
Abstract
In principle, porous physisorbents are attractive candidates for the removal of volatile organic compounds such as benzene by virtue of their low energy for the capture and release of this pollutant. Unfortunately, many physisorbents exhibit weak sorbate–sorbent interactions, resulting in poor selectivity and low uptake when volatile organic compounds are present at trace concentrations. Herein, we report that a family of double-walled metal–dipyrazolate frameworks, BUT-53 to BUT-58, exhibit benzene uptakes at 298 K of 2.47–3.28 mmol g−1 at <10 Pa. Breakthrough experiments revealed that BUT-55, a supramolecular isomer of the metal–organic framework Co(BDP) (H2BDP = 1,4-di(1H-pyrazol-4-yl)benzene), captures trace levels of benzene, producing an air stream with benzene content below acceptable limits. Furthermore, BUT-55 can be regenerated with mild heating. Insight into the performance of BUT-55 comes from the crystal structure of the benzene-loaded phase (C6H6@BUT-55) and density functional theory calculations, which reveal that C–H···X interactions drive the tight binding of benzene. Our results demonstrate that BUT-55 is a recyclable physisorbent that exhibits high affinity and adsorption capacity towards benzene, making it a candidate for environmental remediation of benzene-contaminated gas mixtures.
Similar content being viewed by others

Main
Volatile organic compounds (VOCs) such as benzene are a class of toxic pollutants responsible for both indoor and outdoor air pollution that cause environmental and health issues even at trace concentrations1,2,3,4. Current methods to remove benzene from indoor air include plasma oxidation, photocatalytic oxidation and adsorption by activated carbons5. Oxidation is used to degrade benzene, but secondary pollutants (CO, NOx and O3) are simultaneously generated5. Adsorptive removal of VOCs from polluted air and industrial effluent streams by physisorbents is attractive because of their relatively low recycling energy6,7,8,9,10,11. However, the non-covalent binding forces that typically drive physisorption mean that binding tends to be weak for VOCs, especially at low partial pressure10,11,12.
Metal–organic frameworks (MOFs) are of interest for gas storage and separation as they can offer extra-large surface areas and tunable structures13,14,15,16, and have been studied for the removal of VOCs17,18,19,20,21,22,23,24,25,26. Indeed, some MOFs outperform commercial sorbents such as activated carbon fibres, activated carbon (AC) and zeolites in benzene adsorption6,7,21,22,27. For example, the benzene uptake by MOF-177 (Basolite Z377)22 is 16.8 mmol g−1 at 293 K and relative pressure (P/P0) of 1 (compared with 5.6 mmol g−1 by AC9 and 5.8 mmol g−1 by graphitized biocarbon6). Although there is clear potential for MOFs to capture VOCs for environmental remediation and personal protection, aromatic VOCs such as benzene tend to form weak electrostatic, hydrogen-bonding or coordination-bonding interactions with MOFs, thereby affording poor separation performance. The capacity of MOFs at high pressures (for example,10 kPa) can be high22,23, but this is not a good metric to assess the sorption performance of trace VOCs in air. Several MOFs with high benzene capacity have been reported to perform poorly in simulated air purification in breakthrough experiments21. Another concern with respect to MOFs is that stability is a prerequisite for practical utility28,29,30,31, but many MOFs do not possess good thermal or hydrolytic stability31,32,33,34,35. In addition, humidity can also reduce sorption performance21,32,33. Here, we report a family of stable MOF adsorbents, guided by crystal engineering, that efficiently remove benzene from air even at parts-per-million levels. These metal–dipyrazolate frameworks, designated here as BUT-53 to BUT-58 (BUT = Beijing University of Technology), are constructed from Co2+ or Zn2+ ions and ditopic pyrazolate (dipyrazolate) ligands and exhibit similar framework structures but different pore size and chemistry.
Synthesis and crystal structures
Solvothermal reactions between dipyrazolate ligands (Fig. 1) and Co2+ or Zn2+ salts in N,N-dimethylformamide (DMF) and H2O were optimized, in some cases with acetic acid as a modulator, to yield single crystals of BUT-53 to BUT-58 (Supplementary Fig. 1). Single-crystal X-ray diffraction (SCXRD) revealed that BUT-53 and BUT-54 adopt space group I41/amd, BUT-55, BUT-56 and BUT-58 space group I4122, and BUT-57 space group I2 (Supplementary Figs. 2–7 and Supplementary Table 1).

Schematic representation of the double-walled metal–dipyrazolate frameworks with angular (BUT-53, BUT-54) and linear (BUT-55 to BUT-58) dipyrazolate ligands and Co2+- and Zn2+-based RBBs.
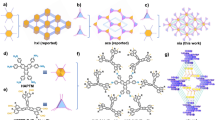
BUT-55 is formed as a three-dimensional (3D) framework structure comprising Co-based rod building blocks (RBBs) and linear BDP2− (H2BDP = 1,4-di(1H-pyrazol-4-yl)benzene) linkers (Fig. 1 and Supplementary Fig. 4). The Co2+ ions exhibit tetrahedral coordination geometry with pyrazolate (pz) groups from four ligands. Alternating Co2+ ions and pz groups generate one-dimensional (1D) RBBs of formula Co(pz)2 (Fig. 2a). Each BDP2− ligand links two adjacent Co(pz)2 moieties. The central benzene and peripheral pyrazolate rings of BDP2− show some bending (Fig. 2b). Adjacent BDP2− ligands pack in criss-cross mode with the central benzene rings eclipsed, resulting in a double-walled network (Fig. 2c). The term ‘double-walled’ is used here as pairs of pz moieties link adjacent metal cations to form undulating RBBs that are cross-linked by pairs of BDP2− ligands (Fig. 2a–c). This contrasts with the supramolecular isomeric structure36 Co(BDP), in which more linear RBBs are cross-linked by BDP2− ligands to form a single-walled arrangement (Fig. 2d–f). BUT-55 exhibits 1D rectangular pores (wall-to-wall distance between ligands ~3.7 Å) and square channels (width ~8.0 Å). These pores, which are key to sorption, lie along the c axis (Supplementary Fig. 4). BUT-58 is the Zn2+ analogue of BUT-55 (Supplementary Fig. 7).

a–c, Zigzag RBB (a), criss-cross stacked BDP2− ligands (b) and double-walled 3D framework structure (c) of BUT-55 and BUT-58. d–f, Chain-like RBB (d), bridging BDP2− ligand (e) and single-walled 3D framework structure (f) of Co(BDP) and Zn(BDP).
The use of the BDP2− ligand in MOFs is exemplified by M(BDP) (M = Zn2+, Ni2+ and Co2+), Fe2(BDP)3 and Ni8(OH)4(H2O)2(BDP)6 reported by the groups of Long and Navarro36,37,38,39. Co(BDP) features structural flexibility and methane storage properties36,40, and Zn(BDP) is its Zn(II) analogue39. BUT-55/BUT-58 and Co(BDP)/Zn(BDP) have the same chemical composition and are supramolecular isomers41 but with different pore structure and chemistry as well as sorption performance.
BUT-56 was obtained by replacing the BDP2− ligand in BUT-55 by DPP2− (H2DPP = 2,5-di(1H-pyrazol-4-yl)pyridine). DPP2− generated pores with a width of 8.0 Å and a wall-to-wall distance between ligands of ~3.8 Å (Supplementary Fig. 5). The longer pyrene-centred PDP2− analogue (H2PDP = 2,7-di(1H-pyrazol-4-yl)pyrene) afforded BUT-57 with enlarged rhombic pores (width ~11.5 Å and wall-to-wall distance ~3.9 Å; Supplementary Fig. 6). The replacement of the dipyrazolate ligands by the angular variants DPB2− (H2DPB = 1,3-di(1H-pyrazol-4-yl)benzene) and DPN2− (H2DPN = 2,7-di(1H-pyrazol-4-yl)naphthalene) afforded BUT-53 and BUT-54. In contrast to the cross-wise packed ligands in BUT-55 to BUT-58, those in BUT-53 and BUT-54 adopt a nose-to-nose arrangement with the central aromatic rings eclipsed (Fig. 1). BUT-54 has larger pores (width ~10.0 Å and wall-to-wall distance ~3.7 Å; Supplementary Fig. 3) than BUT-53 (width ~7.8 Å and wall-to-wall distance ~3.0 Å; Supplementary Fig. 2) as naphthalene is larger than benzene. Ligand substitution did not change the double-walled framework structure but impacted pore size and chemistry as well as benzene adsorption.
Porosity and stability
Supplementary Figs. 8a–13a illustrate that the experimental powder X-ray diffraction (PXRD) patterns of as-synthesized BUT-53 to BUT-58 match their simulated patterns calculated from the SCXRD data, indicating bulk purity. The single-walled Co(BDP) and Zn(BDP) were synthesized and analysed in this study for comparison with the BUT MOFs39,40. The PXRD patterns of Co(BDP) and Zn(BDP) differ from those of BUT-55 and BUT-58, whicn also support phase purity of the BUT MOFs (Supplementary Figs. 14 and 15). N2 adsorption measurements were performed at 77 K to evaluate the porosity of BUT-53 to BUT-58 (Supplementary Fig. 16). Type I adsorption isotherms revealed N2 uptakes of 275, 394, 305, 313, 350 and 279 cm3 g−1 for BUT-53 to BUT-58 at 1 atm, respectively, indicating permanent microporosity. The Brunauer−Emmett−Teller (BET) surface areas determined from the N2 adsorption isotherms were 811, 1,128, 873, 897, 970 and 849 m2 g−1 for BUT-53 to BUT-58, respectively. The pore size distributions, as calculated by density functional theory (DFT), were 8.1, 9.8, 8.4, 8.4, 12.0 and 8.4 Å for BUT-53 to BUT-58, respectively (Supplementary Fig. 17). These results are consistent with the SCXRD structures when van der Waals contacts are considered.
That a MOF must be stable in water, ideally under both acidic and basic aqueous conditions, matters for practical utility. BUT-53 to BUT-58 were thus investigated (Supplementary Figs. 8–13) by treating them with boiling water, aqueous HCl (pH 5) and aqueous NaOH (pH 14) at room temperature for 24 h. BUT-53 to BUT-56 and BUT-58 exhibited phase stability as the PXRD patterns matched their as-synthesized patterns (Supplementary Figs. 8a–11a and 13a). The immersion of BUT-57 in aqueous NaOH (pH 10) for 24 h resulted in reduced PXRD intensity (Supplementary Fig. 12a), implying relatively poor base stability. BUT-53 to BUT-57 retained structural integrity upon exposure to air for 1 year. The N2 sorption isotherms of BUT-53 to BUT-58 were also measured after treatment and confirmed that pore collapse/blockage had not occurred (Supplementary Figs. 8b–13b). Thermogravimetric analysis (TGA) of BUT-53 to BUT-58 revealed stability to 408, 363, 371, 387, 395 and 432 °C, respectively (Supplementary Fig. 18).
Benzene adsorption and capture
BUT-53 to BUT-58 samples were examined for their benzene adsorption properties. Single-component benzene adsorption isotherms collected at 298 K for various equilibration times indicated that a long equilibration time of 1,000 s (with the pressure change less than 0.1% of the average pressure during this interval) was required to reach equilibrium at low pressures (Fig. 3a,b and Supplementary Figs. 19–21). The resulting isotherms revealed that benzene uptake by BUT-53 to BUT-56 and BUT-58 sharply increased at low pressure before gradually increasing at higher pressures (Fig. 3a,b). For BUT-57, two sharp steps were observed: one at low pressure, the other at P/P0 = 0.4 (Fig. 3a). The second step suggests structural flexibility triggered by benzene, a feature of a growing number of MOFs, including Co(BDP). At low benzene pressure, the following adsorption uptakes were observed: 2.47 mmol g−1 at 5.0 Pa for BUT-53, 2.61 mmol g−1 at 9.9 Pa for BUT-54, 3.28 mmol g−1 at 7.3 Pa for BUT-55, 2.98 mmol g−1 at 6.9 Pa for BUT-56, 3.01 mmol g−1 at 9.1 Pa for BUT-57 and 3.09 mmol g−1 at 9.5 Pa for BUT-58 (Fig. 3b).

a, Benzene adsorption isotherms of BUT-53 to BUT-58 at 298 K. b, Logarithmic-scale plots of P/P0 to view the benzene adsorption of BUT-53 to BUT-58 at low partial pressure. c, Benzene uptakes of BUT-53 to BUT-58 compared with representative porous sorbents at low relative pressure. d, Benzene breakthrough curves for BUT-55 and Co(BDP) recorded at different relative humidities at 298 K.
The steps observed in the isotherms of BUT-55 and BUT-58 in the P/P0 range 0.0001–0.001 could indicate a structural change triggered by benzene loading (Fig. 3b). However, in situ PXRD patterns collected from activated BUT-55 at various benzene pressures and temperatures (Fig. 4 and Supplementary Fig. 22) suggest a rigid structure. Therefore, although steps are seen in the isotherms of flexible MOF structures42, we attribute the steps observed here for BUT-55 and BUT-58 to pore filling. BUT-53 to BUT-58 outperform previously studied benzene adsorbents9,18,19,20,21,22,23,24,25,27,39,43,44,45 such as MIL-101(Cr) (0.5 mmol g−1 at 4.9 Pa)18, HKUST-1 (1.0 mmol g−1 at 50.0 Pa)43, PAF-1 (3.30 mmol g−1 at 75.0 Pa)21, BUT-66 (2.83 mmol g−1 at 58.0 Pa)21 and Carboxen 1000 (2.80 mmol g−1 at 53.0 Pa21; Fig. 3c). BUT-53 started to adsorb benzene at the lowest pressure (0.53 Pa), but its uptake of 2.47 mmol g−1 was relatively low, and although BUT-54 exhibited the highest uptake, 4.36 mmol g−1 at 151 Pa, this was achieved at the highest loading pressure. Importantly, BUT-53 to BUT-58 each exhibited strong uptake (>2 mmol g−1) of benzene below 10 Pa (Fig. 3b). Overall, the stability and high uptake of BUT-53 to BUT-58 at low pressure make them candidates for the trace removal of benzene from air.

PXRD patterns collected under benzene pressure from 0 to 8.53 kPa (guest-free to benzene-loaded BUT-55, with structures shown on the right) and after desorption at 120 °C. The data indicate that no structural transformation occurs during the loading and unloading of benzene. The vertical lines shown below the two calculated PXRD patterns are Bragg positions based on the corresponding single crystal structures.
Practically, benzene capture from air will occur in the presence of humidity, which may impact the performance of adsorbents by the co-adsorption of water vapour. Water vapour adsorption experiments at 298 K were performed to assess the hydrophobicity of the MOFs. Supplementary Fig. 23 reveals that each sorbent exhibited S-shaped water adsorption isotherms at >50% relative humidity (RH), except for BUT-56, which exhibited a gradual uptake from 30% RH. That BUT-56 is the most hydrophilic variant is attributed to the pyridyl moiety. For BUT-53 to BUT-55, BUT-57 and BUT-58, a step at P/P0 ≥ 0.6 and hysteresis were observed, suggesting that, except for BUT-56, the BUT family excludes water below 60% RH.
To evaluate the performance of BUT-53 to BUT-58 for capturing trace benzene from ambient air we conducted dynamic gas breakthrough experiments (Supplementary Fig. 24). An air mixture containing trace benzene (10 ppm) was passed through a column packed with ~10 mg of BUT sorbent at a gas flow rate of 10 ml min−1. Control experiments were also performed at RH 50 and 80% at 298 K (Supplementary Figs. 25–29). Supplementary Fig. 25 reveals that benzene broke through the BUT-53 column after ~57 h (5,700 h g−1) for dry gas, a benzene capture capacity of ~1.53 mmol g−1. BUT-53 exhibited reduced capacity (4,600 h g−1 and 1.23 mmol g−1) at RH 50%, and further reduced capacity (2,200 h g−1 and 0.59 mmol g−1) at RH 80%. BUT-54 exhibited a breakthrough time of 3,000 h g−1 (0.80 mmol g−1) under dry conditions and 1,500 h g−1 (0.40 mmol g−1) at RH 50 and 80% (Supplementary Fig. 26). A breakthrough time of 80 h (8,000 h g−1) and a benzene capacity of 2.14 mmol g−1 were recorded for BUT-55 under dry conditions and 50% RH (Fig. 3d), with a reduced adsorption capacity (6,000 h g−1 and 1.61 mmol g−1) at 80% RH. BUT-56 demonstrated the same capacity as BUT-55 under dry conditions, but was more affected by humidity (4,600 h g−1 and 1.23 mmol g−1 at 50% RH, and 2,700 h g−1 and 0.72 mmol g−1 at 80% RH; Supplementary Fig. 27). BUT-57 broke through after 40 h (4,000 h g−1 and 1.07 mmol g−1) under both dry conditions and RH 50%, whereas at 80% RH it occurred at 23 h (2,300 h g−1 and 0.62 mmol g−1; Supplementary Fig. 28). BUT-58 behaved similarly to BUT-55 in the breakthrough experiments (7,900 h g−1 and 2.11 mmol g−1 for dry gas, 7,500 h g−1 and 2.01 mmol g−1 at 50% RH, and 5,000 h g−1 and 1.34 mmol g−1 at 80% RH; Supplementary Fig. 29).
Compared with the results obtained under dry conditions, decreased breakthrough times at RH 80% were observed for BUT-53, BUT-54, BUT-56 and BUT-57 (Supplementary Figs. 25–28). However, BUT-55 (8,000 h g−1 and 2.14 mmol g−1 under dry conditions compared with 6,000 h g−1 and 1.61 mmol g−1 at RH 80%; Fig. 3d) and BUT-58 (7,500 h g−1 and 2.01 mmol g−1 under dry conditions compared with 5,000 h g−1 and 1.34 mmol g−1 at RH 80%; Supplementary Fig. 29) were less affected by humidity and were studied for their benzene adsorption performance after exposure to moisture for 1 week. BUT-55 and BUT-58 exhibited similar isotherms to those obtained from pristine samples (Supplementary Fig. 30).
In addition to humidity, polluted air might contain other VOCs with similar physicochemical properties to benzene, such as cyclohexane and ethanol. BUT-55 exhibited cyclohexane and ethanol uptakes of 2.90 mmol g−1 at 8.4 Pa and 4.10 mmol g−1 at 94 Pa, respectively (Supplementary Figs. 31a,b and 32a,b). The single-component breakthrough curves indicated a cyclohexane adsorption capacity of 1.60 mmol g−1 but little ethanol uptake at 10 ppm (Supplementary Figs. 31c and 32c). In competition tests with benzene (1:1, v/v), the adsorption capacity of cyclohexane decreased to 1.28 mmol g−1, and ethanol broke through immediately (Supplementary Figs. 31d and 32d). Benzene adsorption capacity decreased slightly in the presence of cyclohexane (1.60 mmol g−1) and ethanol (1.60 mmol g−1). These results indicate that the tested VOCs have a minor effect upon the benzene adsorption performance of BUT-55.
The residual benzene in effluent streams flowing through a BUT-55 packed bed was determined to be 2.82 ng l−1 (2.82 ng l−1 = 2.82 μg m−3) by gas chromatography mass spectrometry (GC–MS; Supplementary Figs. 33 and 34 and Supplementary Table 2). These data indicate benzene removal to a level below indoor limits (3–100 μg m−3) set by most countries/regions, including China, the European Union and the United States4,46,47. Recycling breakthrough tests revealed that BUT-55 showed no loss of benzene capture capacity after five cycles (Supplementary Fig. 35), and adsorbed benzene molecules could be removed at 120 °C within 2 h (Supplementary Fig. 36).
The contrast between Co(BDP)/Zn(BDP) and BUT-55/BUT-58 was addressed by preparing Co(BDP)/Zn(BDP) and activating them following the reported approach39,40. Their PXRD patterns and N2 adsorption isotherms matched those in previous reports (Supplementary Figs. 14 and 15). Single-component benzene vapour adsorption and dynamic breakthrough experiments revealed uptake of 1.8 mmol g−1 at 32 Pa (Supplementary Fig. 37) and a benzene capture capacity of 0.17 mmol g−1 for Co(BDP) (Fig. 3d), whereas Zn(BDP) exhibited an uptake of 1.3 mmol g−1 at 1.0 Pa (Supplementary Fig. 38) and a benzene capture capacity of 0.17 mmol g−1 (Supplementary Fig. 29). These values are much lower than those obtained for BUT-55 and BUT-58.
Benzene binding sites
To gain insight into the benzene capture performance of the BUT family we determined the single-crystal structures of several guest-loaded phases, including benzene-loaded C6H6@BUT-53, C6H6@BUT-54, C6H6@BUT-55 and C6H6@BUT-58, water-loaded H2O@BUT-55, and benzene- and water-loaded C6H6/H2O@BUT-55. The crystal structure of C6H6@BUT-55 reveals that the framework was little changed after the capture of benzene, and two types of adsorbed benzene molecules were identified (Fig. 5). One (yellow) resides in the cavity between two neighbouring pairs of BDP2− ligands and two Co(pz)2 RBBs (site I; Fig. 5a,b). This benzene molecule binds to surrounding BDP2− ligands through multiple C–H···X interactions (distances 2.89–3.58 Å): to pyrazolate C–H moieties through C–H(L)···π(B) interactions (L represents BDP2− ligands and B represents benzene guest molecules), to pyrazolate π-electrons through C–H(B)···π(L) interactions and to pyrazolate nitrogen atoms through C–H(B)···N(L) hydrogen bonds (Fig. 5b and Supplementary Table 3)48. The other benzene molecule (red) lies over a pair of packed BDP2− ligands (site II; Fig. 5a,d), and also exhibits multiple C–H···π interactions with adjacent BDP2− ligands (distances 2.78–3.88 Å; Fig. 5d and Supplementary Table 3). In the square unit defined by four rectangular pores and four Co(pz)2 RBBs, the criss-cross packed BDP2− ligand pairs are arranged along the channel walls in a helical mode. Consequently, these (red) benzene molecules form a helical chain. Guest–guest binding occurs through C–H(B)···π(B) interactions (Fig. 5e). Collectively, this plethora of host–guest and guest–guest C–H···X interactions drive the adsorption of benzene by the host framework. The single-crystal structure of C6H6@BUT-55 suggests that benzene uptake in BUT-55 should be 3.43 mmol g−1, consistent with the uptake observed in the adsorption isotherms (3.62 mmol g−1 at P/P0 = 1; Fig. 3a,b).

a, Adsorbed benzene molecules lie in pores, as viewed along the c axis. b,d, Binding sites of adsorbed benzene molecules at site I (b) and site II (d), with C–H···π interactions presented in blue and C–H···N interactions in pink. e, Benzene–benzene interactions occur between the two types of adsorbed benzene molecules. c,f, Isosurfaces of the charge density difference for the interactions of site I (c) and site II (f) with adsorbed benzene molecules in BUT-55. Yellow indicates increased electronic density and light blue indicates depletion.
To further understand the observed adsorption behaviour, DFT calculations were conducted on the basis of the experimental single-crystal structures. Isosurfaces of charge density differences before and after benzene loading in BUT-55 are shown in Fig. 5c,f and Supplementary Fig. 39. Figure 5c,f reveals that benzene molecules interact more strongly in site I through electron accumulation on benzene and depletion on the ligand. Preferential benzene adsorption at site I is supported by a higher calculated binding energy of −110.06 kJ mol−1 (−72.99 kJ mol−1 at site II). At lower pressures of benzene, benzene molecules may first bind to BUT-55 at site I. Similar results were obtained for BUT-53, BUT-54 and BUT-58 (Supplementary Table 3 and Supplementary Figs. 40–42). That BUT-53 was calculated to exhibit the strongest benzene binding at site I agrees with its experimental adsorption profile at ultralow pressure (that is, the first few data points in Fig. 3b and Supplementary Fig. 43a). However, the isotherm for BUT-55 crosses that of BUT-53 at low pressure and offers better benzene sorption performance than BUT-53 at higher loading (Supplementary Fig. 43a and Supplementary Section 7). The performance of BUT-55 could be attributed to slightly faster kinetics of loading compared with BUT-53 (Supplementary Fig. 43b), but we also note that benzene binding induced a minor structural change, ligand swing, in BUT-53 (Supplementary Fig. 43c).
The impact of moisture on the benzene adsorption ability of BUT-55 was addressed by analysis of both the single-crystal structural data of H2O@BUT-55 and C6H6/H2O@BUT-55 and calculated data (Supplementary Fig. 44). In the single-crystal structure of H2O@BUT-55, water molecules are located at site I (Supplementary Fig. 44a,b), but no water molecules are observed at site II because of unresolvable crystallographic disorder. In the structure of C6H6/H2O@BUT-55, water molecules are located at site I and benzene molecules are found at site II (Supplementary Fig. 44d). We also calculated the energy of interaction between BUT-55 and water molecules. DFT calculations indicate the binding energy for water molecules to be −31.51 to −43.52 kJ mol−1, much lower than for benzene (−110.06 kJ mol−1 at site I and −72.99 kJ mol−1 at site II). Preferential benzene binding in the co-adsorption of benzene and moisture would therefore be expected in breakthrough experiments performed under humid conditions. These results provide insight as to why moisture has a relatively small effect on the benzene adsorption performance of BUT-55 (Fig. 3d).
In conclusion, a family of double-walled pyrazolate MOFs, BUT-53 to BUT-58, capture trace benzene from air at 298 K owing to benzene-selective binding sites resulting from a plethora of weak non-covalent interactions, including C–H···X interactions. BUT-55 is notable because it performs well even under humid conditions, which we attribute to its relatively low affinity for water molecules. BUT-55 can therefore reduce trace benzene concentrations in air to levels below acceptable limits and, as a physisorbent, requires relatively low energy for recycling. This work further highlights that the tuning of pore chemistry and pore size can afford tight binding sites that enable physisorbents to remove even trace contaminants from air with advantageously long breakthrough times.
Methods
Materials and instruments
All reagents and solvents (AR grade) were purchased commercially and used as received. 1H NMR spectra were recorded on a BRUKER AVANCE III HD 400 MHz spectrometer. PXRD patterns were recorded on a Rigaku Smartlab 3 X-ray powder diffractometer using a Cu sealed tube (λ = 1.54178 Å) at room temperature. In situ variable-pressure PXRD patterns were collected on a Rigaku Smart Lab instrument at 40 kV and 40 mA using an Anton Paar TTK 600 accessory. TGA data were obtained on a SHIMADZU TGA-50 thermogravimetric analyser at a heating rate of 10 °C min–1 under air. Gas adsorption–desorption isotherms were obtained using a Micrometrics ASAP 2020 or BEL BELSORP-maxII instrument. Column breakthrough experiments were carried out using a typical breakthrough system consisting of a gas source, adsorption bed and gas analyser. The residual benzene in effluent was determined using a Clarus 600 GC–MS instrument (Perkin Elmer) coupled with a Turbomatrix350 thermal detector (Perkin Elmer).
Synthesis of ligands
The synthetic procedures for the ligands H2DPB, H2DPN, H2BDP, H2DPP and H2PDP are described in detail in Supplementary Section 1 and Supplementary Figs. 45–54).
Synthesis of BUT-53 (CoDPB)
H2DPB (0.14 mmol, 30 mg) and Co(OAc)2·4H2O (0.24 mmol, 60 mg) were dissolved under ultrasound in 10 ml DMF in a 20-ml glass vial. Next, 0.10 ml acetic acid and 8.0 ml deionized water were added to the mixture. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting suspension was heated in a traditional oven at 150 °C for 12 h. After cooling naturally to room temperature, bluish violet crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (15 mg of activated sample, 49% yield based on the H2DPB ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Synthesis of BUT-54 (CoDPN)
H2DPN (0.090 mmol, 24 mg) and Co(OAc)2·4H2O (0.16 mmol, 40 mg) were dissolved under ultrasound in 8.0 ml DMF in a 20-ml glass vial. Next, 0.080 ml acetic acid and 2.4 ml deionized water were added to the mixture. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting suspension was heated in a traditional oven at 150 °C for 24 h. After cooling naturally to room temperature, wine-red crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (19 mg of activated sample, 65% yield based on the H2DPN ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Synthesis of BUT-55 (CoBDP)
H2BDP (0.11 mmol, 24 mg) and Co(OAc)2·4H2O (0.19 mmol, 48 mg) were dissolved under ultrasound in 8.0 ml DMF in a 20-ml glass vial. Next, 4.0 ml deionized water was added to the mixture. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting suspension was heated in a traditional oven at 150 °C for 12 h. After cooling naturally to room temperature, wine-red crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (23 mg of activated sample, 75% yield based on the H2BDP ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Synthesis of BUT-56 (CoDPP)
H2DPP (0.11 mmol, 24 mg) and Co(OAc)2·4H2O (0.19 mmol, 48 mg) were dissolved under ultrasound in 8.0 ml DMF in a 20-ml glass vial. Next, 4.0 ml deionized water was added to the mixture. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting suspension was heated in a traditional oven at 150 °C for 12 h. After cooling naturally to room temperature, wine-red crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (22 mg of activated sample, 72% yield based on the H2DPP ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Synthesis of BUT-57 (CoPDP)
H2PDP (0.090 mmol, 30 mg) and Co(OAc)2·4H2O (0.24 mmol, 60 mg) were dissolved under ultrasound in 10 ml DMF in a 20-ml glass vial. Next, 0.050 ml acetic acid and 1.5 ml deionized water were added to the mixture. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting suspension was heated in a traditional oven at 120 °C for 12 h. After cooling naturally to room temperature, wine-red crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (21 mg of activated sample, 60% yield based on the H2PDP ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Synthesis of BUT-58 (ZnBDP)
H2BDP (0.090 mmol, 30 mg) and Zn(NO3)2·6H2O (0.24 mmol, 60 mg) were dissolved under ultrasound in 10 ml DMF in a 20-ml glass vial. Next, 7.0 ml deionized water was added to the mixture. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting suspension was heated in a traditional oven at 100 °C for 6 h. After cooling naturally to room temperature, colourless crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (17 mg of activated sample, 44% yield based on the H2BDP ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Synthesis of Co(BDP)
H2BDP (0.14 mmol, 30 mg) and Co(NO3)2·4H2O (0.23 mmol, 60 mg) were dissolved under ultrasound in 10 ml DMF in a 20-ml glass vial. Next, 2.0 ml deionized water was added to the solution. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting solution was heated in a traditional oven at 130 °C for 12 h. After cooling naturally to room temperature, blue crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (18 mg of activated sample, 47% yield based on the H2BDP ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Synthesis of Zn(BDP)
H2BDP (0.14 mmol, 30 mg) and Zn(NO3)2·6H2O (0.23 mmol, 60 mg) were dissolved under ultrasound in 10 ml DMF in a 20-ml glass vial. Next, 2.0 ml deionized water was added to the solution. The vial was then tightly sealed and the mixture sonicated for another 15 min. The resulting solution was heated in a traditional oven at 80 °C for 6 h. After cooling naturally to room temperature, colourless crystals were filtered off and washed with DMF (2 × 15 ml) and acetone (9 × 15 ml) with short cycles of ultrasonic treatment to remove amorphous solids. The crystals (18 mg of activated sample, 46% yield based on the H2BDP ligand) were collected by filtration and dried under vacuum at 100 °C for 5 h.
Single-crystal X-ray diffraction
The diffraction data of as-synthesized BUT-53 to BUT-58 samples and guest-loaded phases were collected on a Rigaku Supernova CCD diffractometer (mirror monochromator, Cu Kα source, λ = 1.54184 Å).
To obtain single crystals of the benzene-loaded phases, open vials each containing an activated BUT-53 to BUT-58 sample were loaded into a bottle containing an open vial of benzene and the bottle was then sealed. H2O@BUT-55 was obtained by loading activated BUT-55 in an open vial into a bottle containing an open vial of water and the bottle was sealed. For the C6H6/H2O@BUT-55 phase, an open vial containing activated BUT-55 was loaded into a bottle containing open vials of both water and benzene, and the bottle was then sealed. After 24 h, single crystals of C6H6@BUT-53, C6H6@BUT-54, C6H6@BUT-55, C6H6@BUT-58, H2O@BUT-55 and C6H6/H2O@BUT-55 were harvested and studied by SCXRD. Both H2O@BUT-55 and C6H6/H2O@BUT-55 were obtained under 100% RH. The specific binding sites of benzene could hardly be determined in the single-crystal structure of C6H6@BUT-56, and single crystals of C6H6@BUT-57 were of insufficient quality to be characterized by SCXRD.
Sample activation and gas adsorption
Sample activation and gas adsorption measurements were performed according to ref. 49 and the details are reproduced here for completeness. Prior to the gas adsorption tests, samples of BUT-53 to BUT-58 (~80 mg of each) were immersed in 15 ml DMF at 60 °C for 24 h; the solvent was exchanged for fresh DMF after 12 h. The samples were then harvested carefully by decanting and next soaked in 15 ml acetone at room temperature for another 72 h, during which time the solvent was exchanged for fresh acetone three times a day. After this time, the samples were loaded into a sample tube and degassed under high vacuum at an optimal temperature of 100 °C for 5 h.
N2 and water vapour adsorption isotherms were collected on a Micromeritics ASAP 2020 surface area and pore analyser, which was customized with all stainless-steel parts inside the manifold. Benzene vapour adsorption isotherms were recorded on a Micromeritics ASAP 2020 or BELSORP-maxII vapour adsorption instrument. The adsorption temperature was controlled using a liquid nitrogen bath (77 K) or a water bath (298 K).
Breakthrough experiments
Breakthrough experiments were performed according to ref. 21 and the details are reproduced here for completeness. A calibration gas (~20 ppm benzene balanced with dry air) and a set-up as shown in Supplementary Fig. 24 were used. Quartz tubes (3.0 mm outer diameter, 1 mm inner diameter and 50 mm length) were packed with the samples. For dry gas breakthrough experiments, a mixture of the calibration gas and dry air (1:1, v/v) was passed through the packed tube at a flow rate of 10 ml min−1. The 50% RH breakthrough experiments were performed similarly to the dry gas experiments except that the dry air flow was presaturated with water vapour by bubbling through deionized water before being mixed with the calibration gas flow. For the 80% RH experiments, a calibration gas containing 50 ppm benzene was used, and the ratio of calibration gas to humid air was set to 1:4 (v/v) with a total flow rate of 10 ml min−1. The breakthrough experiments were all conducted with a feed stream containing 10 ppm benzene. The flow rate of the gases was controlled by a mass flow controller (Alicat). The concentration of benzene in the gases passing through the packed columns was monitored using a Hiden HPR20 mass spectrometer gas analysis system.
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2085589 (BUT-53), 2085590 (BUT-54), 2085591 (BUT-55), 2085592 (BUT-56), 2085593 (BUT-57), 2109769 (BUT-58), 2085594 (C6H6@BUT-55), 2122629 (C6H6@BUT-54), 2122630 (C6H6@BUT-53), 2122631 (C6H6@BUT-58), 2122645 (H2O@BUT-55) and 2122646 (C6H6/H2O@BUT-55). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the article and its Supplementary Information. Source data are provided with this paper.
References
He, C. et al. Recent advances in the catalytic oxidation of volatile organic compounds: a review based on pollutant sorts and sources. Chem. Rev. 119, 4471–4568 (2019).
Poschl, U. & Shiraiwa, M. Multiphase chemistry at the atmosphere–biosphere interface influencing climate and public health in the anthropocene. Chem. Rev. 115, 4440–4475 (2015).
Guo, Y., Wen, M., Li, G. & An, T. Recent advances in VOC elimination by catalytic oxidation technology onto various nanoparticles catalysts: a critical review. Appl. Catal. B 281, 119447 (2021).
World Health Organization WHO Guidelines for Indoor Air Quality: Selected Pollutants (WHO Regional Office for Europe, 2010).
Tsuji, M. et al. Efficient removal of benzene in air at atmospheric pressure using a side-on type 172 nm Xe2 excimer lamp. Environ. Sci. Pollut. Res. 25, 18980–18989 (2018).
Zhu, M., Tong, Z., Zhao, Z., Jiang, Y. & Zhao, Z. A microporous graphitized biocarbon with high adsorption capacity toward benzene volatile organic compounds (VOCs) from humid air at ultralow pressures. Ind. Eng. Chem. Res. 55, 3765–3774 (2016).
Wang, C. et al. Remarkable adsorption performance of MOF-199 derived porous carbons for benzene vapor. Environ. Res. 184, 109323 (2020).
Lv, S. W. et al. Recent advances on porous organic frameworks for the adsorptive removal of hazardous materials. J. Environ. Sci. 80, 169–185 (2019).
Baytar, O., Sahin, O., Horoz, S. & Kutluay, S. High-performance gas-phase adsorption of benzene and toluene on activated carbon: response surface optimization, reusability, equilibrium, kinetic, and competitive adsorption studies. Environ. Sci. Pollut. Res. 27, 26191–26210 (2020).
Zhang, D., Liu, J., Liu, M., Liu, L. & Do, D. D. On the capture of ultralow-level benzene in indoor environments: experiments, modeling and molecular simulation. Sep. Purif. Technol. 251, 117306 (2020).
Szulejko, J. E., Kim, K.-H. & Parise, J. Seeking the most powerful and practical real-world sorbents for gaseous benzene as a representative volatile organic compound based on performance metrics. Sep. Purif. Technol. 212, 980–985 (2019).
Szulejko, J. E. & Kim, K.-H. Is the maximum adsorption capacity obtained at high VOC pressures (>1000 Pa) really meaningful in real-world applications for the sorptive removal of VOCs under ambient conditions (<1 Pa)? Sep. Purif. Technol. 228, 115729 (2019).
Wang, H., Lustig, W. P. & Li, J. Sensing and capture of toxic and hazardous gases and vapors by metal–organic frameworks. Chem. Soc. Rev. 47, 4729–4756 (2018).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 1230444 (2013).
Moosavi, S. M. et al. Understanding the diversity of the metal-organic framework ecosystem. Nat. Commun. 11, 4068 (2020).
Lin, R.-B. et al. Molecular sieving of ethylene from ethane using a rigid metal–organic framework. Nat. Mater. 17, 1128–1133 (2018).
Macreadie, L. K. et al. Enhancing multicomponent metal–organic frameworks for low pressure liquid organic hydrogen carrier separations. Angew. Chem. Int. Ed. 59, 6090–6098 (2020).
Jhung, S. H. et al. Microwave synthesis of chromium terephthalate MIL-101 and its benzene sorption ability. Adv. Mater. 19, 121–124 (2007).
Britt, D., Tranchemontagne, D. & Yaghi, O. M. Metal-organic frameworks with high capacity and selectivity for harmful gases. Proc. Natl Acad. Sci. USA 105, 11623–11627 (2008).
Yang, K., Sun, Q., Xue, F. & Lin, D. Adsorption of volatile organic compounds by metal–organic frameworks MIL-101: influence of molecular size and shape. J. Hazard. Mater. 195, 124–131 (2011).
Xie, L.-H., Liu, X.-M., He, T. & Li, J.-R. Metal-organic frameworks for the capture of trace aromatic volatile organic compounds. Chem 4, 1911–1927 (2018).
Gwardiak, S., Szczęśniak, B., Choma, J. & Jaroniec, M. Benzene adsorption on synthesized and commercial metal–organic frameworks. J. Porous Mater. 26, 775–783 (2018).
Zhu, M., Hu, P., Tong, Z., Zhao, Z. & Zhao, Z. Enhanced hydrophobic MIL(Cr) metal-organic framework with high capacity and selectivity for benzene VOCs capture from high humid air. Chem. Eng. J. 313, 1122–1131 (2017).
Belarbi, H., Gonzales, P., Basta, A. & Trens, P. Comparison of the benzene sorption properties of metal organic frameworks: influence of the textural properties. Environ. Sci. Process. Impacts 21, 407–412 (2019).
Macreadie, L. K. et al. CUB-5: a contoured aliphatic pore environment in a cubic framework with potential for benzene separation applications. J. Am. Chem. Soc. 141, 3828–3832 (2019).
Woellner, M. et al. Adsorption and detection of hazardous trace gases by metal–organic frameworks. Adv. Mater. 30, 1704679 (2018).
Veerapandian S. K. P., De Geyter, N., Giraudon, J.-M., Lamonier, J.-F. & Morent, R. The use of zeolites for VOCs abatement by combining non-thermal plasma, adsorption, and/or catalysis: a review. Catalysts 9, 98 (2019).
Choi, H. J., Dincă, M., Dailly, A. & Long, J. R. Hydrogen storage in water-stable metal–organic frameworks incorporating 1,3- and 1,4-benzenedipyrazolate. Energy Environ. Sci. 3, 117–123 (2010).
He, T. et al. Kinetically controlled reticular assembly of a chemically stable mesoporous Ni(II)-pyrazolate metal–organic framework. J. Am. Chem. Soc. 142, 13491–13499 (2020).
Jin, J. K. et al. Building a pyrazole–benzothiadiazole–pyrazole photosensitizer into metal–organic frameworks for photocatalytic aerobic oxidation. J. Am. Chem. Soc. 143, 21340–21349 (2021).
Rieth, A. J., Wright, A. M. & Dincă, M. Kinetic stability of metal–organic frameworks for corrosive and coordinating gas capture. Nat. Rev. Mater. 4, 708–725 (2019).
Liu, X., Wang, X. & Kapteijn, F. Water and metal–organic frameworks: from interaction toward utilization. Chem. Rev. 120, 8303–8377 (2020).
Wang, C., Liu, X., Demir, N. K., Chen, J. P. & Li, K. Applications of water stable metal–organic frameworks. Chem. Soc. Rev. 45, 5107–5134 (2016).
Jayaramulu, K. et al. Hydrophobic metal–organic frameworks. Adv. Mater. 31, 1900820 (2019).
Mukherjee, S. et al. An ultrahydrophobic fluorous metal–organic framework derived recyclable composite as a promising platform to tackle marine oil spills. Chem. Eur. J. 22, 10937–10943 (2016).
Choi, H. J., Dinca, M. & Long, J. R. Broadly hysteretic H2 adsorption in the microporous metal–organic framework Co(1,4-benzenedipyrazolate). J. Am. Chem. Soc. 130, 7848–7850 (2008).
Padial, N. M. et al. Highly hydrophobic isoreticular porous metal–organic frameworks for the capture of harmful volatile organic compounds. Angew. Chem. Int. Ed. 52, 8290–8294 (2013).
Herm, Z. R. et al. Separation of hexane isomers in a metal-organic framework with triangular channels. Science 340, 960–964 (2013).
Galli, S. et al. Adsorption of harmful organic vapors by flexible hydrophobic bis-pyrazolate based MOFs. Chem. Mater. 22, 1664–1672 (2010).
Mason, J. A. et al. Methane storage in flexible metal–organic frameworks with intrinsic thermal management. Nature 527, 357–361 (2015).
Moulton, B. & Zaworotko, M. J. From molecules to crystal engineering: supramolecular isomerism and polymorphism in network solids. Chem. Rev. 101, 1629–1658 (2001).
Tanaka, D. et al. Kinetic gate-opening process in a flexible porous coordination polymer. Angew. Chem. Int. Ed. 47, 3914–3918 (2008).
Zhao, Z. et al. Competitive adsorption and selectivity of benzene and water vapor on the microporous metal organic frameworks (HKUST-1). Chem. Eng. J. 259, 79–89 (2015).
Wang, C.-M., Chang, K.-S., Chung, T.-W. & Wu, H. Adsorption equilibria of aromatic compounds on activated carbon, silica gel, and 13X seolite. J. Chem. Eng. Data 49, 527–531 (2004).
Barea, E., Montoro, C. & Navarro, J. A. R. Toxic gas removal – metal–organic frameworks for the capture and degradation of toxic gases and vapours. Chem. Soc. Rev. 43, 5419–5430 (2014).
Settimo, G., Manigrasso, M. & Avino, P. Indoor air quality: a focus on the European legislation and state-of-the-art research in Italy. Atmosphere 11, 370 (2020).
Sekar, A., Varghese, G. K. & Ravi Varma, M. K. Analysis of benzene air quality standards, monitoring methods and concentrations in indoor and outdoor environment. Heliyon 5, e02918 (2019).
Yang, Y. et al. Ethylene/ethane separation in a stable hydrogen-bonded organic framework through a gating mechanism. Nat. Chem. 13, 933–939 (2021).
He, T. et al. Unique T-shaped ligand as a new platform for metal–organic frameworks. Cryst. Growth Des. 19, 430–436 (2018).
Acknowledgements
J.-R.L. acknowledges support by the National Natural Science Foundation of China (22038001). J.-R.L. and Z.-R.N. acknowledge support by the National Natural Science Foundation of China (51621003). M.J.Z. acknowledges support from Science Foundation Ireland (16/IA/4624) and the European Research Council (ADG 885695). T.H. acknowledges support by the Postdoctoral Science Foundation of Chaoyang District (2020ZZ-8). We also thank Q. Chen, X. Lin, T.-C. Li and L. Tang from Beijing University of Technology and N. Kumar from the University of Limerick for their assistance with in situ PXRD and GC–MS measurements.
Author information
Authors and Affiliations
Contributions
J.-R.L., M.J.Z., Z.-R.N., T.H. and X.-J.K. conceived the research idea and designed the experiments. T.H. and X.-J.K. performed the synthetic experiments. T.H., X.-J.K., Z.-X.B., Y.-Z.Z., G.-R.S., X.-Q.W. and Z.C. conducted the bulk characterizations and gas sorption measurements. T.H., X.-J.K., L.-H.X., M.J.Z., J.-R.L. and H.H. analysed the experimental data. H.H. conducted the DFT calculations. J.-R.L., M.J.Z., Z.-R.N., X.-H.B., T.H. and X.-J.K. discussed, wrote and revised the manuscript. All authors have given their approval to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Materials thanks Jorge Rodriguez Navarro and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary methods, general characterizations and additional tables and figures.
Source data
Source Data Fig. 3
Source data for Fig. 3.
Source Data Fig. 4
Source data for Fig. 4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, T., Kong, XJ., Bian, ZX. et al. Trace removal of benzene vapour using double-walled metal–dipyrazolate frameworks. Nat. Mater. 21, 689–695 (2022). https://doi.org/10.1038/s41563-022-01237-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41563-022-01237-x
This article is cited by
-
Engineering porous crystals to do different things
Nature Materials (2024)
-
Double-walled Al-based MOF with large microporous specific surface area for trace benzene adsorption
Nature Communications (2024)
-
Enhanced carbon capture with motif-rich amino acid loaded defective robust metal-organic frameworks
Nano Research (2024)
-
Incorporation of multiple supramolecular binding sites into a robust MOF for benchmark one-step ethylene purification
Nature Communications (2023)
-
Temperature-dependent rearrangement of gas molecules in ultramicroporous materials for tunable adsorption of CO2 and C2H2
Nature Communications (2023)
