
Abstract
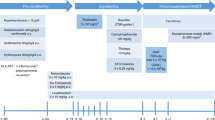
Sickle cell disease (SCD) and transfusion-dependent β-thalassemia (TDT) are the most prevalent monogenic disorders worldwide. Trial HGB-205 (NCT02151526) aimed at evaluating gene therapy by autologous CD34+ cells transduced ex vivo with lentiviral vector BB305 that encodes the anti-sickling βA-T87Q-globin expressed in the erythroid lineage. HGB-205 is a phase 1/2, open-label, single-arm, non-randomized interventional study of 2-year duration at a single center, followed by observation in long-term follow-up studies LTF-303 (NCT02633943) and LTF-307 (NCT04628585) for TDT and SCD, respectively. Inclusion and exclusion criteria were similar to those for allogeneic transplantation but restricted to patients lacking geno-identical, histocompatible donors. Four patients with TDT and three patients with SCD, ages 13–21 years, were treated after busulfan myeloablation 4.6–7.9 years ago, with a median follow-up of 4.5 years. Key primary endpoints included mortality, engraftment, replication-competent lentivirus and clonal dominance. No adverse events related to the drug product were observed. Clinical remission and remediation of biological hallmarks of the disease have been sustained in two of the three patients with SCD, and frequency of transfusions was reduced in the third. The patients with TDT are all transfusion free with improvement of dyserythropoiesis and iron overload.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
Data in the published article have been presented where possible in aggregated form. Any data presented from individual patients have been de-identified. The study protocol and statistical plan are available at https://clinicaltrials.gov/ct2/show/NCT02151526, and the datasets generated and/or analyzed during HGB-205 and follow-up studies LTF-303 for TDT and LTF-307 for SCD are available from the study sponsor, Bluebird Bio (clinicaltrials@bluebirdbio.com), for trial and LTF datasets, and from the corresponding authors (M.C. or P.L.) for investigator-provided data, as applicable and upon reasonable request, although restrictions might apply due to patient privacy and the General Data Protection Regulation. Source data are provided with this paper.
References
Cao, A. & Galanello, R. Beta-thalassemia. Genet. Med. 12, 61–76 (2010).
Pasricha, S. R. & Drakesmith, H. Hemoglobinopathies in the fetal position. N. Engl. J. Med. 379, 1675–1677 (2018).
Shah, F. T., Sayani, F., Trompeter, S., Drasar, E. & Piga, A. Challenges of blood transfusions in β-thalassemia. Blood Rev. 37, 100588 (2019).
Cappellini, M. D. et al. A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N. Engl. J. Med. 382, 1219–1231 (2020).
Niihara, Y. et al. A phase 3 trial of ʟ-glutamine in sickle cell disease. N. Engl. J. Med. 379, 226–235 (2018).
Vichinsky, E. et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N. Engl. J. Med. 381, 509–519 (2019).
Ataga, K. I. et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N. Engl. J. Med. 376, 429–439 (2017).
Bolanos-Meade, J. et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood 120, 4285–4291 (2012).
Magrin, E., Miccio, A. & Cavazzana, M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood 134, 1203–1213 (2019).
Imren, S. et al. Permanent and panerythroid correction of murine β thalassemia by multiple lentiviral integration in hematopoietic stem cells. Proc. Natl Acad. Sci. USA 99, 14380–14385 (2002).
Pawliuk, R. et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 294, 2368–2371 (2001).
Imren, S. et al. High-level β-globin expression and preferred intragenic integration after lentiviral transduction of human cord blood stem cells. J. Clin. Invest. 114, 953–962 (2004).
Ronen, K. et al. Distribution of lentiviral vector integration sites in mice following therapeutic gene transfer to treat β-thalassemia. Mol. Ther. 19, 1273–1286 (2011).
Negre, O. et al. Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell disease. Curr. Gene Ther. 15, 64–81 (2015).
Takekoshi, K. J., Oh, Y. H., Westerman, K. W., London, I. M. & Leboulch, P. Retroviral transfer of a human beta-globin/delta-globin hybrid gene linked to beta locus control region hypersensitive site 2 aimed at the gene therapy of sickle cell disease. Proc. Natl Acad. Sci. USA 92, 3014–3018 (1995).
Srinivasulu, S. et al. Pair-wise interactions of polymerization inhibitory contact site mutations of hemoglobin-S. Protein J. 25, 503–516 (2006).
Cavazzana-Calvo, M. et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 467, 318–322 (2010).
Thompson, A. A. et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N. Engl. J. Med. 378, 1479–1493 (2018).
Ribeil, J. A. et al. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 376, 848–855 (2017).
Nagel, R. L. et al. Hematologically and genetically distinct forms of sickle cell anemia in Africa. The Senegal type and the Benin type. N. Engl. J. Med. 312, 880–884 (1985).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423 (1948). 623–656.
Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270 (1984).
Hebert, N. et al. Individual red blood cell fetal hemoglobin quantification allows to determine protective thresholds in sickle cell disease. Am. J. Hematol. 95, 1235–1245 (2020).
Eaton, W. A., Hofrichter, J. & Ross, P. D. Editorial: Delay time of gelation: a possible determinant of clinical severity in sickle cell disease. Blood 47, 621–627 (1976).
Imren, S. et al. Permanent and panerythroid correction of murine beta thalassemia by multiple lentiviral integration in hematopoietic stem cells. Proc. Natl Acad. Sci. USA 99, 14380–14385 (2002).
Henry, E. R. et al. Allosteric control of hemoglobin S fiber formation by oxygen and its relation to the pathophysiology of sickle cell disease. Proc. Natl Acad. Sci. USA 117, 15018–15027 (2020).
Ilboudo, Y. et al. A common functional PIEZO1 deletion allele associates with red blood cell density in sickle cell disease patients. Am. J. Hematol. 93, E362–E365 (2018).
Philippidis, A. After analysis, Bluebird Bio says vector ‘very unlikely’ cause of acute myeloid leukemia. Hum. Gene Ther. 32, 332–334 (2021).
Tisdale, J. F. et al. Updated results from HGB-206 LentiGlobin for Sickle Cell Disease Gene Therapy Study: Group C data and Group A AML case investigation. Abstract 196. American Society of Gene & Cell Therapy Annual Meeting (2021).
Seminog, O. O., Ogunlaja, O. I., Yeates, D. & Goldacre, M. J. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J. R. Soc. Med. 109, 303–309 (2016).
Brunson, A. et al. Increased risk of leukemia among sickle cell disease patients in California. Blood 130, 1597–1599 (2017).
Jones, R. J. & DeBaun, M. R. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood 138, 942–947 (2021).
Shimoni, A. et al. Secondary malignancies after allogeneic stem-cell transplantation in the era of reduced-intensity conditioning; the incidence is not reduced. Leukemia 27, 829–835 (2013).
Thompson, A. A. et al. Resolution of serious vaso-occlusive pain crises and reduction in patient-reported pain intensity: results from the ongoing phase 1/2 HGB-206 group C study of lentiglobin for sickle cell disease (bb1111) gene therapy. https://ash.confex.com/ash/2020/webprogram/Paper134940.html (2020).
Brusson, M. et al. Novel lentiviral vectors for gene therapy of sickle cell disease combining gene addition and gene silencing strategies. https://ash.confex.com/ash/2021/webprogram/Paper151076.html (2021).
Thompson, A. A. et al. Favorable outcomes in pediatric patients in the phase 3 Hgb-207 (Northstar-2) and Hgb-212 (Northstar-3) studies of betibeglogene autotemcel gene therapy for the treatment of transfusion-dependent β-thalassemia. Blood 136, 52–54 (2020).
Nualkaew, T. et al. Coordinated β-globin expression and α2-globin reduction in a multiplex lentiviral gene therapy vector for β-thalassemia. Mol. Ther. 29, 2841–2853 (2021).
Acknowledgements
We thank all the patients who participated in this study and staff members at the clinical and laboratory sites and at Bluebird Bio, including D. Davidson, P. Gregory, G. Veres and M. Finer. We also thank R. Pawliuk, K. A. Westerman, K. J. Takekoshi, C. J. Eaves, R. K. Humphries, S. P. Goff, R. Dorazio, E. Gluckman, F. Bernaudin, F. Galactéros, V. Jolaine and R. L. Maas for their contributions to this project through the years. This work was supported by Bluebird Bio; by grants from France’s Agence Nationale de la Recherche under ‘Investissements d’avenir’ program (ANR-10-IAHU-01, M.C. and A. Miccio) and the Paris Ile de France Region under the ‘DIM Thérapie génique’ initiative (M.C., P. Bartolucci and A. Miccio); by Assistance Publique-Hôpitaux de Paris, INSERM, Etablissement Français du Sang (P. Bartolucci and W.E.N.) and the Imagine Institute (M.C. and A. Miccio); by Commissariat à l’Energie Atomique et aux Energies Alternatives (P.L. and E.P.); by the Dior chair for tailored medicine to M.C.; and by ANR’s Chair of Excellence and Industrial Chair of Excellence and Industrial Chair grants to P.L. from the Agence National de la Recherche. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript, except Bluebird Bio, the study sponsor. This article is dedicated to the memory of Irving London, Dorothy Tuan Lo, Ronald Nagel, Arthur Bank and Bill Solomon.
Author information
Authors and Affiliations
Contributions
M.C. was the principal investigator and P.L. the scientific director of the trial, which they designed together with Bluebird Bio, the sponsor, and with contributions from Y.B. and E.P. P.L. designed the βA-T87Q-globin gene, the LentiGlobin BB305 vector and the packaging plasmids; P.L. also led preclinical studies and regulatory filing. J.A.R., M. Semeraro and M.C. supervised the trial on behalf of the sponsor, with contributions from M.A. and C.W. M. Semeraro and L.J. were involved in patient inclusion and follow-up. E.P. was involved in preclinical studies and designed key biological assays, with contributions from O.N. F.T., E.M. and A.M. performed vector transduction of patient cells and some biological analysis in the follow-up. E.M. was in charge of production, and A.M. was in charge of quality control at the hospital site. F.L., J.-S.D., B.N., I.F.-B., S.R., V.B., C.P. and S.B. performed transplantation and/or clinical services during hospitalization. M. Semeraro, L.J. and M.d.M. provided clinical follow-up. N.H., A.C., L.K., P. Bourget, W.E.N., A. Miccio, P. Bartolucci, P.L. and M.C. performed biological assays and/or analyzed and interpreted data. M. Schmidt, E.S. and A.D. performed the analysis of integration sites. E.M., M. Semeraro, N.H., A. Miccio, P. Bartolucci, M.C. and P.L. wrote the manuscript. All authors provided input to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.L. is a scientific founder of Bluebird Bio, with stock options. P.L. is an inventor of awarded patents that claim the βA-T87Q-globin gene, the LentiGlobin BB305 vector and packaging plasmids. O.N., M.A., C.W. and J.A.R. are current or former employees of Bluebird Bio, with stock options. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. Anna Maria Ranzoni was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Integration site analysis (ISA) for the patients with SCD.
ISA kinetics for each of the 3 SCD patients, showing relative abundance of vector-marked cell clones. Each of the most represented unique integration sites (UISs) is indicated by a different color, with white showing the cumulative proportion of all other UISs and their names as a side table. The total number of UISs is indicated at the top of each column. SCD-2 M54 provided post datacut.
Extended Data Fig. 2 Integration site analysis (ISA) for the patients with TDT.
Same legend as for Supplementary Fig. 2. TDT-2 M84, TDT-3 M60, and TDT-4 M60 provided post datacut.
Extended Data Fig. 3 VCN kinetics (c/dg) in SCD patients.
(a) In blood cell sub-populations. The values are investigator’s provided and are lower than the qualified values in Extended Data Table 1, due to different standards; they are provided here to compare VCN in cell sub-fractions in a time-dependent manner. (b) In erythroid burst-forming units (BFU-E). (c) In granulocyte-macrophage colony-forming units (CFU-GM).
Extended Data Fig. 4 Flow cytometry analyses of RBCs in SCD patients.
(a) Controls showing that the Pacific Blue (PB) labeled anti-HbA monoclonal antibody (Rockland) does not cross react with HbS in individualized RBCs by flow cytometry analysis. S/S RBC exhibit low level of fluorescence because the anti-HbA antibody also reacts with HbA2 (data from Rockland). However, the anti-HbA antibody does not cross react with HbF (data from Rockland). Because the epitope recognized by the anti-HbA antibody is also present on HbAT87Q, the latter is equally well recognized (data not shown). (b) Distribution of γ-globin and βA-T87Q-globin expressing RBCs in SCD patients’ blood by flow cytometry using antibodies that recognize either βA-T87Q-globin (anti-HbA PB-labeled) or γ-globin (anti-HbF PE-labeled). Co-stainings performed at M36, M14 and M15 for SCD-1, SCD-2 and SCD-3, respectively, showing reduced levels of HbF in high containing HbAT87Q RBCs, and inversely, for SCD-2 and SCD-3. (c) Distribution of βA-T87Q-globin vs. γ-globin expressing RBCs in SCD patients’ blood by flow cytometry using antibodies that recognize either βA-T87Q-globin (left) or γ-globin (right). M, months after GT; PB, Pacific Blue; PE, phycoerythrin. Corresponding datasets are represented in Tables 2a and b.
Extended Data Fig. 5
VCN kinetics (c/dg) in blood cell sub-populations of TDT patients.
Extended Data Fig. 6 Non-invasive in vivo quantification of tissue iron levels by 1.5 Tesla magnetic resonance imaging (MRI).
(a) In SCD patients’ liver (R2*/T2* relaxometry) (left) and heart (T2*) (right). (b) In TDT patients, as in (A).
Supplementary information
Supplementary Material
Supplementary Tables 1–9 and Supplementary Figs. 1–3
Supplementary Data
NOTE: Source data for Supplementary Fig. 2 are the same as for Extended Data Figs. 1 and 2
Source data
Source Data Fig. 1
Statistical Source Data
Source Data Fig. 2
Statistical Source Data
Source Data Fig. 3
Statistical Source Data
Source Data Fig. 4
Statistical Source Data
Source Data for Extended Data Figs. 1 and 2
Statistical Source Data
Source Data for Extended Data Fig. 3
Statistical Source Data
Source Data Extended Data Fig. 4
Statistical Source Data
Source Data Extended Data Fig. 5
Statistical Source Data
Source Data Extended Data Fig. 6
Statistical Source Data
Rights and permissions
About this article

Cite this article
Magrin, E., Semeraro, M., Hebert, N. et al. Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: the HGB-205 trial. Nat Med 28, 81–88 (2022). https://doi.org/10.1038/s41591-021-01650-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01650-w
This article is cited by
-
Successes and challenges in clinical gene therapy
Gene Therapy (2023)
-
Base-editing-mediated dissection of a γ-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression
Nature Communications (2022)
