)]PF6 (1 mol%) and 2 equiv of benzenethiol, and subsequently with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), allosecurinine (2) was obtained in 55% yield along with securinine (1, 14% yield). The use of 2 equiv of benzenethiol was necessary as one equivalent of it underwent 1,6-conjugate addition to the substrate. DBU was the optimal base for the E1cB elimination of benzenethiol. The prevalence of C2-epimeric pairs within securinega alkaloids family hints at further general transformations between those epimeric pairs.

The light-mediated HAT-based epimerization of piperidines enabled the interconversion between secu’amamine E and ent-virosine B, the neosecurinane derivatives of allosecurinine and securinine, respectively. Direct conversion of securinine to allosecurinine was also possible by the iridium-catalyzed HAT-based epimerization reaction by employing 2 equiv of benzenethiol and subsequent DBU-mediated E1cB reaction. DBU 1,8-diazabicyclo[5.4.0]undec-7-ene.
Total synthesis of phyllanthine and its C2-epimer
The Mannich reaction-based union of the piperidine precursor (A-ring) and the menisdaurilide derivative 23 has proven to be a robust method to set the S-configuration at the C2 position (Figs. 4 and 5)19. Peixoto and coworkers could obtain a diastereomeric mixture of compounds with R- and S-configurations at C2 via their aldol reaction-based fragments coupling and subsequent reductive amination strategy31. We also showed the versatility of HAT approach for the epimerization of this stereogenic center (Figs. 6 and 7). However, direct and selective access to the R-configuration at C2 via a C2–C9 bond formation between the piperidine precursor and the menisdaurilide derivative 23 has been historically elusive31. Hence, we initiated the exploration of an orthogonal C2–C9 bond-forming strategy that selectively sets the R-configuration at the C2 position. To our pleasure, we found that the vinylogous Michael addition of the lithium dienolate derivative of 23 to enone 45, accessed by dehydrogenation of 1-Boc-4-piperidone43, resulted in the diastereoselective formation of tricycle 46 in 79% yield (Fig. 8a). We reasoned that the sterically bulky TBDPS moiety in the dienolate derivative of 23 governs its facial selectivity and sets the stereochemistry at C9 (Fig. 8b). The steric clash between the C6-methylene of 45 and the menisdaurilide backbone renders the si-face approach of the nucleophile disfavorable, hence, leading to the diastereoselective formation of 46 with the R-configuration at C2 (Fig. 8b).

a Reagents and conditions: (a) 45 (1.5 equiv), LiHMDS (1.2 equiv), THF, –78 °C to –40 °C, 79%; (b) K-selectride (1.2 equiv), THF, –78 °C, 84%; (c) 4-nitrobenzoic acid (2.0 equiv), PPh3 (1.5 equiv), DIAD (2.0 equiv), THF, 0 °C to 23 °C; (d) K2CO3 (3.0 equiv), MeOH, –78 °C, 99% (2 steps); (e) Me3OBF4 (3.0 equiv), proton-sponge (4.0 equiv), CH2Cl2, 0 °C to 23 °C, 91%; (f) TBAF (1.0 equiv), THF, 23 °C, 84%; (g) TFA:CH2Cl2 (1:1 v/v), 23 °C; (h) Et3N:MeOH (1:2 v/v), 50 °C, 93% (2 steps); (i) MsCl (3.0 equiv), Et3N (6.0 equiv), CH2Cl2, 0 °C, 90%; (j) [Ir{dF(CF3)ppy}2(dtbpy)]PF6 (0.01 equiv), PhSH (1.0 equiv), blue LED (427 nm), MeOH, 23 °C, 51 63% and 50 15%; (k) MsCl (3.0 equiv), Et3N (6.0 equiv), CH2Cl2, 0 °C, 72%; b Rationale for the diastereoselective formation of 46. LiHMDS lithium bis(trimethylsilyl)amide, DIAD diisopropyl azodicarboxylate.
With robust access to compound 46, we envisioned converting it to phyllanthine (5). 46 was stereoselectively reduced to alcohol 47 in 84% yield upon reaction with K-selectride. Less sterically hindered sodium borohydride resulted in the same stereochemical outcome. The C4 stereochemistry in 47 was inverted via a two-step Mitsunobu reaction-mediated protocol to yield alcohol 48 (99% yield over 2 steps). Ensuing methylation of alcohol 48 with Meerwein salt, consequent desilylation, Boc deprotection, and base-mediated intramolecular aza-Michael addition of the resulting tricycle 49 afforded tetracyclic compound 50. Mesylation of the alcohol moiety in 50 induced the 1,2-amine shift to yield phyllanthine (5) in 90% yield. As expected, application of previously explored C2-epimerization reaction conditions to 50 resulted in thermodynamically more stable compound 51 in 63% yield along with 15% of 50. DFT-calculated difference of the solution-phase free energy between 50 and 51 was in line with the favorable formation of 51 over 50. Final mesylation of alcohol 51 produced 4-epi-securitinine (52, 72% yield), a presumed natural product yet to be discovered44.
Discussion
To sum up, we described a unified synthetic approach to C4-oxygenated securinine-type alkaloids. Securingines A (7b), C (8b), D (9b), securitinine (3), secu’amamine D (6), 4-epi-phyllanthine (4), and phyllanthine (5) could be chemically synthesized in this study. Keys to our success were; (1) the scalable synthetic route to the menisdaurilide derivative 23, (2) the discovery of catalytic reaction conditions for asymmetric borylation and epoxidation of α,β-unsaturated lactam, (3) the mechanism-based substrate design of N-oxide derivatives favoring either 1,2-Meisenheimer rearrangement or Cope elimination, (4) the VO(acac)2-mediated Ei (syn) selective Polonovski reaction, (5) the application of Ellman’s light-mediated HAT-based piperidine epimerization, and (6) the diastereoselective Michael addition of the lithium dienolate derivative of menisdaurilide to piperidone 45. These findings have built the foundation to access all known C4-oxygenated securinine-type alkaloids. Furthermore, our discoveries en route to monomeric high-oxidation state securinine-type alkaloids synthesis would serve as the basis for the exploration of even more complex high-order and high-oxidation state securinine-type alkaloids6,8,11. Those will be the subject of our forthcoming reports.
Methods
Experimental instrumentations
All reactions were performed in oven-dried or flame-dried round-bottomed flasks and vials. Unless otherwise noted, the flasks were fitted with rubber septa and reactions were conducted under a positive pressure of argon, and vials were tightly sealed with plastic septa and parafilm. Stainless steel syringes or cannula were used to transfer air- and moisture-sensitive liquids. Flash column chromatography was performed as described by Still et al. using silica gel (60-Å pore size, 40–63 µm, 4–6% H2O content, Merck). Analytical thin-layer chromatography (TLC) was performed using glass plates pre-coated with 0.25 mm silica gel impregnated with a fluorescent indicator (254 nm). Thin-layer chromatography plates were visualized by exposure to ultraviolet light, an aqueous ceric ammonium molybdate (CAM) solution, and/or an aqueous potassium permanganate (KMnO4) solution.
Reagents and solvents
Unless otherwise stated, all commercial reagents and solvents were used without additional purification with the following exceptions as indicated below. Dichloromethane and tetrahydrofuran were purchased from Merck and Daejung Inc., respectively and were purified by the method of Grubbs et al. under positive argon pressure.
Data analysis
1H and 13C nuclear magnetic resonance spectra were recorded with Bruker AVANCE NEO (500 MHz), Bruker AVANCE III HD Nanobay (400 MHz), Bruker AVANCE III HD (400 MHz), or Bruker AVANE NEO Nanobay (400 MHz) and calibrated by using the residual undeuterated chloroform (δH = 7.24 ppm) and CDCl3 (δC = 77.23 ppm) or monodeuterated dichloromethane (δH = 5.32 ppm) as internal references. Data are reported in the following manners: chemical shift in ppm [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = quintet, m = multiplet, app = apparent, br = broad), coupling constant(s) in Hertz, integration]. The NMR solvent CDCl3 was taken from a stock containing anhydrous K2CO3 to remove residual DCl. High resolution mass spectra were obtained from KAIST Analysis Center for Research Advancement (Daejeon) by using ESI ionization method. Specific rotation was obtained by JASCO P-2000 polarimeter.
Detailed experimental procedures
The detailed experimental procedures were provided in Supplementary Information.
Cartesian coordinates and vibrational frequencies
Cartesian coordinates of the optimized geometries and vibrational frequencies of the optimized structures were provided in Supplementary Data 1.
Data availability
Crystallographic data for the structure reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 2170237 (9b). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
References
Muravieva, V. I. & Bankovsky, A. I. A chemical study of alkaloids of Securinega suffruticosa. Dokl. Akad. Nauk SSSR 110, 998–1000 (1956).
Chirkin, E., Atkatlian, W. & Porée, F.-H. The securinega alkaloids. Alkaloids Chem. Biol. 74, 1–120 (2015).
Babady-Bila, Gedris, T. E. & Herz, W. Niruroidine, a norsecurinine-type alkaloid from Phyllanthus niruroides. Phytochemistry 41, 1441–1443 (1996).
Zhou, M., Zhu, H., Wang, K., Wei, W. & Zhang, Y. Isolation and X-ray crystal structure of a securinega-type alkaloid from Phyllanthus niruri Linn. Nat. Prod. Res. 26, 762–764 (2012).
Diallo, M. S. T. et al. Ethnomedical, phytochemical and biological investigations of Margaritaria discoidea (Baill.) webster, a plant species widely used in Guinean traditional medicine. J. Plant Sci. 3, 40–46 (2015).
Qin, S., Liang, J.-Y., Gu, Y.-C. & Guo, Y.-W. Suffruticosine, a novel octacyclic alkaloid with an unprecedented skeleton from Securinega suffruticosa (Pall.) Rehd. Tetrahedron Lett. 49, 7066–7069 (2008).
Luo, X.-K. et al. Fluvirosaones A and B, two indolizidine alkaloids with a pentacyclic skeleton from Flueggea virosa. Org. Lett. 20, 991–994 (2018).
Wu, Z.-L. et al. Flueggeacosines A–C, dimeric securinine-type alkaloid analogues with neuronal differentiation activity from Flueggea suffruticosa. Org. Lett. 20, 7703–7707 (2018).
Park, K. J. et al. Securinega alkaloids from the twigs of Securinega suffruticosa and their biological activities. J. Nat. Prod. 82, 1345–1353 (2019).
Fan, Z. et al. Discovery and biosynthesis of ascorbylated Securinega alkaloids. ACS Catal. 11, 8818–8828 (2021).
He, Q.-F. et al. Discovery of neuritogenic Securinega alkaloids from Flueggea suffruticosa by a building blocks-based molecular network strategy. Angew. Chem. Int. Ed. 60, 19609–19613 (2021).
Atwi, R. et al. An automated framework for high-throughput predictions of NMR chemical shifts within liquid solutions. Nat. Comput. Sci. 2, 112–122 (2022).
Kang, G., Baik, M.-H. & Han, S. Calculation-assisted stereochemical analysis of securingine A. Bull. Korean Chem. Soc. 42, 486–488 (2021).
Novitskiy, I. M. & Kutateladze, A. G. DU8ML: machine learning-augmented density functional theory nuclear magnetic resonance computations for high-throughput in silico solution structure validation and revision of complex alkaloids. J. Org. Chem. 87, 4818–4828 (2022).
Weinreb, S. M. Total synthesis of the Securinega alkaloids. Nat. Prod. Rep. 26, 758–775 (2009).
Wehlauch, R. & Gademann, K. Securinega alkaloids: complex structures, potent bioactivities, and efficient total syntheses. Asian J. Org. Chem. 6, 1146–1159 (2017).
Kang, G., Park, S. & Han, S. The chemistry of high‐oxidation state securinega alkaloids. Eur. J. Org. Chem. 2021, 1508–1520 (2021).
Liras, S., Davoren, J. E. & Bordner, J. An approach to the skeleton of the Securinega alkaloids. The total synthesis of (±)-securinine. Org. Lett. 3, 703–706 (2001).
Bardaji, G. G. et al. Diastereoselective synthesis of allosecurinine and viroallosecurinine from menisdaurilide. J. Org. Chem. 73, 7657–7662 (2008).
Horii, Z. et al. Isolation of securinol A, B, and C from Securinega suffruticosa REHD. and the structures of securinol A and B. Chem. Pharm. Bull. 13, 1307–1311 (1965).
Magnus, P., Rodriguez-Lopez, J., Mulholland, K. & Matthews, I. Biomimetic synthesis of the pentacyclic alkaloid (±)-nirurine and possible biogenetic rearrangement of a precursor into (±)-norsecurinine. J. Am. Chem. Soc. 114, 382–383 (1992).
Wehlauch, R. et al. Investigating biogenetic hypotheses of the Securinega alkaloids: Enantioselective total syntheses of secu’amamine E/ent-virosine A and bubbialine. Org. Lett. 19, 548–551 (2017).
Horii, Z. et al. Structure of phyllantidine. Tetrahedron Lett. 13, 1877–1880 (1972).
Carson, C. A. & Kerr, M. A. Total synthesis of (+)-phyllantidine. Angew. Chem. Int. Ed. 45, 6560–6563 (2006).
Lambert, K. M. et al. Total synthesis of (±)-phyllantidine: development and mechanistic evaluation of a ring expansion for installation of embedded nitrogen‐oxygen bonds. Angew. Chem. Int. Ed. 132, 9844–9853 (2020).
Han, G., LaPorte, M. G., Folmer, J. J., Werner, K. M. & Weinreb, S. M. A new enantioselective approach to total synthesis of the securinega alkaloids: application to (−)-norsecurinine and phyllanthine. Angew. Chem. Int. Ed. 39, 237–240 (2000).
Han, G., LaPorte, M. G., Folmer, J. J., Werner, K. M. & Weinreb, S. M. Total syntheses of the Securinega alkaloids (+)-14,15-dihydronorsecurinine, (−)-norsecurinine, and phyllanthine. J. Org. Chem. 65, 6293–6306 (2000).
Cantó, M. et al. First synthesis of (+)-rengyolone and (+)-and (−)-menisdaurilide. Tetrahedron Asymmetry 13, 455–459 (2002).
Urakawa, Y., Sugimoto, T., Sato, H. & Ueda, M. Enantioselective synthesis of phyllanthurinolactone, a leaf-closing substance of Phyllanthus urinaria L., and its analogs toward the development of molecular probes. Tetrahedron Lett. 45, 5885–5888 (2004).
Kato, N. et al. Enantio-differential approach using fluorescence-labeled phyllanthurinolactone, a leaf-closing factor of Phyllanthus urinaria L. Tetrahedron Lett. 48, 7702–7705 (2007).
Antien, K. et al. Bio-inspired total synthesis of twelve Securinega alkaloids: Structural reassignments of (+)-virosine B and (−)-episecurinol A. Chem. Eur. J. 25, 11574–11580 (2019).
Lee, S. et al. Biosynthetically inspired synthesis of secu’amamine A, fluvirosaones A and B. Angew. Chem. Int. Ed. 59, 6894–6901 (2020).
Zhao, Y., Rodrigo, J., Hoveyda, A. H. & Snapper, M. L. Enantioselective silyl protection of alcohols catalysed by an amino-acid-based small molecule. Nature 443, 67–70 (2006).
Manville, N., Alite, H., Haeffner, F., Hoveyda, A. H. & Snapper, M. L. Enantioselective silyl protection of alcohols promoted by a combination of chiral and achiral Lewis basic catalysts. Nat. Chem. 5, 768–774 (2013).
Feng, X. & Yun, J. Catalytic enantioselective boron conjugate addition to cyclic carbonyl compounds: a new approach to cyclic β-hydroxy carbonyls. Chem. Commun. 6577–6579 (2009).
Jang, S. & Kim, H. Chiral 1H NMR analysis of carbonyl compounds enabled by cationic cobalt complex. Org. Lett. 22, 4185–4189 (2020).
Nakano, T., Terao, S., Lee, K. H., Saeki, Y. & Durham, L. J. Some observations on the oxidation of virosecurinine with monoperphthalic acid. J. Org. Chem. 31, 2274–2279 (1966).
Sousa, C. A. D., Sampaio-Dias, I. E., García-Mera, X., Lima, C. F. R. A. & Rodríguez-Borges, J. E. On the scope of oxidation of tertiary amines: Meisenheimer rearrangements versus Cope elimination in 2-(cyanoethyl)-2-azanorbornanes. Org. Chem. Front. 3, 1624–1634 (2016).
Wu, X.-Y., She, X. & Shi, Y. Highly enantioselective epoxidation of α,β-unsaturated esters by chiral dioxirane. J. Am. Chem. Soc. 124, 8792–8793 (2002).
Wang, Y., Carder, H. M. & Wendlandt, A. E. Synthesis of rare sugar isomers through site-selective epimerization. Nature 578, 403–408 (2020).
Shen, Z. et al. General light-mediated, highly diastereoselective piperidine epimerization: from most accessible to most stable stereoisomer. J. Am. Chem. Soc. 143, 126–131 (2021).
Lim, H., Seong, S., Kim, Y., Seo, S. & Han, S. Biopatterned reorganization of alkaloids enabled by ring-opening functionalization of tertiary amines. J. Am. Chem. Soc. 143, 19966–19974 (2021).
Diao, T. & Stahl, S. S. Synthesis of cyclic enones via direct palladium-catalyzed aerobic dehydrogenation of ketones. J. Am. Chem. Soc. 133, 14566–14569 (2011).
Hetzler, B. E., Trauner, D. & Lawrence, A. L. Natural product anticipation through synthesis. Nat. Rev. Chem. 6, 170–181 (2022).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIT) (No. NRF-2021R1A2C2011203). We also acknowledge support by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2018R1A5A1025208).
Author information
Authors and Affiliations
Contributions
S.H., S.P., and G.K. conceived the study. S.H. supervised the project. S.P. played a key role in experimentations. G.K. conducted computational studies. C.K. played a supportive role in experimentations. D.K. performed the single-crystal X-ray diffraction analysis. S.H., S.P., and G.K. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, S., Kang, G., Kim, C. et al. Collective total synthesis of C4-oxygenated securinine-type alkaloids via stereocontrolled diversifications on the piperidine core. Nat Commun 13, 5149 (2022). https://doi.org/10.1038/s41467-022-32902-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32902-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
