
Abstract
Primary pleomorphic adenomas (PAs) and mucoepidermoid carcinomas (MECs) of the breast are vanishingly rare. Here we sought to determine whether breast PAs and MECs would be underpinned by the fusion genes reported to occur in their salivary gland counterparts. Our study included three breast PAs and one breast MEC, which were subjected to RNA sequencing (PAs, n = 2; MEC, n = 1) or to Archer FusionPlex sequencing (PA, n = 1). Our analyses revealed the presence of the HMGA2-WIF1 fusion gene in breast PA3, the CTNNB1-PLAG1 fusion gene in breast PA2, and the CRTC1-MAML2 fusion gene in the breast MEC analyzed (1/1). No oncogenic fusion genes were detected in breast PA1, and no additional oncogenic fusion genes were detected in the cases studied. The presence of the fusion genes identified was validated by fluorescence in situ hybridization (n = 1), reverse transcription-PCR (n = 1), or by both methods (n = 1). Taken together, our findings indicate that PAs and MECs arising in the breast resemble their salivary gland counterparts not only phenotypically but also at the genetic level. Furthermore, our data suggest that the molecular analysis of breast PAs and MECs might constitute a useful tool to aid in their differential diagnosis.
Similar content being viewed by others


Introduction
Pleomorphic adenomas (PAs) and mucoepidermoid carcinomas (MECs) of the breast are rare tumors that histologically resemble their salivary gland counterparts1. PAs are characterized by an admixture of epithelial and myoepithelial cells immersed in myxochondroid stroma1,2. Cartilaginous and osseous metaplasia may be present1,2. Breast PAs usually have a benign course, although local recurrences have been described1. PAs arising in the salivary glands, skin, and soft tissue harbor recurrent gene rearrangements involving PLAG1 or HMGA2 (refs. 3,4). MECs, although common in the salivary glands, are vanishingly rare in the breast1,5. MECs are characterized by the admixture of different cell types, including mucinous, intermediate, and squamous cells1. MECs arising in the salivary gland, lung, and cervix have been shown to harbor the t(11,19)(q14–21; p12–13) translocation that results in the CRTC1-MAML2 fusion gene6,7,8, or less frequently the CRTC3-MAML2 fusion gene9.
A subset of salivary gland-like tumors arising in the breast have been shown to be underpinned by the same genetic alterations as their salivary glands counterparts1,10. For instance, adenoid cystic carcinomas arising either in the breast or in the salivary glands harbor MYB or MYBL1 rearrangements, regardless of their anatomic origin10,11,12, and secretory carcinomas of the breast and mammary analog secretory carcinomas of the salivary glands are characterized by the ETV6-NTRK3 fusion gene10,13,14. The genetic underpinning of other breast salivary gland-like tumors, such as acinic cell carcinomas, however, is different than that of their salivary gland counterparts. Salivary gland acinic cell carcinomas are characterized by the t(4;9)(q13;q31) translocation, which results in a rearrangement involving the nuclear transcription factor NR4A3 (ref. 15), whereas breast acinic cell carcinomas seem not to harbor a pathognomonic genetic alteration and consistently display complex genomes and TP53 mutations16,17. Here we sought to determine whether breast PAs and MECs arising in the breast would be underpinned by fusion genes, in particular those reported in their salivary gland counterparts.
Results
Cases
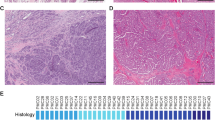
Three PAs and one MEC arising in the breast were included in this study. The breast PAs (n = 3) included in our series were well-circumscribed lesions (Fig. 1a), composed of epithelial cells and myoepithelial cells arranged in cords, clusters, and forming glands immersed in a myxochondroid stroma (Fig. 1b, c). Myoepithelial cells floating in the stroma as single stellate cells (Fig. 1b) and focal areas of solid growth were frequently observed (Fig. 1d). No cellular atypia or increased mitotic index were detected (Fig. 1d) The breast MEC studied here corresponded to a low-grade MEC with well-circumscribed borders (Fig. 1e), composed of mucinous, intermediate, and squamous cells arranged in cords, sheets, and clusters (Fig. 1e). No cytologic atypia or necrosis were observed (Fig. 1e, f).

a–d Representative photomicrographs of hematoxylin and eosin (H&E)-stained sections of breast pleomorphic adenomas (PAs) included in this study. Breast PAs were well-circumscribed lesions (a), composed of epithelial and myoepithelial cells in a myxochondroid stroma (b, c). Focal areas with solid growth were also identified (d). e, f Representative photomicrographs of H&E-stained sections of the breast mucoepidermoid carcinoma (MEC) displaying squamous, mucinous, and intermediate cells (e), and absence of atypia or necrosis (f). g Heatmap depicting fusion genes identified in breast pleomorphic adenomas (BPAs; n=3) and a breast mucoepidermoid carcinoma (BMEC2; n=1). Cases are shown in columns and fusion genes in rows. Tumor type and detection method are shown (top). Scale bars, 200μm (a), 100μm (b, e), 50μm (c, d), and 20μm (f). FISH fluorescence in situ hybridization.
To determine whether the breast PAs (n = 3) and the MEC (n = 1) included in our series would harbor fusion genes, akin to their counterparts arising in the salivary glands, we subjected two breast PAs (BPA1 and BPA3) and one MEC (BMEC2) to RNA sequencing (Fig. 1g, Supplementary Table 1). RNA-sequencing data were analyzed using a validated bioinformatics pipeline for de novo discovery of fusion genes as previously described18,19. RNA derived from one breast PA (BPA2) with insufficient material for RNA sequencing was subjected to Archer FusionPlex analysis, a targeted RNA-based assay, for fusion gene detection (Fig. 1g, Supplementary Table 1). Our analyses revealed the presence of fusion genes in two of the three breast PAs and in the breast MEC analyzed (Fig. 1g, Supplementary Table 1).
HMGA2 and PLAG1 rearrangements in breast pleomorphic adenomas
Our analyses revealed the presence of an HMGA2-WIF1 fusion gene in BPA3 by RNA sequencing (Figs. 1g and 2a, Supplementary Table 1). The HMGA2-WIF1 fusion gene identified has been previously reported in PAs of the salivary gland20,21. HMGA2 encodes for a transcriptional regulator of the TGF-β signaling pathway22, and WIF1 for a tumor suppressor modulating signaling via the Wnt pathway20. The HMGA2-WIF1 fusion gene detected in BPA3 resulted in a chimeric transcript composed of exons 1–3 of HMGA2 and exon 10 of WIF1, and was predicted to be translated in the initial segment of HMGA2 fused to the C′-terminus of WIF1, with the loss of its active WIF domain20 (Fig. 2a). The HMGA2-WIF1 fusion gene identified in BPA3 was validated by reverse transcription (RT)-PCR, followed by Sanger sequencing (Fig. 2b). As predicted, the HMGA2-WIF1 fusion resulted in retained expression of HMGA2 exons 1–3 and decreased expression of exons 4 and 5 (Fig. 2c). We identified the presence of a CTNNB1-PLAG1 fusion gene in BPA2 by Archer FusionPlex targeted RNA sequencing (Figs. 1g and 2d, Supplementary Table 1). The CTNNB1-PLAG1 fusion gene was predicted to result in a chimeric transcript encompassing exon 1 of CTNNB1 and exons 4 and 5 of PLAG1 (Fig. 2d) and has been described in PAs of the salivary glands and of the lacrimal glands23,24. The CTNNB1-PLAG1 fusion identified in BPA2 was validated by fluorescence in situ hybridization (FISH) using PLAG1 dual-color break-apart probes (Fig. 2e). No additional likely pathogenic fusion genes were identified in the breast PAs analyzed (Supplementary Tables 1 and 2).

a Schematic representation of the HMGA2-WIF1 fusion transcript detected in the pleomorphic adenoma BPA3 by RNA sequencing, depicting the exons and protein domains involved (top). Vertical lines show the breakpoints of HMGA2 and WIF1. The spanning reads found to cross the genomic breakpoint of the HMGA2-WIF1 chimeric transcript are shown aligned to the predicted junction sequence (bottom). b Representative Sanger sequencing electropherogram of cDNA validating the HMGA2-WIF1 fusion junction in BPA3. c Schematic representation showing the reads per kilobase per million (RPKM) mapped read counts of each HMGA2 exon in the pleomorphic adenoma BPA3. The HMGA2 fusion breakpoint is represented as a red dashed line. d Schematic representation of the CTNNB1-PLAG1 fusion transcript identified in the pleomorphic adenoma BPA2 by Archer FusionPlex, depicting the exons and protein domains involved. Vertical lines show the breakpoints of CTNNB1 and PLAG1. e Representative photomicrographs of a hematoxylin and eosin-stained section (left) and fluorescence in situ hybridization using PLAG1 dual-color break-apart probes (red, 5′ PLAG1, green, 3′ PLAG1; right) of BPA2. Scale bar, 20 μm (e). AcD acidic domain, DBD DNA-binding domain, SpD spacer domain, Arm Armadillo repeat, zf-H2C2 Zinc finger double domain, zf-C2H2 Zinc finger C2H2 type.
Given that HMGA2 and PLAG1 rearrangements are the hallmark genetic alterations of PAs, regardless of their anatomic location3,4, we subjected the breast PA included in this study in which no fusion genes were identified by RNA sequencing (BPA1) to FISH analysis using HMGA2 and PLAG1 dual-color break-apart probes, which confirmed the absence of HMGA2 or PLAG1 rearrangements (Supplementary Table 1). Notably, BPA1 did not differ histologically from the PAs found to harbor fusion genes.
Breast adenomyoepitheliomas and PAs may show histologic and genetic overlap25. Indeed, we have reported on a bona fide breast adenomyoepithelioma displaying focal myxoid matrix and harboring an HMGA2-WIF1 fusion gene26. Hence, we sought to determine whether the breast PAs of this series subjected to RNA sequencing would harbor hotspot mutations27 affecting HRAS, PIK3CA, and AKT1 as in breast adenomyoepitheliomas28. Upon manual inspection of the HRAS Q61 hotspot locus, PIK3CA H1047, E545, E542, G118, G1050, and G106 hotspot loci and AKT1 E17 hotspot locus in the RNA sequencing BAM files of BPA1 and BPA3 using the Integrative Genomics Viewer (IGV)29, no hotspot mutations affecting these genes were identified.
CRTC1-MAML2 fusion gene in a breast mucoepidermoid carcinoma
RNA sequencing analysis of BMEC2, a MEC arising in the breast, revealed the presence of a CRTC1-MAML2 fusion gene (Figs. 1g and 3a), the hallmark genetic alteration of MECs arising in other anatomic origins6,7,8. These findings are in agreement with the study by Bean et al.30, who reported on two breast MECs harboring the CRTC1-MAML2 fusion gene. The fusion gene identified in BMEC2 was predicted to result in a chimeric transcript encompassing exon 1 of CRTC1 and exons 2–5 of MAML2 (Fig. 3a, b), akin to MECs in other anatomic locations and to the two breast MECs previously reported6,7,8. As predicted, the CRTC1-MAML2 fusion resulted in increased expression of exons 2–5 and decreased expression of exon 1 of MAML2 (Fig. 3c), and its presence was confirmed by RT-PCR and FISH using a three-color break-apart probe (Fig. 3b, d).

a Schematic representation of the CRTC1-MAML2 fusion transcript detected in the breast mucoepidermoid carcinoma BMEC2 by RNA sequencing, depicting the exons and protein domains involved (top). Vertical lines show the breakpoints of CRTC1 and MAML2. The spanning reads found to cross the genomic breakpoint of the CRTC1-MAML2 chimeric transcript are shown aligned to the predicted junction sequence (bottom). b Representative Sanger sequencing electropherogram of cDNA validating the CRTC1-MAML2 fusion junction in BMEC2. c Schematic representation showing the reads per kilobase per million (RPKM) mapped read counts of each MAML2 exon. The MAML2 fusion breakpoint is represented as a red dashed line. d Representative photomicrographs of a hematoxylin and eosin-stained section (left) and fluorescence in situ hybridization using three-color break-apart probes (green, CRTC1; 5′ MAML2, red; 3′ MAML2, orange; right) of BMEC2. Scale bar, 20 μm (d). AcD acidic domain, BD basic domain, CBD CREB-binding domain, NES nuclear export signal, NLS nuclear localization signal, SD splicing domain, TAD transactivation domain.
Discussion
Given that various salivary gland-like tumors arising in the breast have been shown to harbor the same genetic alterations as their salivary gland counterparts10, we posited that breast PAs and MECs might be underpinned by the oncogenic fusion genes described in PAs and MECs arising in the salivary glands. Our analyses resulted in the identification of an HMGA2-WIF1 and a CTNNB1-PLAG1 fusion gene in breast PAs, and of a CRTC1-MAML2 fusion gene in a breast MEC. The HMGA2 and PLAG1 rearrangements, and the CRTC1-MAML2 fusion genes identified in the breast PAs and breast MECs of this study, have been reported in PAs and MECs arising in the salivary gland and in other anatomic locations6,7,8,20,21,23,24. Moreover, our findings on breast MECs are in agreement with the study by Bean et al.30, who reported on the presence of the CRTC1-MAML2 fusion gene in two breast MECs.
HMGA2 is an oncogene with diverse oncogenic mechanisms, as it has been shown to induce the activity of E2F1 and AP1, result in inactivation of p53-dependent apoptosis, and in the activation of the TGF-β signaling pathway22,31. WIF1 is a tumor suppressor that encodes for a secreted antagonist that binds Wnt proteins hampering ligand–receptor interactions32,33. The HMGA2-WIF1 fusion gene identified in BPA3 results in the loss of the 3′ UTR regulatory sites of HMGA2, resulting in its overexpression and in increased signaling via TGF-β22,34. It is also predicted to result in the loss of the WIF domain of WIF1, required for its tumor suppressor activities, with ensuing Wnt signaling activation32,33. We have recently reported the presence of an HMGA2-WIF1 fusion gene in an estrogen receptor-positive breast adenomyoepithelioma (AME) lacking HRAS mutations26. Although the HMGA2 and WIF1 genomic breakpoints in the HMGA2-WIF1 fusion gene identified in the HRAS-wild type AME were different than those identified in BPA3, they resulted in the loss of the 3′UTR of HMGA2 and of the WIF domain of WIF1 (ref. 26), akin to the fusion gene present in BPA3 reported here. Notably, the previously reported AME harboring the HMGA2-WIF1 fusion gene26, despite having the cardinal diagnostic features of AMEs, displayed focal myxochondroid stroma, akin to that found in PAs26. We identified a CTNNB1-PLAG1 fusion gene in BPA2, described in PAs of the salivary gland. This fusion gene results in promoter swapping between PLAG1 and CTNNB1, leading to increased PLAG1 expression and reduction of CTNNB1 expression35. Taken together, these findings suggest that the spectrum of histologic features of breast PAs and AMEs overlaps, despite the differences in their repertoire of somatic genetic alterations (i.e. HMGA2 and PLAG1 fusion genes in the majority of PAs, in contrast with mutations affecting HRAS, PIK3CA, AKT1, and PIK3R1 in breast AMEs)3,4,28.
MECs of the salivary glands and two breast MECs previously reported are characterized by the t(11:19) (q21;p13) translocation, which results in the CRTC1-MAML2 fusion gene6,30. CRTC1 encodes for the CREB-regulated transcriptional coactivator 1 (ref. 36) and MAML2 for the NOTCH/RBPJ mastermind-like 2. MAML2 forms DNA-binding complexes with Rbp-j and NotchIC and augments Notch signaling37. The CRTC1-MAML2 fusion gene identified in BMEC2 has been shown to result in activation of the Notch signaling pathway, be tumorigenic in vitro, and silencing of its fusion product reportedly inhibits tumor growth38.
Given their rarity, the differential diagnosis of PAs and MECs arising in the breast from other breast tumors, such as metaplastic carcinomas and squamous cell carcinomas, respectively, might be challenging1. The absence of peripheral myoepithelial cell layer between the epithelium and stroma in PAs together with their infiltrative appearances, at least focally, makes their distinction from metaplastic carcinoma of the matrix producing type challenging, particularly in core biopsies. Such distinction may have significant management implications. Similarly, the distinction between MECs and the more aggressive metaplastic squamous cell carcinoma or the benign papilloma with squamous metaplasia can be difficult and an additional genomic testing may help in their accurate diagnosis. Reanalysis of the RNA sequencing results by Piscuoglio et al.39 and Weigelt et al.40 revealed that none of the metaplastic breast cancers analyzed in those studies harbored any of the fusion genes identified in breast PAs and MECs (data not shown). Our findings suggest that the assessment of rearrangements involving PLAG1 or HMGA2 in breast PAs, and of MAML2 rearrangements in breast MECs, might be used as ancillary tools to aid in the diagnosis of these rare breast tumors.
Our study has important limitations, including the small sample size due to the rarity of PAs and MECs arising in the breast. In addition, we could only perform transcriptomic analyses of the samples included here due to limited available material. Finally, no fusion genes were identified in one of the breast PAs analyzed, and its driver has yet to be identified. Despite these limitations, our findings support the notion that PAs and MECs arising in the breast harbor genetic alterations akin to those described in their salivary gland counterparts, and constitute additional examples of genotypic–phenotypic correlations in breast tumors. These findings may also constitute the basis for the development of ancillary methods for the diagnosis of these tumors in the breast.
Methods
Ethics
This study was approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board (IRB) and local research ethics committees of the authors’ institutions. Patient consent was obtained if required by the approved IRB protocols. Formalin-fixed paraffin-embedded (FFPE) tissue blocks of three breast PAs and one breast MEC were retrieved from the pathology archives of the authors’ institutions. All samples were anonymized before analysis.
Cases and RNA extraction
All tumors included in this study were centrally reviewed by four pathologists (F.P., M.V., F.C.G., J.S.R.-F.) for diagnosis confirmation, following the diagnostic criteria put forward by the World Health Organization (WHO)41. The three breast PAs and one breast MEC were microdissected from eight-micron-thick histologic sections under a stereomicroscope (Olympus SZ61) to ensure a tumor cell content >80%, and RNA was extracted using the RNeasy FFPE Kit.
RNA sequencing and identification of fusion genes
FFPE-derived RNA from two breast PA and one breast MEC were subjected to paired-end RNA sequencing following the standard protocols used at the Integrated Genomics Operation at MSKCC42, as previously described19,26,43. INTEGRATE44, deFuse45, and FusionCatcher46 were used to identify read pairs supporting chimeric transcripts, followed by exclusion of candidate fusion transcripts and read-through candidates identified in a set of 297 normal breast tissues from the TCGA47. The Bayesian probability to constitute drivers of the remaining candidate fusion genes supported by at least two spanning reads was assessed using OncoFuse (v1.0.9b2)48. Reads per kilobase million (RPKM) values were calculated from raw counts after normalization for sequencing depth and following adjustment for length of genes. Data are available as described on the Data availability statement49,50.
ARCHER FusionPlex
One breast PA with insufficient material for RNA sequencing was subjected to Archer FusionPlex assay, an RNA-based panel that uses Archer Anchored Multiplex PCT technology and next-generation sequencing for the identification of fusion genes51 at the Integrated Genomics Operation at MSKCC, as previously described52. Archer FusionPlex data were analyzed using Archer Software (v4.0.10).
RT-PCR for fusion gene validation
RNA was reverse-transcribed using SuperScript VILO Master Mix (ThermoFisher Scientific), following the manufacturers’ instructions. PCR amplification of 10 ng cDNA was conducted using fusion gene-specific primers sets designed based on the identified breakpoints (Supplementary Table 3). PCR fragments were purified with ExoSAP-IT (ThermoFisher Scientific) and subjected to Sanger Sequencing. Sequence electropherograms of the forward and reversed strands were manually inspected.
Fluorescence in situ hybridization
Cases in this study with available material were subjected to FISH analysis for rearrangements involving HMGA2 and PLAG1 or for the presence of the CRTC1-MAML2 fusion gene following validated protocols at the Molecular Cytogenetics Core of MSKCC, as previously described19. HMGA2 and PLAG1 rearrangements were assessed using dual-color break-apart probes consisting of bacterial artificial chromosome (BAC) clones mapping to 5′ HMGA2 (RP11-230G5, RP11-662G15, red) and 3′ HMGA2 (RP11-937C6, RP11-167E10, green) and to 5′ PLAG1 (RP11-92A9, RP11-111I18, red) and 3′ PLAG1 (RP11-144E19, RP11-246A9, green), respectively. The presence of CRTC1-MAML2 fusion gene was assessed using a three-color break-apart probe consisting of BAC clones mapping to CRTC1 (RP11-282P1, RP11-908B10, green), 5′MAML2 (RP11-385G6, RP11-8N17, orange), and 3′MAML2 (RP11-277H22, RP11-111I14, orange). FISH analysis was conducted by observers blinded to the results of the RNA sequencing and ARCHER FusionPlex analyses. Cases were considered positive if the rearrangement was identified in >15% tumor cells.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data generated and analyzed during this study are described in the following data record: https://doi.org/10.6084/m9.figshare.12199781 (ref. 49). The RNA sequencing data have been deposited in the NCBI Sequence Read Archive under the project accession number SRP257886 (https://identifiers.org/ncbi/insdc.sra:SRP257886)50. The histologic images, fluorescence in situ hybridization, and Sanger sequencing data will be provided upon reasonable request to Dr. Fresia Pareja. The Archer FusionPlex data will be provided upon reasonable request to Dr. Arnaud Da Cruz Paula.
References
Foschini, M. P. et al. The morphological spectrum of salivary gland type tumours of the breast. Pathology 49, 215–227 (2017).
Pia-Foschini, M., Reis-Filho, J. S., Eusebi, V. & Lakhani, S. R. Salivary gland-like tumours of the breast: surgical and molecular pathology. J. Clin. Pathol. 56, 497–506 (2003).
Stenman, G. Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head. Neck Pathol. 7(Suppl 1), S12–S19 (2013).
Bahrami, A., Dalton, J. D., Krane, J. F. & Fletcher, C. D. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer 51, 140–148 (2012).
Basbug, M. et al. Mucoepidermoid carcinoma in a breast affected by burn scars: comprehensive literature review and case report. Breast Care (Basel) 6, 293–297 (2011).
Tonon, G. et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet. 33, 208–213 (2003).
Behboudi, A. et al. Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer 45, 470–481 (2006).
Lennerz, J. K., Perry, A., Mills, J. C., Huettner, P. C. & Pfeifer, J. D. Mucoepidermoid carcinoma of the cervix: another tumor with the t(11;19)-associated CRTC1-MAML2 gene fusion. Am. J. Surg. Pathol. 33, 835–843 (2009).
Fehr, A. et al. A new type of MAML2 fusion in mucoepidermoid carcinoma. Genes Chromosomes Cancer 47, 203–206 (2008).
Pareja, F. et al. Triple-negative breast cancer: the importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer 2, 16036 (2016).
Persson, M. et al. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 106, 18740–18744 (2009).
Kim, J. et al. MYBL1 rearrangements and MYB amplification in breast adenoid cystic carcinomas lacking the MYB-NFIB fusion gene. J. Pathol. 244, 143–150 (2018).
Tognon, C. et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2, 367–376 (2002).
Skalova, A. et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am. J. Surg. Pathol. 34, 599–608 (2010).
Haller, F. et al. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat. Commun. 10, 368 (2019).
Beca, F. et al. Whole-exome sequencing and RNA sequencing analyses of acinic cell carcinomas of the breast. Histopathology 75, 931–937 (2019).
Guerini-Rocco, E. et al. The repertoire of somatic genetic alterations of acinic cell carcinomas of the breast: an exploratory, hypothesis-generating study. J. Pathol. 237, 166–178 (2015).
Pareja, F. et al. The Genomic landscape of mucinous breast cancer. J. Natl Cancer Inst. 111, 737–741 (2019).
Kim, S. H. et al. Identification of recurrent FHL2-GLI2 oncogenic fusion in sclerosing stromal tumors of the ovary. Nat. Commun. 11, 44 (2020).
Queimado, L., Lopes, C. S. & Reis, A. M. WIF1, an inhibitor of the Wnt pathway, is rearranged in salivary gland tumors. Genes Chromosomes Cancer 46, 215–225 (2007).
Stenman, G., Andersson, M. K. & Andren, Y. New tricks from an old oncogene: gene fusion and copy number alterations of MYB in human cancer. Cell Cycle 9, 2986–2995 (2010).
Fusco, A. & Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 7, 899–910 (2007).
Kas, K. et al. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat. Genet. 15, 170–174 (1997).
Asahina, M. et al. Clinicopathological effect of PLAG1 fusion genes in pleomorphic adenoma and carcinoma ex pleomorphic adenoma with special emphasis on histological features. Histopathology 74, 514–525 (2019).
McLaren, B. K., Smith, J., Schuyler, P. A., Dupont, W. D. & Page, D. L. Adenomyoepithelioma: clinical, histologic, and immunohistologic evaluation of a series of related lesions. Am. J. Surg. Pathol. 29, 1294–1299 (2005).
Pareja, F. et al. Assessment of HMGA2 and PLAG1 rearrangements in breast adenomyoepitheliomas. NPJ Breast Cancer 5, 6 (2019).
Chang, M. T. et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 (2016).
Geyer, F. C. et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat. Commun. 9, 1816 (2018).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinformatics 14, 178–192 (2013).
Bean, G. R. et al. CRTC1-MAML2 fusion in mucoepidermoid carcinoma of the breast. Histopathology 74, 463–473 (2019).
Morishita, A. et al. HMGA2 is a driver of tumor metastasis. Cancer Res. 73, 4289–4299 (2013).
Malinauskas, T., Aricescu, A. R., Lu, W., Siebold, C. & Jones, E. Y. Modular mechanism of Wnt signaling inhibition by Wnt inhibitory factor 1. Nat. Struct. Mol. Biol. 18, 886–893 (2011).
Hsieh, J. C. et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature 398, 431–436 (1999).
Mayr, C., Hemann, M. T. & Bartel, D. P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315, 1576–1579 (2007).
Astrom, A. K. et al. Conserved mechanism of PLAG1 activation in salivary gland tumors with and without chromosome 8q12 abnormalities: identification of SII as a new fusion partner gene. Cancer Res. 59, 918–923 (1999).
Conkright, M. D. et al. TORCs: transducers of regulated CREB activity. Mol. Cell 12, 413–423 (2003).
Kitagawa, M. Notch signalling in the nucleus: roles of Mastermind-like (MAML) transcriptional coactivators. J. Biochem. 159, 287–294 (2016).
Komiya, T. et al. Sustained expression of Mect1-Maml2 is essential for tumor cell growth in salivary gland cancers carrying the t(11;19) translocation. Oncogene 25, 6128–6132 (2006).
Piscuoglio, S. et al. Genomic and transcriptomic heterogeneity in metaplastic carcinomas of the breast. NPJ Breast Cancer 3, 48 (2017).
Weigelt, B. et al. Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected]. Mod. Pathol. 28, 340–351 (2015).
WHO Classification of Tumors Editorial Board. Breast Tumours. WHO Classification of Tumors. 5th edn (IARC, Lyon, 2019).
Weinreb, I. et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat. Genet. 46, 1166–1169 (2014).
Pareja, F. et al. Loss-of-function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat. Commun. 9, 3533 (2018).
Zhang, J. et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 26, 108–118 (2016).
McPherson, A. et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 7, e1001138 (2011).
Edgren, H. et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 12, R6 (2011).
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Shugay, M., Ortiz de Mendibil, I., Vizmanos, J. L. & Novo, F. J. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 29, 2539–2546 (2013).
Pareja, F. et al. Pleomorphic adenomas and mucoepidermoid carcinomas of the breast are underpinned by fusion genes. figshare. https://doi.org/10.6084/m9.figshare.12199781 (2020).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP257886 (2020).
Zheng, Z. et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 20, 1479–1484 (2014).
Arias-Stella, J. A. 3rd et al. Novel PLAG1 gene rearrangement distinguishes a subset of uterine myxoid leiomyosarcoma from other uterine myxoid mesenchymal tumors. Am. J. Surg. Pathol. 43, 382–388 (2019).
Acknowledgements
This study was partially funded by the Breast Cancer Research Foundation. Research reported in this publication was partly funded by a Cancer Center Support Grant of the National Institutes of Health (NIH)/National Cancer Institute (grant No P30CA008748). F.P. is partially funded by an NIH K12 CA184746 grant, and B.W. is partially funded by a Cycle for Survival grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
J.S.R.-F. and B.W. conceived the study. H.Y.W., Z.V., E.B., and E.A.R. contributed with cases. F.P., M.V., F.C.G., and J.R.S.-F. reviewed the cases. F.P., A.D.C.P., R.G.-M., A.L., F.C.G., E.M.dS., and G.N. analyzed and interpreted the data. F.P. wrote the first manuscript, which was reviewed by all coauthors.
Corresponding authors
Ethics declarations
Competing interests
J.S.R.-F. reports receiving personal/consultancy fees from Goldman Sachs and REPARE Therapeutics, membership of the scientific advisory boards of VolitionRx and Page.AI, and ad hoc membership of the scientific advisory boards of Roche Tissue Diagnostics, Ventana Medical Systems, Novartis, Genentech, and InVicro, outside the scope of this study. All other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pareja, F., Da Cruz Paula, A., Gularte-Mérida, R. et al. Pleomorphic adenomas and mucoepidermoid carcinomas of the breast are underpinned by fusion genes. npj Breast Cancer 6, 20 (2020). https://doi.org/10.1038/s41523-020-0164-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-020-0164-0
This article is cited by
-
Genomics and tumor microenvironment of breast mucoepidermoid carcinoma based on whole-exome and RNA sequencing
Diagnostic Pathology (2024)
-
Molecular pathology in breast disease: diagnostic, prognostic, and therapeutic tools
Virchows Archiv (2024)
-
High-grade HER2-positive mucoepidermoid carcinoma of the breast: a case report and review of the literature
Journal of Medical Case Reports (2023)
-
Clinical-pathologic characteristics and response to neoadjuvant chemotherapy in triple-negative low Ki-67 proliferation (TNLP) breast cancers
npj Breast Cancer (2022)
-
Triple-negative breast carcinomas of low malignant potential: review on diagnostic criteria and differential diagnoses
Virchows Archiv (2022)
