
Abstract
The fetal-to-adult switch in hemoglobin production is a model of developmental gene control with relevance to the treatment of hemoglobinopathies. The expression of transcription factor BCL11A, which represses fetal β-type globin (HBG) genes in adult erythroid cells, is predominantly controlled at the transcriptional level but the underlying mechanism is unclear. We identify HIC2 as a repressor of BCL11A transcription. HIC2 and BCL11A are reciprocally expressed during development. Forced expression of HIC2 in adult erythroid cells inhibits BCL11A transcription and induces HBG expression. HIC2 binds to erythroid BCL11A enhancers to reduce chromatin accessibility and binding of transcription factor GATA1, diminishing enhancer activity and enhancer–promoter contacts. DNA-binding and crystallography studies reveal direct steric hindrance as one mechanism by which HIC2 inhibits GATA1 binding at a critical BCL11A enhancer. Conversely, loss of HIC2 in fetal erythroblasts increases enhancer accessibility, GATA1 binding and BCL11A transcription. HIC2 emerges as an evolutionarily conserved regulator of hemoglobin switching via developmental control of BCL11A.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
The ChIP–seq, RNA-seq, ATAC-seq, CUT&RUN and Capture-C data have been deposited into the Gene Expression Omnibus database (accession no. GSE173587). The X-ray structures of the HIC2 ZF2–5 domain with bound DNA have been submitted to PDB (accession no. 7TXC). Publicly available datasets used in this manuscript: structure of DNA binding by GATA3: PDB, accession no. 4HC9; Pol II ChIP–seq, accession no. GSE36994; RNA-seq of fetal and adult erythroblasts, accession nos. GSE102201 and GSE90878; RNA-seq of erythroid differentiation, accession no. GSE53983; G1E-ER4 GATA1 ChIP–seq, accession no. GSE36029. Source data are provided with this paper.
Code availability
Codes used in the present study were obtained from published pipelines and packages.
Change history
31 July 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41588-023-01488-7
References
Orkin, S. H. Globin gene regulation and switching: circa 1990. Cell 63, 665–672 (1990).
Sankaran, V. G. & Orkin, S. H. The switch from fetal to adult hemoglobin. Cold Spring Harb. Perspect. Med. 3, a011643 (2013).
Huang, P. et al. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 31, 1704–1713 (2017).
Liu, N. et al. Transcription factor competition at the γ-globin promoters controls hemoglobin switching. Nat. Genet. 53, 511–520 (2021).
Xu, J. et al. Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev. Cell 23, 796–811 (2012).
Doerfler, P. A. et al. Activation of γ-globin gene expression by GATA1 and NF-Y in hereditary persistence of fetal hemoglobin. Nat. Genet. 53, 1177–1186 (2021).
Forget, B. G. Molecular basis of hereditary persistence of fetal hemoglobin. Ann. N.Y. Acad. Sci. 850, 38–44 (1998).
Driscoll, M. C., Dobkin, C. S. & Alter, B. P. Gamma delta beta-thalassemia due to a de novo mutation deleting the 5’ beta-globin gene activation-region hypersensitive sites. Proc. Natl Acad. Sci. USA 86, 7470–7474 (1989).
Kioussis, D., Vanin, E., deLange, T., Flavell, R. A. & Grosveld, F. G. β-Globin gene inactivation by DNA translocation in γβ-thalassaemi. Nature 306, 662–666 (1983).
Topfer, S. K. et al. Disrupting the adult globin promoter alleviates promoter competition and reactivates fetal globin gene expression. Blood 139, 2107–2118 (2022).
Menzel, S. et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 39, 1197–1199 (2007).
Lettre, G. et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc. Natl Acad. Sci. USA 105, 11869–11874 (2008).
Martyn, G. E. et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 50, 498–503 (2018).
Liu, N. et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173, 430–442.e17 (2018).
Lee, Y. T. et al. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood 122, 1034–1041 (2013).
Basak, A. et al. Control of human hemoglobin switching by LIN28B-mediated regulation of BCL11A translation. Nat. Genet. 52, 138–145 (2020).
de Vasconcellos, J. F. et al. IGF2BP1 overexpression causes fetal-like hemoglobin expression patterns in cultured human adult erythroblasts. Proc. Natl Acad. Sci. USA 114, E5664–E5672 (2017).
Sankaran, V. G. et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842 (2008).
Lessard, S., Beaudoin, M., Orkin, S. H., Bauer, D. E. & Lettre, G. 14q32 and let-7 microRNAs regulate transcriptional networks in fetal and adult human erythroblasts. Hum. Mol. Genet. 27, 1411–1420 (2018).
Khandros, E. et al. Understanding heterogeneity of fetal hemoglobin induction through comparative analysis of F and A erythroblasts. Blood 135, 1957–1968 (2020).
Zhou, D., Liu, K., Sun, C.-W., Pawlik, K. M. & Townes, T. M. KLF1 regulates BCL11A expression and γ- to β-globin gene switching. Nat. Genet. 42, 742–744 (2010).
Canver, M. C. et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015).
Grevet, J. D. et al. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290 (2018).
Huang, P. et al. The HRI-regulated transcription factor ATF4 activates BCL11A transcription to silence fetal hemoglobin expression. Blood 135, 2121–2132 (2020).
Qin, K. et al. Dual function NFI factors control fetal hemoglobin silencing in adult erythroid cells. Nat. Genet. 54, 874–884 (2022).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Skene, P. J. & Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856 (2017).
Kurita, R. et al. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE 8, e59890 (2013).
Lan, X. et al. ZNF410 uniquely activates the NuRD component CHD4 to silence fetal hemoglobin expression. Mol. Cell 81, 239–254.e8 (2021).
Dykes, I. M. et al. HIC2 Is a novel dosage-dependent regulator of cardiac development located within the distal 22q11 deletion syndrome region. Circ. Res. 115, 23–31 (2014).
Vinjamur, D. S. et al. ZNF410 represses fetal globin by singular control of CHD4. Nat. Genet. 53, 719–728 (2021).
An, X. et al. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 123, 3466–3477 (2014).
Davies, J. O. et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods 13, 74–80 (2016).
Peslak, S. A. et al. HRI depletion cooperates with pharmacologic inducers to elevate fetal hemoglobin and reduce sickle cell formation. Blood Adv. 4, 4560–4572 (2020).
Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013).
Blobel, G. A., Nakajima, T., Eckner, R., Montminy, M. & Orkin, S. H. CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc. Natl Acad. Sci. USA 95, 2061–2066 (1998).
Letting, D. L., Rakowski, C., Weiss, M. J. & Blobel, G. A. Formation of a tissue-specific histone acetylation pattern by the hematopoietic transcription factor GATA-1. Mol. Cell. Biol. 23, 1334–1340 (2003).
Vakoc, C. R. et al. Proximity among distant regulatory elements at the beta-globin locus requires GATA-1 and FOG-1. Mol. Cell 17, 453–462 (2005).
Welch, J. J. et al. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104, 3136–3147 (2004).
Wu, W. et al. Dynamic shifts in occupancy by TAL1 are guided by GATA factors and drive large-scale reprogramming of gene expression during hematopoiesis. Genome Res. 24, 1945–1962 (2014).
Patel, A., Hashimoto, H., Zhang, X. & Cheng, X. Characterization of how DNA modifications affect DNA binding by C2H2 zinc finger proteins. Methods Enzymol. 573, 387–401 (2016).
Patel, A. et al. DNA conformation induces adaptable binding by tandem zinc finger proteins. Cell 173, 221–233.e12 (2018).
Luscombe, N. M., Laskowski, R. A. & Thornton, J. M. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 29, 2860–2874 (2001).
Chen, Y. et al. DNA binding by GATA transcription factor suggests mechanisms of DNA looping and long-range gene regulation. Cell Rep. 2, 1197–1206 (2012).
Tunyasuvunakool, K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021).
Shin, H. Y. et al. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 48, 904–911 (2016).
Dykes, I. M., van Bueren, K. L. & Scambler, P. J. HIC2 regulates isoform switching during maturation of the cardiovascular system. J. Mol. Cell. Cardiol. 114, 29–37 (2018).
Crossley, M. & Orkin, S. H. Phosphorylation of the erythroid transcription factor GATA-1. J. Biol. Chem. 269, 16589–16596 (1994).
Andrews, N. C. & Faller, D. V. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19, 2499 (1991).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinf. 22, 433 (2021).
Yang, Y. et al. Structural basis for human ZBTB7A action at the fetal globin promoter. Cell Rep. 36, 109759 (2021).
Otwinowski, Z., Borek, D., Majewski, W. & Minor, W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59, 228–234 (2003).
Adams, P. D. et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 (2002).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Acknowledgements
We thank the CHOP flow cytometry core for assistance with cell sorting and members of the Blobel laboratory for helpful comments and discussions. We thank R. Kurita and Y. Nakamura for sharing HUDEP1 and HUDEP2 cells. This work was supported by National Institutes of Health (NIH) grants from the National Heart, Lung, and Blood Institute (grant no. HL119479) and research funding from Pfizer (to G.A.B.); the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; grant no. R24DK106766 to G.A.B. and R.C.H.); NIH/NIDDK Mentored Clinical Scientist Research Career Development Award (no. K08 DK129716), the Doris Duke Charitable Foundation Physician Scientist Fellowship grant (no. 2020062), and American Society of Hematology Scholar Award (to S.A.P.); a fellowship by the Cooley’s Anemia Foundation (to X.L.); T32 training grant (no. HL007150-42) and an American Society of Hematology Research Training Award for Fellows (to E.K.); a National Health and Medical Research Council Australia grant (to M.C.); an Australian government Research Training Program Scholarship (toH.W.B.); the St. Jude Children’s Research Hospital Collaborative Research Consortium on Novel Gene Therapies for Sickle Cell Disease; and the National Institute of General Medical Sciences grant (no. R35GM134744) and the Cancer Prevention & Research Institute of Texas grant (no. RR160029) (to X.C.). Cooperative Centers of Excellence in Hematology at Fred Hutchinson Cancer Research Center was supported by NIDDK (DK106829). We thank the DiGaetano family for their generous support.
Author information
Authors and Affiliations
Contributions
P.H. and G.A.B. conceived the project. P.H., S.A.P., E.K., K.Q., X.L., M.S., C.A.K., B.G., H.W.B., O.A., S.T.C., J.S. and R.C.H. conducted the experiments. R.R. and J.R.H. performed protein purification, DNA-binding, X-ray data collection and structure determination. J.R.H. performed computational modeling. X.C. (who is a CPRIT Scholar in Cancer Research) organized and designed the scope of the structural study. P.H., M.C., X.C. and G.A.B. designed the experiments, performed data analyses and wrote the manuscript. All authors contributed to editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Swee Lay Thein, Sjaak Philipsen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Developmental regulation of BCL11A occurs primarily at the transcriptional level.
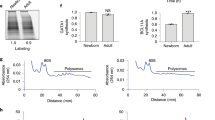
a, PolII ChIP-seq tracks from primary human fetal and adult erythroblasts of the BCL11A locus. Results were obtained from published data. The BCL11A erythroid enhancers +55, +58 and +62 are highlighted in pink. b, HIC2, BCL11A and ZBTB7A mRNA levels in primary human fetal and adult erythroblasts. RNA-seq data were obtained from previously published results. FPKM, fragments per kilobase per million; F, fetal; A, adult. c, Representative tracks of ATAC-seq, H3K27ac and GATA1 CUT&RUN from primary human fetal and adult erythroblasts of the β-globin locus. Results are normalized to reads in peaks. d, MA plot of read counts of ATAC-seq of fetal and adult erythroblasts. BCL11A enhancers are highlighted in red. BCL11A promoter, HBG1/2, HBB and HS2 from LCR (locus control region) enhancer are highlighted in blue. e, MA plot of read counts of H3K27ac CUT&RUN of fetal and adult erythroblasts. BCL11A promoter and enhancers are highlighted in red. HBG1/2 and HBB are highlighted in blue. f, MA plot of read counts of GATA1 CUT&RUN of fetal and adult erythroblasts. BCL11A enhancers are highlighted in red. HS2 from LCR is highlighted in blue.
Extended Data Fig. 2 A CRISPR screen identifies HIC2 as a BCL11A repressor and HBG activator.
a, Scatter plot of CRISPR screen in HUDEP2 cells. Each dot represents a sgRNA. sgRNAs targeting to HIC2 are highlighted in red. RPM, reads per million reads. b, Cell fitness scores of non-targeting and HIC2 sgRNAs. Each dot represents a sgRNA. Fitness scores were calculated according to the abundance of the sgRNAs in the cells of Day13 compared to Day3 post sgRNA transduction. Statistical test compares non-targeting and HIC2 sgRNAs, P = 0.8931 by two-tailed Student’s t-test. c, HBB (sgRNA #1, P = 0.8051; sgRNA #2, P = 0.9997), HBG (sgRNA #1, P < 0.0001; sgRNA #2, P < 0.0001), BCL11A (sgRNA #1, P = 0.0112; sgRNA #2, P = 0.0118) and ZBTB7A (sgRNA #1, P = 0.1816; sgRNA #2, P = 0.1502) mRNA measured by RT-qPCR in HIC2 depleted HUDEP2 cells. Results are normalized to AHSP and shown as mean ± s.d. (n = 3). Statistical tests compare HIC2 sgRNAs and parental samples. P-values were calculated by one-way ANOVA test. d, Western blot of extracts from HUDEP2 cells upon HIC2 depletion. Results are representative of three biological replicates. e, Quantitative of HIC2 protein levels of WB blots in primary human erythroblasts from three sequential developmental stages. Results are normalized to ACTIN and shown as mean ± s.d. f, HIC2 expression levels during erythropoiesis. Results were obtained from published RNA-seq data. FPKM, fragments per kilobase per million. g, HIC2, HBB (low, P = 0.3948; high, P = 0.7553), BCL11A primary transcript (low, P = 0.0058; high, P = 0.0023), ZBTB7A (low, P>0.9999; high, P = 0.0918) and percentage of HBG mRNA measured by RT-qPCR in HIC2 OE HUDEP2 cells. Results are normalized to AHSP and shown as mean ± s.d. (n = 3). Statistical tests compare HIC2 OE and parental samples. P-values were calculated by one-way ANOVA test.
Extended Data Fig. 3 HIC2 overexpression represses BCL11A transcription and augments fetal globin production in primary erythroid cells.
a, Representative flow cytometry results of surface markers CD71 and CD235a staining in primary human adult erythroblasts. b, Percentage of each population in panel a). Results are shown as mean ± s.d. (n = 3). Statistical tests compare parental and HIC2 OE samples; P1, P = 0.0605; P2, P = 0.0222; P3, P = 0.0314 by two-tailed Student’s t-test. c, ZBTB7A (low, P = 0.2160; high, P = 0.0031), HBB (low, P = 0.0133; high, P < 0.0001) and percentage of HBG (low, P = 0.1596; high, P = 0.0001) measured by RT-qPCR in primary adult human erythroblasts. Results are normalized to AHSP and shown as mean ± s.d. (n = 3). Statistical tests compare HIC2 OE and parental samples. P-values were calculated by one-way ANOVA test. d, Scatter plot of the RNA-seq results in primary human adult erythroblasts. FPKM, fragments per kilobase per million. e, ZBTB7A (P1, P = 0.9319; P2, P = 0.0948; P3, P = 0.0889), BCL11A (P1, P = 0.0441; P2, P = 0.0298; P3, P = 0.0040), HBG (P1, P = 0.0243; P2, P = 0.0063; P3, P = 0.0055) and percentage of HBG (P1, P = 0.0040; P2, P = 0.0028; P3, P = 0.0011) measured by RT-qPCR in the stage-matched populations in panel a). Results are normalized to AHSP and shown as mean ± s.d. (n = 3). Statistical tests compare parental and HIC2 OE samples. P-values were calculated by two-tailed Student’s t-test. f, NBSGW mice xenotransplantation experiment. Results are shown as mean ± s.e.m., each symbol represents one mouse (Vector, n = 4 and HIC2 OE, n = 3).
Extended Data Fig. 4 HIC2 overexpression represses BCL11A transcription, augments fetal globin production, and reduces SCD cell sickling.
a, HBG (P < 0.0001), BCL11A mature (P < 0.0001) and primary transcript (P = 0.0043), ZBTB7A (P = 0.2917) and percentage of HBG mRNA (P < 0.0001) measured by RT-qPCR in primary human erythroblasts derived from SCD patient CD34+ cells. Results are normalized to AHSP and shown as mean ± s.d. (3 independent donors with 2 technical replicates for each donor). Statistical tests compare HIC2 OE and parental samples. P-values were calculated by two-tailed Student’s t-test. b, Western blot of extracts from primary human erythroblasts derived from SCD patient CD34+ cells upon HIC2 OE. Results are representative of three biological replicates. c, Flow cytometry results of HbF staining in erythroblasts derived from SCD patient HSPCs. Results are shown as mean ± s.d. (n = 3). Statistical tests compare HIC2 OE and parental samples by two-tailed Student’s t-test, P = 0.0007. d, Representative flow cytometry results of HbF staining in erythroblasts derived from SCD patient HSPCs. e, Representative brightfield images of terminally differentiated SCD patient-derived erythroid cells following incubation at 2.5% O2 for 1 hour followed by fixation. Sickled cells are indicated by red arrowheads. f, Percentage of sickled cells quantified following low-O2 sickling assay. Results are mean ± s.d. (3 independent donors with 2 technical replicates for each donor). Statistical tests compare HIC2 OE and parental samples by two-tailed Student’s t-test, P < 0.0001.
Extended Data Fig. 5 Chromatin binding profiles of HIC2 and its effects on GATA1 occupancy and chromatin accessibility.
a, Heatmaps of endogenous HIC2 ChIP–seq in primary newborn erythroblasts and HIC2-ER ChIP-seq in HUDEP2 cells, and the most enriched motif. b, Tracks of endogenous HIC2 ChIP-seq in primary newborn erythroblasts, HIC2-ER and ATAC-seq in HUDEP2 cells of the β-globin locus. The HIC2 and HIC2-ER ChIP-seq were normalized to total reads. LCR, locus control region. c, Scatter plots of ATAC-seq and GATA1 ChIP-seq in HUDEP2 and HUDEP1 cells upon HIC2 perturbations. BCL11A enhancers are highlighted in red. HBG1, HBG2, HBB and HS2 from LCR are highlighted in blue. RPM, reads per million reads.
Extended Data Fig. 6 HIC2 imparts developmental control onto the BCL11A enhancers.
a, The core sequence of human and mouse BCL11A +55 enhancer. The point mutations in the luciferase reporter constructs in panels b) and c), and CRISPR-mediated knock-in mutations in HUDEP2 cells are highlighted in red. b, Luciferase reporter assay of human BCL11A +55 enhancers in HUDEP2 cells. The two HIC2 motifs in (a) are indicated by green boxes. Red X-crosses indicate the mutated HIC2 motifs. Results are mean ± s.d. (n = 3). Statistical tests compare GFP control and HIC2 OE samples by two-tailed Student’s t-test; WT, P < 0.0001; m1, P = 0.0004; m2, P < 0.0001; m3, P < 0.0001. c, Luciferase reporter assay of human BCL11A +55 enhancers in HUDEP2 cells. The two GATA motifs in panel a) are indicated by blue boxes. Red X-crosses indicate the mutated GATA motifs. Results are mean ± s.d. (n = 3). Statistical tests compare GFP control and HIC2 OE samples by two-tailed Student’s t-test; WT, P < 0.0001; m4, P = 0.0002; m5, P = 0.0087. d, Representative Sanger sequencing traces of WT and HIC2 motifs edited HUDEP2 single cell clones. Mutations are indicated by red arrows. e, ChIP-qPCR of HIC2-HA in representative WT and HIC2 motifs edited HUDEP2 single cell clones. Results are mean ± s.d. (n = 3). Statistical tests compare WT clone #1 and Mutant clone #1 samples by two-tailed Student’s t-test; BCL11A+55, P = 0.0040; BCL11A+58, P = 0.0833; EPDR1, P = 0.7585; CD4, P = 0.0778. f, BCL11A and HBG mRNA measured by RT-qPCR in independent WT and HIC2 motifs edited HUDEP2 single cell clones. Results are normalized to AHSP and shown as mean ± s.d. (n = 3). Statistical tests compare GFP control and HIC2 OE samples by two-tailed Student’s t-test; BCL11A (WT clone #1, P = 0.0016; WT clone #2, P < 0.0001; Mutant clone #1, P = 0.0003; Mutant clone #2, P = 0.0192; Mutant clone #3, P < 0.0001); HBG (WT clone #1, P < 0.0001; WT clone #2, P < 0.0001; Mutant clone #1, P < 0.0001; Mutant clone #2, P < 0.0001; Mutant clone #3, P = 0.0001).
Extended Data Fig. 7 The repression of BCL11A by HIC2 is conserved between human and mouse.
a, Heatmaps of HIC2-HA ChIP-seq in induced mouse G1E-ER4 cells and the most enriched motif. b, ChIP-seq tracks of hHIC2-HA and GATA1 in G1E-ER4 cells. The mBcl11a enhancers homologous to human +55 and +62 are highlighted in pink. GATA1 ChIP-seq of WT G1E-ER4 cells was obtained from published data. c, Bcl11a (P = 0.0002), Hbb (P = 0.0076), Hbb-y (P = 0.0002) and Hbb-bh1 (P = 0.0001) mRNA levels as measured by RT-qPCR in the hHIC2 OE G1E-ER4 cells. Results are normalized to Actb and shown as mean ± s.d. (n = 3). Statistical tests compare HIC2 OE and parental samples. P-values were calculated by two-tailed Student’s t-test.
Extended Data Fig. 8 HIC2 developmentally controls HBG by repressing BCL11A transcription.
a, Western blot with indicated antibodies of extracts from HUDEP1 cells upon HIC2 depletion. Results are representative of two biological replicates. b, ZBTB7A mRNA (sgRNA #1, P = 0.4015; sgRNA #2, P = 0.0110) measured by RT-qPCR in HUDEP1 cells upon HIC2 depletion. Results are normalized to AHSP and shown as mean ± s.d. (n = 3). Statistical tests compare HIC2 sgRNAs and parental samples. P-values were calculated by one-way ANOVA test. c, Purified recombinant proteins used in the crystallography and fluorescence polarization experiments. Results were performed with one biological replicate. d, The FAM-labeled oligonucleotides of HIC2 binding motif-1 and motif-2 at BCL11A +55 enhancer. e, Comparison of binding affinities between HIC2 ZF1-5 and ZF2-5, and ZNF410 ZF1-5 at the two HIC2 binding motifs. f, EMSA of GATA1 and HIC2 using probes from the BCL11A +55 enhancer, wild type or with mutated binding motifs. Red arrowheads indicate the super-shifted bands; asterisks indicate non-specific bands. Results were performed with one biological replicate.
Extended Data Fig. 9 Model of HIC2 regulating BCL11A transcription during development.
At fetal stage, HIC2 binds to BCL11A enhancers and blocks GATA1 binding. At adult stage, HIC2 is downregulated and GATA1 binds to the enhancers promoting BCL11A transcription.
Supplementary information
Supplementary Tables
Supplementary Tables 1–9.
Source data
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 6
Unprocessed western blots and gels.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 8
Unprocessed western blotsand EMSA gels.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, P., Peslak, S.A., Ren, R. et al. HIC2 controls developmental hemoglobin switching by repressing BCL11A transcription. Nat Genet 54, 1417–1426 (2022). https://doi.org/10.1038/s41588-022-01152-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-022-01152-6
