
Abstract
The STEP 5 trial assessed the efficacy and safety of once-weekly subcutaneous semaglutide 2.4 mg versus placebo (both plus behavioral intervention) for long-term treatment of adults with obesity, or overweight with at least one weight-related comorbidity, without diabetes. The co-primary endpoints were the percentage change in body weight and achievement of weight loss of ≥5% at week 104. Efficacy was assessed among all randomized participants regardless of treatment discontinuation or rescue intervention. From 5 October 2018 to 1 February 2019, 304 participants were randomly assigned to semaglutide 2.4 mg (n = 152) or placebo (n = 152), 92.8% of whom completed the trial (attended the end-of-trial safety visit). Most participants were female (236 (77.6%)) and white (283 (93.1%)), with a mean (s.d.) age of 47.3 (11.0) years, body mass index of 38.5 (6.9) kg m–2 and weight of 106.0 (22.0) kg. The mean change in body weight from baseline to week 104 was −15.2% in the semaglutide group (n = 152) versus −2.6% with placebo (n = 152), for an estimated treatment difference of −12.6 %-points (95% confidence interval, −15.3 to −9.8; P < 0.0001). More participants in the semaglutide group than in the placebo group achieved weight loss ≥5% from baseline at week 104 (77.1% versus 34.4%; P < 0.0001). Gastrointestinal adverse events, mostly mild-to-moderate, were reported more often with semaglutide than with placebo (82.2% versus 53.9%). In summary, in adults with overweight (with at least one weight-related comorbidity) or obesity, semaglutide treatment led to substantial, sustained weight loss over 104 weeks versus placebo. NCT03693430
Similar content being viewed by others


Main
Behavioral intervention incorporating modifications in diet and physical activity remains the foundation of treatment for overweight and obesity. However, because behavioral intervention is often not associated with clinically meaningful and sustainable weight loss, pharmacotherapy is recommended as an additional tool for long-term weight management in people with a body mass index (BMI) of at least 30 kg m–2, or at least 27 kg m–2 in those with weight-related comorbidities1.
Semaglutide is a glucagon-like peptide-1 (GLP-1) analog approved for the treatment of type 2 diabetes (oral semaglutide and subcutaneous semaglutide) and for reducing the risk of cardiovascular events in people with type 2 diabetes and cardiovascular disease (subcutaneous semaglutide only)2,3,4,5. At a dose of 2.4 mg once-weekly, subcutaneous semaglutide was approved in the United States, Europe, the United Kingdom and Canada for weight management in adults with overweight (BMI ≥ 27 kg m–2 with at least one weight-related comorbidity) or obesity (BMI ≥ 30 kg m–2)2,3,4,5, based on results from the Semaglutide Treatment Effect in People with Obesity (STEP) clinical trial program. In the STEP 1 and 3 trials in participants without type 2 diabetes, average placebo-subtracted weight losses of 12.4% and 10.3%, respectively, were seen with semaglutide 2.4 mg at week 68 (refs. 6,7).
Previous studies in the STEP trial program have been limited to treatment durations of up to 68 weeks6,7,8. The 2-year STEP 5 study reported herein was conducted to evaluate the long-term effect of once-weekly subcutaneous semaglutide 2.4 mg compared with placebo, as an adjunct to behavioral intervention, on body weight and cardiometabolic risk factors, in adults with obesity (BMI ≥ 30 kg m–2), or with overweight (BMI ≥ 27 kg m–2) and at least one weight-related comorbidity, without diabetes (Extended Data Fig. 1). This phase 3, randomized, double-blind, placebo-controlled, multinational trial represents the longest study of the use of semaglutide for weight management to date. Co-primary endpoints were percentage change in body weight from baseline to week 104 and achievement of weight loss of at least 5% of baseline weight at week 104.
Results
Participants and treatment
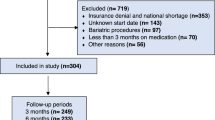
From 5 October 2018 to 1 February 2019, 304 participants were randomly assigned to semaglutide 2.4 mg (n = 152) or placebo (n = 152) and included in the full analysis set (all randomized participants according to the intention-to-treat principle). Observation periods included the in-trial period (that is, while in the trial, regardless of treatment discontinuation or rescue intervention) and the on-treatment period (with trial product). Overall, of 304 participants, 282 (92.8%) completed the trial (attended the end-of-trial safety visit), 272 (89.5%) had a body weight assessment at the end-of-treatment visit at week 104, and 243 (79.9%) adhered to treatment (were on-treatment at the end-of-treatment visit) (Fig. 1).

s.c., subcutaneous.
Demographics and baseline characteristics were similar between groups (Table 1). Most participants were female (236 (77.6%) of 304) and most were white (283 (93.1%) of 304). Mean age was 47.3 years. Mean body weight was 106.0 kg and mean BMI was 38.5 kg m–2.
Two estimands were employed for the assessment of efficacy endpoints—estimands assess treatment efficacy from different perspectives and account for intercurrent events (for example, discontinuation of trial product or initiation of other weight loss interventions) and missing data differently. The ‘treatment policy’ estimand quantified the treatment effect for the in-trial period among all randomly assigned participants, regardless of treatment discontinuation or rescue intervention, based on the intention-to-treat principle, and was used as the primary analysis method. The ‘trial product’ estimand quantified the average treatment effect for the on-treatment period in all randomly assigned participants, assuming that the drug or placebo was taken as intended, and was used as the secondary analysis method (Methods).
Efficacy endpoint results for the treatment policy estimand
Mean observed change in body weight over time during the in-trial period is shown as percentage change in Fig. 2a and as absolute change (kg) in Extended Data Fig. 2. Based on the treatment policy estimand, the estimated mean (standard error (s.e.)) change in body weight from baseline to week 104 was –15.2% (0.9) with semaglutide and –2.6% (1.1) with placebo (co-primary endpoint; estimated treatment difference (ETD) –12.6 percentage points, 95% confidence interval (CI) –15.3 to –9.8, P < 0.0001). Semaglutide-treated participants, compared with placebo, were more likely to lose at least 5% of baseline body weight at week 104 (co-primary endpoint; odds ratio (OR) 5.0, 95% CI 3.0 to 8.4; P < 0.0001). At week 104, 111 (77.1%) versus 44 (34.4%) participants in the semaglutide and placebo groups, respectively, were observed to have achieved this endpoint (in-trial period data; among 144 participants for semaglutide and 128 for placebo) (Table 2 and Fig. 2b). As statistical superiority for both co-primary endpoints was demonstrated for semaglutide versus placebo, the prespecified criteria for a positive trial were met, indicating a significant benefit of semaglutide versus placebo.

a, Observed mean percentage change from baseline in body weight over time for participants in the full analysis set during the in-trial observation period (error bars are standard error of the mean; numbers below the panels are the number of participants contributing to the mean) and estimated treatment difference for the percentage change from baseline to week 104 in body weight based on the treatment policy estimand. b, Observed proportions of participants and OR for achieving weight loss of at least 5% from baseline at week 104 in the full analysis set during the in-trial observation period, based on the treatment policy estimand. *Estimated means in percent are from the primary analysis. The in-trial observation period was the time from random assignment to last contact with a trial site, regardless of treatment discontinuation or rescue intervention. The treatment policy estimand assesses treatment effect regardless of treatment discontinuation or rescue intervention; see Extended Data Fig. 6 for corresponding data for the trial product estimand (which assesses treatment effect assuming all participants adhered to treatment and did not receive rescue intervention). The change in body weight analysis was conducted with the use of the analysis-of-covariance method, with randomized treatment as a factor and baseline body weight as a covariate. The achievement of at least 5% weight loss analysis was conducted with the use of logistic regression, with the same factor and covariate. A multiple imputation approach was used for missing data. The results were accompanied by two-sided 95% CIs and corresponding P values (significance defined as P < 0.05). As co-primary endpoints, the analyses were controlled for multiple comparisons.
Semaglutide-treated participants, compared with placebo, were also more likely to lose at least 10%, 15% or 20% of baseline body weight at week 104 (P < 0.0001 for the OR for the 10% and 15% thresholds (both were confirmatory secondary endpoints); the 20% threshold (a supportive secondary endpoint) was not part of statistical testing hierarchy). For the in-trial observation period, these weight loss thresholds were achieved by 89 (61.8%), 75 (52.1%) and 52 (36.1%) of 144 participants in the semaglutide group versus 17 (13.3%), nine (7.0%) and three (2.3%) of 128 participants in the placebo group, respectively (Table 2 and Extended Data Fig. 3 for cumulative distribution of change from baseline).
Semaglutide was associated with greater reductions from baseline to week 104 in waist circumference (–14.4 cm (0.9) with semaglutide versus –5.2 cm (1.2) with placebo; ETD –9.2 cm, 95% CI –12.2 to –6.2, P < 0.0001) and systolic blood pressure (–5.7 mmHg (1.1) with semaglutide versus –1.6 (1.2) with placebo; ETD –4.2 mmHg, 95% CI –7.3 to –1.0; P = 0.01) (both were confirmatory secondary endpoints; Table 2, Fig. 2 and Extended Data Fig. 4a,b). Compared with placebo, semaglutide also led to improvements in diastolic blood pressure, glycated hemoglobin (HbA1c), fasting plasma glucose, fasting serum insulin, C-reactive protein, total cholesterol, low-density lipoprotein cholesterol, very-low-density lipoprotein cholesterol and triglycerides (all were supportive secondary endpoints; Table 2 and Extended Data Fig. 4c,d).
Of the participants with prediabetes at baseline who also had a glycemic status assessment at week 104, 59 (79.7%) of 74 treated with semaglutide reverted to normoglycemia at week 104, compared with 20 (37.0%) of 54 participants on placebo (an exploratory endpoint; Table 2 and Extended Data Fig. 5). Of the participants with normoglycemia at baseline who also had a glycemic status assessment at week 104, one (1.4%) of 71 treated with semaglutide had prediabetes at week 104, compared with 10 (13.0%) of 77 participants on placebo. Among participants with a week 104 assessment, none in the semaglutide group and three in the placebo group had type 2 diabetes at week 104 (one had normoglycemia at baseline and two had prediabetes at baseline). The proportion of participants with changes in the use of lipid-lowering and antihypertensive medication (among those receiving such medications during the trial) is reported in Table 2 (both were exploratory endpoints).
Efficacy endpoint results for the trial product estimand
Mean observed change in body weight over time during the on-treatment period is shown in Extended Data Fig. 6a. For the trial product estimand, the estimated mean (s.e.) change in body weight from baseline to week 104 was –16.7% (0.9) with semaglutide and –0.6% (0.9) for placebo (ETD –16.0 percentage points, 95% CI –18.6 to –13.5). Semaglutide-treated participants, compared with placebo, were more likely to lose at least 5% of baseline body weight at week 104 (OR 18.1 (95% CI 10.0 to 32.5). At week 104, 110 (83.3%) versus 38 (34.9%) participants in the semaglutide and placebo groups, respectively, were observed to have achieved this endpoint (on-treatment period data; among 132 participants for semaglutide and 109 for placebo) (Supplementary Table 1 and Extended Data Fig. 6b). Results of analyses of the confirmatory and selected supportive secondary endpoints for the trial product estimand, are provided in Supplementary Table 1.
Safety and tolerability
Adverse events leading to discontinuation of trial product were reported by nine participants (5.9%) in the semaglutide group and seven participants (4.6%) in the placebo group (Table 3).
Gastrointestinal disorders, namely nausea, diarrhea, vomiting and constipation, were the most frequently reported adverse events and occurred in more participants treated with semaglutide than with placebo (125 (82.2%) of 152 versus 82 (53.9%) of 152, respectively) (Table 3). Most gastrointestinal adverse events were mild-to-moderate and transient, and such events led to permanent treatment discontinuation in six (3.9%) participants in the semaglutide group and one (0.7%) participant in the placebo group (Table 3 and Extended Data Fig. 7).
Serious adverse events were reported by 12 (7.9%) of 152 participants in the semaglutide group and 18 (11.8%) of 152 participants in the placebo group (Table 3). One death was reported in the semaglutide group and was considered by the independent external event adjudication committee to be unrelated to the trial product (Table 3). In the semaglutide versus placebo groups, gallbladder-related disorders were reported by four (2.6%) versus two (1.3%) participants and malignant neoplasms were reported by two (1.3%) versus four (2.6%), respectively (Table 3; details on malignant neoplasms are shown in Supplementary Table 2). There were no reports of pancreatitis in either treatment group. Additional safety variables are described in Table 3 and Supplementary Table 3. COVID-19 infection was reported by 16 (10.5%) of 152 participants in the semaglutide group versus eight (5.3%) of 152 participants in the placebo group, with very few cases in each group classed as serious and none requiring temporary or permanent interruption of semaglutide treatment.
Discussion
In STEP 5, once-weekly treatment with semaglutide 2.4 mg as an adjunct to behavioral intervention in adults with overweight (with at least one weight-related comorbidity) or obesity led to a substantial initial reduction in weight, which plateaued after approximately week 60 and was maintained for the remainder of the study. At week 104, participants in the semaglutide group had achieved a mean weight loss of 15.2% from baseline—a difference of 12.6 percentage points versus placebo plus behavioral intervention. This weight loss is comparable to the mean reduction of 14.9% (placebo-corrected weight loss of 12.4 percentage points) seen at week 68 in the STEP 1 trial of semaglutide 2.4 mg versus placebo (both plus behavioral intervention)7. Thus, our findings indicate that the substantial weight losses reported during 68 weeks’ treatment with semaglutide 2.4 mg in prior STEP trials6,7,9 can be maintained with continued semaglutide treatment up to at least 104 weeks. The mean weight loss of ~15% achieved with semaglutide 2.4 mg at week 104 in STEP 5 exceeds weight loss reported at similar time points in trials with other pharmacotherapies for weight management in adults with overweight or obesity10,11,12,13,14.
Weight loss of ≥5%, a threshold widely used to indicate a clinically meaningful response to therapy15, was achieved by >75% of participants in the semaglutide group at week 104. Moreover, 61.8% of participants on semaglutide lost ≥10% of baseline weight, and over a third of participants had achieved at least 20% weight loss at week 104 in the semaglutide group. As was seen in prior studies6,7,9,16, while the vast majority of participants receiving semaglutide 2.4 mg had lost weight at the end of the STEP 5 study, a small proportion of participants experienced weight gain. We do not know how weight would have changed in these participants had they not been receiving the drug; notably, the proportion of patients with weight gain during the study was substantially higher in the placebo group. There is marked variability in weight change in patients on weight management treatments; the reason for this is still unclear and likely involves complex biological and societal influences.
Obesity is a chronic, relapsing disease that requires continuous effort to control6,17. With all nonsurgical interventions and to some extent with bariatric surgery, weight regain after initial weight loss is common10,11,12,13,14,18,19,20,21,22. In contrast to findings with behavioral20,21,22 and other pharmacological interventions10,12,13, the similar mean weight loss achieved with semaglutide 2.4 mg in STEP 5 at weeks 52 and 104 (–15.6% and –15.2%, respectively) suggests that, on average, there is minimal weight regain over 104 weeks when once-weekly semaglutide therapy is continued. When interpreted together with the findings of the STEP 4 withdrawal trial and STEP 1 off-treatment extension study, which both showed weight regain after semaglutide discontinuation (after 20 weeks’ treatment in STEP 4 and 68 weeks’ treatment in STEP 1)23,24, these results support the benefit of continued semaglutide treatment for sustained weight loss.
Prior 68-week trials in adults with overweight or obesity have reported cardiometabolic improvements with semaglutide 2.4 mg (refs. 6,7,9,16). Consistent with these findings, in STEP 5 semaglutide treatment improved a range of cardiometabolic risk parameters, including waist circumference, systolic and diastolic blood pressure, HbA1c levels, total cholesterol, low-density lipoprotein cholesterol, very-low-density lipoprotein cholesterol and triglycerides. Collectively, these results indicate a beneficial effect of treatment on overall patient health. In addition, semaglutide treatment reduced C-reactive protein levels, a marker of systemic inflammation that is known to be elevated in patients with obesity25,26. The reduction in fasting insulin and glucose with semaglutide is indicative of an increase in insulin sensitivity. Similar to the findings of other studies in the STEP trial program7,27, exploratory outcomes showed that in the semaglutide group 80% of participants with prediabetes at baseline reverted to normoglycemia by the end of the trial (compared with 37% of those receiving placebo), while 99% of participants with normoglycemia at baseline maintained normoglycemia at the end of the trial (compared with 86% with placebo). These findings suggest a potential beneficial effect of semaglutide on glycemic status, but whether semaglutide treatment delays or prevents progression to type 2 diabetes requires confirmation. In the 68-week trials7,9, reductions in weight, waist circumference, blood pressure and HbA1c appeared to plateau around week 60 with semaglutide. STEP 5 shows that the changes in these parameters were sustained through 104 weeks’ treatment.
The safety profile of semaglutide 2.4 mg in STEP 5 was consistent with that in other STEP program trials6,7,9,16,23, and with the GLP-1 receptor agonist class in general28. Gastrointestinal disorders were the most common adverse events with semaglutide, typically transient, of mild-to-moderate severity, occurring during dose escalation, and infrequently leading to treatment discontinuation.
Strengths of STEP 5 include the high rates of adherence to treatment and completion of the trial (which contributed to consistency in findings between the two estimands). Limitations include the low proportion of nonwhite participants and the preponderance of female participants. In addition, while the homogenous nature of the prescribed dietary intake deficit, physical activity goal and counseling frequency provided consistency, it may not fully reflect the need for approaches tailored to the health profiles of individuals or to different populations in clinical practice; however, beyond adherence to the stipulated criteria for counseling on diet and physical activity, behavioral intervention was delivered by each study site with no further direction, allowing a degree of local tailoring and aiding real-world applicability.
In conclusion, treatment with once-weekly subcutaneous semaglutide in conjunction with behavioral intervention in adults with overweight (with at least one weight-related comorbidity) or obesity (without diabetes) was associated with clinically impactful and sustained weight loss of 15.2% at week 104, along with improvements in weight-related cardiometabolic risk factors.
Methods
Trial design and participants
This phase 3, randomized, double-blind, placebo-controlled study was conducted at 41 sites across five countries (Canada, Italy, Hungary, Spain and the United States), as described in a previous publication8 and listed in the Supplementary information. Most investigators specialized in endocrinology and internal medicine, with others specializing in family medicine, psychiatry and clinical psychology. The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol was approved by independent ethics committees or institutional review boards at each study site (a redacted protocol is provided separately).
Participants were eligible to be included in the trial only if all of the following criteria applied:
-
Informed consent obtained before any trial-related activities. Trial-related activities were any procedures that were carried out as part of the trial, including activities to determine suitability for the trial.
-
Male or female, aged ≥18 years at the time of signing informed consent.
-
BMI ≥ 30.0 kg m–2 or ≥27.0 kg m–2 with the presence of at least one of the following weight-related comorbidities (treated or untreated): hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease.
-
History of at least one self-reported unsuccessful dietary effort to lose body weight.
Participants were excluded from the trial if any of the following criteria applied:
Glycemia-related
-
HbA1c ≥ 48 mmol mol–1 (6.5%) as measured by the central laboratory at screening.
-
History of type 1 or type 2 diabetes.
-
Treatment with glucose-lowering agent(s) within 90 days before screening.
Obesity-related
-
A self-reported change in body weight >5 kg (11 lbs) within 90 days before screening irrespective of medical records.
-
Treatment with any medication for the indication of obesity within the past 90 days before screening.
-
Previous or planned (during the trial period) obesity treatment with surgery or a weight loss device. However, the following were allowed: (1) liposuction and/or abdominoplasty, if performed >1 year before screening; (2) lap banding, if the band had been removed >1 year before screening; (3) intragastric balloon, if the balloon had been removed >1 year before screening; or (4) duodenal-jejunal bypass sleeve, if the sleeve had been removed >1 year before screening.
-
Uncontrolled thyroid disease, defined as thyroid-stimulating hormone >6.0 mIU l–1 or <0.4 mIU l–1 as measured by the central laboratory at screening.
Mental health
-
History of major depressive disorder within 2 years before screening.
-
Diagnosis of other severe psychiatric disorder (for example, schizophrenia, bipolar disorder).
-
A Patient Health Questionnaire-9 score of ≥15 at screening.
-
A lifetime history of a suicidal attempt.
-
Suicidal behavior within 30 days before screening.
-
Suicidal ideation corresponding to type 4 or 5 on the Columbia-Suicide Severity Rating Scale within the past 30 days before screening.
General safety
-
Presence of acute pancreatitis within the past 180 days before the day of screening.
-
History or presence of chronic pancreatitis.
-
Calcitonin ≥100 ng l–1 as measured by the central laboratory at screening.
-
Personal or first-degree relative(s) history of multiple endocrine neoplasia type 2 or medullary thyroid carcinoma.
-
Renal impairment measured as estimated glomerular filtration rate value of <15 ml min 1.73 m–2 as defined by KDIGO 2012 (ref. 30) by the central laboratory at screening.
-
History of malignant neoplasms within the past 5 years before screening. Basal and squamous cell skin cancer and any carcinoma in situ were allowed.
-
Any of the following: myocardial infarction, stroke, hospitalization for unstable angina or transient ischemic attack within the past 60 days before screening.
-
Participant classified as being in New York Heart Association Class IV.
-
Surgery scheduled for the duration of the trial, except for minor surgical procedures, in the opinion of the investigator.
-
Known or suspected abuse of alcohol or recreational drugs.
-
Known or suspected hypersensitivity to trial product(s) or related products.
-
Previous participation in the trial. Participation was defined as signed informed consent.
-
Participation in another clinical trial within 90 days before screening.
-
Other person(s) from the same household participating in any semaglutide trial.
-
Female who was pregnant, breast-feeding, or intended to become pregnant, or was of child-bearing potential and not using a highly effective contraceptive method.
-
Any disorder, unwillingness or inability not covered by any of the other exclusion criteria which, in the investigator’s opinion, might have jeopardized the participant’s safety or compliance with the protocol.
Randomization and masking
Randomization (1:1) to semaglutide 2.4 mg or placebo was done centrally by the clinical research organization (Parexel) in a double-blind manner using an interactive web-based response system (IWRS) with a fixed-size blocking schema, without stratification. The IWRS generated the randomization list and assigned patients to the next available treatment according to the randomization schedule. The IWRS allocated dispensing unit numbers for each patient, with the trial product dispensed by the site investigator or study coordinator at the trial site visits. The active product and corresponding placebo product were visually identical to maintain masking of participants and site staff. The people analyzing the data were blinded to treatment/group assignment until breaking the blinding at database lock.
Procedures
Participants received subcutaneous semaglutide 2.4 mg or placebo once-weekly for 104 weeks, in addition to standard behavioral intervention, followed by 7 weeks without treatment. Semaglutide was initiated at 0.25 mg per week for the first 4 weeks via a pre-filled pen injector, escalating in a fixed-dose regimen every 4 weeks to reach the maintenance dose of 2.4 mg by week 16 (lower maintenance doses were permitted if participants were unable to tolerate 2.4 mg) (Extended Data Fig. 1). Behavioral intervention consisted of counseling by a dietitian or similarly qualified healthcare professional every 4 weeks via in-person visits or telephone on adherence to a reduced-calorie diet (500 kcal deficit a day relative to the energy expenditure estimated at randomization) and increased physical activity (150 minutes a week encouraged, for example, walking), both recorded daily (via a diary, app or other tools, which were reviewed during counseling sessions); beyond these criteria for behavioral intervention, no further standardization of behavioral intervention was applied across study sites. Participants discontinuing treatment prematurely remained in the trial and were encouraged to attend scheduled visits, particularly those at weeks 104 and 111.
Body weight, waist circumference and vital signs (systolic and diastolic blood pressure and pulse) were measured at baseline; these measurements were repeated every 4 weeks until week 20, and every 8 weeks thereafter, until week 100 and week 104 (within 3 days either side of scheduled visit day). These parameters were also measured at the end-of-trial visit at week 111 (within 5 days either side of scheduled visit day). Height was measured at screening. HbA1c, fasting plasma glucose, lipids and C-reactive protein were measured at baseline and weeks 20, 52, 84, and 104; electrocardiograms were also performed at these time points. Fasting serum insulin was measured at baseline and week 104. Physical examinations were performed at screening and weeks 52 and 104. Hematology and biochemistry laboratory parameters were measured at screening and weeks 20, 52, 84 and 104. Adverse events were recorded at each visit. Control of eating was assessed in a subset of participants from the United States and Canada; these results will be presented in a separate manuscript.
Given the emergence of COVID-19 in the second year of the study, trial visits were permitted to be conducted via telephone, during which counseling was provided and safety-related information was collected; endpoint assessments were not performed during telephone visits. Assessment data were collected at the next possible in-person visit.
Outcomes
Co-primary endpoints were percentage change in body weight from baseline to week 104 and achievement of weight loss of at least 5% of baseline weight at week 104. These were tested first in the statistical testing hierarchy, followed by the confirmatory secondary endpoints, which were tested in the following order: achievement of weight loss of at least 10% or 15% at week 104; and change from baseline to week 104 in waist circumference and systolic blood pressure.
Supportive secondary endpoints were not included in the statistical testing hierarchy and were: achievement of weight loss of ≥20% at week 104; change from baseline to week 104 in body weight (in kg), BMI, HbA1c, fasting plasma glucose, fasting serum insulin, diastolic blood pressure, lipids (total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, very-low-density lipoprotein cholesterol, free fatty acids and triglycerides) and C-reactive protein; change from baseline to week 52 in body weight (percentage change and kg change), BMI and waist circumference; and achievement of weight loss of ≥5%, ≥10%, ≥15% and ≥20% at week 52.
Exploratory endpoints reported herein include change from baseline to week 104 in glycemic category, antihypertensive medication use and lipid-lowering medication use. Glycemic category (normoglycemia, prediabetes or type 2 diabetes) was determined by investigators on the basis of available information (for example, medical records, concomitant medication, and blood glucose variables) and in accordance with American Diabetes Association criteria30, which for prediabetes includes fasting plasma glucose levels of 100 mg dl–1 (5.6 mmol l–1) to 125 mg dl–1 (6.9 mmol l–1) or HbA1c levels of 5.7–6.4% (39–47 mmol l–1), and for type 2 diabetes includes fasting plasma glucose levels of ≥126 mg dl–1 (7.0 mmol l–1) or HbA1c levels ≥6.5% (48 mmol l–1). The allowance for investigators to use all available information (for example, concomitant medication) to assess glycemic category was primarily included to account for scenarios in which glucose-lowering medications were initiated during the trial that would confound glycemic category assessment if based purely on fasting plasma glucose or HbA1c levels (for example, if a patient developed diabetes during the study and received a glucose-lowering drug that resulted in their glucose level being below the American Diabetes Association threshold for type 2 diabetes diagnosis). Additional exploratory endpoints for which data are not reported were: permanent discontinuation of trial product between baseline and week 104; time to permanent discontinuation of trial product; and Control of Eating Questionnaire scores from the four domains and 19 individual items (applicable for United States and Canada only).
Safety endpoints included the number of treatment-emergent adverse events and serious adverse events, assessed between baseline and week 111; and change from baseline to week 104 in pulse, amylase, lipase and calcitonin. An independent external event adjudication committee reviewed cardiovascular events, acute pancreatitis and deaths.
Statistical analysis
A sample size of 300 participants provided an effective power of at least 96% for the two co-primary endpoints, and at least 43% for all confirmatory secondary endpoints, which were tested in a predefined hierarchical order (Supplementary Table 4). The two co-primary endpoints were analyzed independently of each other, and for the trial to be considered to be positive (indicating a significant benefit of semaglutide versus placebo), statistical superiority for both co-primary endpoints was required to be demonstrated.
Efficacy endpoints were analyzed using the full analysis set (all randomized participants according to the intention-to-treat principle). Safety endpoints were analyzed using the safety analysis set of all randomized participants exposed to at least one dose of randomized treatment. Observation periods included the in-trial period (that is, while in the trial, regardless of treatment discontinuation or rescue intervention) and the on-treatment period (with trial product). All results from statistical analyses of confirmatory endpoints were accompanied by two-sided 95% CIs and corresponding P values (significance defined as P < 0.05). Supportive secondary endpoint analyses were not controlled for multiple comparisons and should not be used to infer definitive treatment effects.
Two estimands were employed to assess treatment efficacy from different perspectives and accounted for intercurrent events and missing data differently, as described in a previous publication31. The treatment policy estimand quantified the treatment effect among all randomly assigned participants, regardless of treatment discontinuation or rescue intervention (participants in trial; intention to treat). This estimand was used to assess the superiority of semaglutide versus placebo for the co-primary and confirmatory secondary endpoints in a predefined hierarchical order.
For the treatment policy estimand, continuous endpoint analyses were conducted with the use of the analysis-of-covariance method, with randomized treatment as a factor and baseline endpoint value as a covariate. Analyses of categorical endpoints were conducted with the use of logistic regression, with the same factor and covariate. A multiple imputation approach was used to handle missing data31, with imputation based on available data from participants in the same treatment arm with the same treatment status (on-treatment or discontinued). Imputation was performed using a linear regression model, with sex, baseline BMI and timing of last observation as factors, and baseline value and last observation value as covariates. One thousand complete datasets were generated for analysis, with results combined using Rubin’s formula.
The trial product estimand addressed the average treatment effect in all randomly assigned participants, assuming that the drug or placebo was taken as intended (participants on treatment). For the trial product estimand, continuous endpoint analyses were conducted using a mixed model for repeated measures with randomized treatment as a factor and baseline endpoint value as a covariate. Analyses of categorical endpoints were conducted with the use of logistic regression, with categorization for missing data based on values predicted from the mixed model for repeated measures. Analyses of endpoints for the trial product estimand were not adjusted for multiplicity.
Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc.). Additional details on analytic methods per endpoint are in Supplementary Table 4. Exploratory endpoints were assessed with descriptive statistics based on observed data.
The trial is closed and completed. The study is registered with ClinicalTrials.gov, NCT03693430.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data will be shared with bona fide researchers submitting a research proposal approved by the independent review board. The research proposal must outline: the scientific rationale and relevance of the proposed research; a short lay summary intended for public disclosure; research methodology and data; statistical analysis plan and publication plan. Data must not be used for commercial purposes. Data will be made available after research completion, and approval of the product and product use in the European Union and the USA. Individual participant data will be shared in datasets in a de-identified and anonymized format. Access request proposals can be found at novonordisk-trials.com.
References
Jensen, M. D. et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation 129, S102–S138 (2014).
Wegovy prescribing information. Updated June 2021. US Food and Drug Administration https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215256s000lbl.pdf (2022).
Wegovy summary of product characteristics. European Medicines Agency https://www.ema.europa.eu/en/documents/product-information/wegovy-epar-product-information_en.pdf (2022).
Regulatory decision summary – Wegovy. Health Canada https://hpr-rps.hres.ca/reg-content/regulatory-decision-summary-detail.php?lang=en&linkID=RDS00896 (2022).
Wegovy summary of product characteristics. Medicines and Healthcare Products Regulatory Agency https://mhraproducts4853.blob.core.windows.net/docs/ee95471f9265e346688fce45e8c9fd8bd5e4e3d3 (2022).
Wadden, T. A. et al. STEP 3 Investigators. Effect of subcutaneous semaglutide vs placebo as an adjunct to intensive behavioral therapy on body weight in adults with overweight or obesity: the STEP 3 randomized clinical trial. JAMA 325, 1403–1413 (2021).
Wilding, J. P. H. et al. STEP 1 Study Group. Once-weekly semaglutide in adults with overweight or obesity. N. Engl. J. Med. 384, 989–1002 (2021).
Kushner, R. F. et al. Semaglutide 2.4 mg for the treatment of obesity: key elements of the STEP trials 1 to 5. Obesity (Silver Spring) 28, 1050–1617 (2020).
Davies, M. et al. STEP 2 Study Group. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet 397, 971–984 (2021).
Sjöström, L. et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet 352, 167–72. (1998).
Garvey, W. T. et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am. J. Clin. Nutr. 95, 297–308 (2012).
Torgerson, J. S. et al. XENical in the Prevention of Diabetes in Obese Subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 27, 155–161 (2004).
Astrup, A. et al. NN8022-1807 Investigators. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J. Obes. (Lond.) 36, 843–854 (2012).
le Roux, C. W. et al. SCALE Obesity and Prediabetes NN8022-1839 Study Group. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double-blind trial. Lancet 389, 1399–409. (2017).
Williamson, D. A., Bray, G. A. & Ryan, D. H. Is 5% weight loss a satisfactory criterion to define clinically significant weight loss? Obesity (Silver Spring) 23, 2319–2320 (2015).
Rubino, D. M. et al. STEP 8 Investigators. Effect of weekly subcutaneous semaglutide vs daily liraglutide on body weight in adults with overweight or obesity without diabetes: the STEP 8 randomized clinical trial. JAMA 327, 138–150 (2022).
Bray, G. A., Kim, K. K. & Wilding, J. P. H. World Obesity Federation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 18, 715–723 (2017).
Smith, S. R. et al. Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N. Engl. J. Med. 363, 245–256 (2010).
MacLean, P. S. et al. NIH working group report: innovative research to improve maintenance of weight loss. Obesity (Silver Spring) 23, 7–15 (2015).
Svetkey, L. P. et al. Comparison of strategies for sustaining weight loss: the weight loss maintenance randomized controlled trial. JAMA 299, 1139–1148 (2008).
Wadden, T. A. et al. Four-year weight losses in the Look AHEAD study: factors associated with success. Obesity (Silver Spring) 19, 1987–1998 (2011).
Wing, R. R., Tate, D. F., Gorin, A. A., Raynor, H. A. & Fava, J. L. A self-regulation program for maintenance of weight loss. N. Engl. J. Med. 355, 1563–1571 (2006).
Rubino, D. et al. STEP 4 Investigators. Effect of continued weekly subcutaneous semaglutide vs placebo on weight loss maintenance in adults with overweight or obesity: the STEP 4 randomized clinical trial. JAMA 325, 1414–1425 (2021).
Wilding, J. P. et al. Weight regain and cardiometabolic effects after withdrawal of semaglutide: the STEP 1 trial extension. Diabetes Obes. Metab. 24, 1553–1564 (2022).
Visser, M., Bouter, L. M., McQuillan, G. M., Wener, M. H. & Harris, T. B. Elevated C-reactive protein levels in overweight and obese adults. JAMA 282, 2131–2135 (1999).
Timpson, N. J. et al. C-reactive protein levels and body mass index: elucidating direction of causation through reciprocal Mendelian randomization. Int J. Obes. (Lond.) 35, 300–308 (2011).
Perreault, L. et al. Changes in glucose metabolism and glycemic status with once-weekly subcutaneous semaglutide 2.4 mg among participants with prediabetes in the STEP Program. Diabetes Care 45, 2396–2405 (2022).
Nauck, M. A., Quast, D. R., Wefers, J. & Meier, J. J. GLP-1 receptor agonists in the treatment of type 2 diabetes - state-of-the-art. Mol. Metab. 46, 101102 (2021).
American Diabetes Association. 2. Classification and diagnosis of diabetes. Diabetes Care 40, S11–S24 (2017).
Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 3, 1–150 (2013).
Wharton, S. et al. Estimating and reporting treatment effects in clinical trials for weight management: using estimands to interpret effects of intercurrent events and missing data. Int J. Obes. (Lond.) 45, 923–933 (2021).
Acknowledgments
We thank the study participants, and the investigators and study site staff who conducted the study. In addition, we thank N. Beadle of Axis, a division of Spirit Medical Communications Group Limited, for medical writing and editorial assistance (funded by Novo Nordisk A/S, Denmark). The study was funded by Novo Nordisk. The funder designed the trial, oversaw its conduct, monitored trial sites, and collected and analyzed the data; investigators were responsible for trial-related medical decisions and data collection. This article was drafted under the guidance of the authors, with medical writing and editorial support paid for by the funder.
Author information
Authors and Affiliations
Consortia
Contributions
W.T.G. contributed to acquisition, analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. R.L.B. contributed to analysis or interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. M.B. contributed to analysis and interpretation of data; drafting of the manuscript; and critical revision of manuscript for important intellectual content. S.B. contributed to concept and design; acquisition, analysis or interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. L.N.C. contributed to analysis and interpretation of data; critical revision of manuscript for important intellectual content; and statistical analysis. J.P.F. contributed to acquisition, analysis or interpretation of data; critical revision of the manuscript for important intellectual content; and supervision. E.J. contributed to acquisition, analysis or interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. K.K. contributed to analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; statistical analysis; and administrative, technical or material support. G.R. contributed to interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. T.A.W. contributed to interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. S.W. contributed to acquisition, analysis or interpretation of data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. Investigators were responsible for data collection, and the sponsor undertook site monitoring, data collation and analysis. All authors had full access to aggregated study data and to unaggregated data on request from the sponsor; participated in the data interpretation, presentation and manuscript drafting (assisted by a sponsor-funded medical writer); approved its submission, and vouched for data accuracy and fidelity to the protocol.
Corresponding author
Ethics declarations
Competing interests
W.T.G. reports a grant from Novo Nordisk; serving as site principal investigator for the current clinical trial, which was sponsored by his university during the conduct of the study; and receiving grants to serve as site principal investigator for other university-sponsored clinical trials funded by Eli Lilly & Company, Lexicon, Epitomee and Pfizer outside the submitted work. He also served as a compensated consultant on advisory committees for Alnylam, Amgen, Boehringer Ingelheim, Fractyl and Novo Nordisk, and a volunteer uncompensated consultant on advisory committees for Boehringer Ingelheim, Jazz Pharmaceuticals, Novo Nordisk and Pfizer. R.L.B. reports research grant support, on behalf of their institution, from Novo Nordisk and advisory/consultancy fees from Boehringer Ingelheim, Eli Lilly & Company, Gila Therapeutics Inc, GLW-01, International Medical Press, Novo Nordisk, Pfizer and ViiV. M.B. is an employee of Novo Nordisk A/S. S.B. served as site principal investigator for the clinical trial (he received no financial compensation, nor was there a financial relationship) and reports advisory/consulting fees and/or other support from Boehringer Ingelheim, Eli Lilly & Company, Guidotti Laboratories, Menarini Diagnostics, Novo Nordisk and Therascience Lignaform. L.N.C. is an employee of Novo Nordisk A/S. J.P.F. reports research support grants from Akero, AstraZeneca, Boehringer Ingelheim, BMS, 89bio, Eli Lilly & Company, Intercept, IONIS, Janssen, Madrigal, Metacrine, Merck, NorthSea Therapeutics, Novartis, Novo Nordisk, Oramed, Pfizer, Poxel and Sanofi; and advisory/consultancy fees from Akero, Altimmune, Axcella Health, Becton Dickenson, Boehringer Ingelheim, Carmot Therapeutics, Echosens, 89bio, Eli Lilly & Company, Gilead, Intercept, Metacrine, Merck, Novo Nordisk, Pfizer and Sanofi. E.J. reports grants from Amgen, AstraZeneca, Boehringer Ingelheim, FAES, Janssen, Eli Lilly & Company, MSD, Novo Nordisk, Pfizer, Sanofi, Shire and UCB; personal fees from Amgen, AstraZeneca, FAES, Helios-Fresenius, Italfármaco, Eli Lilly & Company, MSD, Mundipharma, Novo Nordisk, UCB and Viatris. K.K. is an employee of Novo Nordisk A/S. G.R. reports personal (advisory/consultancy and lecture) fees and nonfinancial support from iNova Pharmaceuticals, Nestle HealthScience and Novo Nordisk; personal (lecture) fees from Johnson & Johnson, Medtronic (formerly Covidien), Merck Sharpe & Dohme, ReShape Lifesciences (formerly Apollo-Endosurgery and Allergan Australia) and W.L. Gore Device Technologies. T.A.W. serves on advisory boards for Novo Nordisk and WW (formerly Weight Watchers), and has received grant support, on behalf of the University of Pennsylvania, from Novo Nordisk and from Epitomee Medical Ltd (the latter outside of the submitted work). S.W. reports research funding, advisory/consulting fees and/or other support from AstraZeneca, Bausch Health Inc., Boehringer Ingelheim, CIHR, Janssen, Eli Lilly & Company and Novo Nordisk.
Peer review
Peer review information
Nature Medicine thanks George Bray, Amanda Adler, Victor Volovici and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Jennifer Sargent, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Trial design for STEP 5 clinical study.
s.c., subcutaneous.
Extended Data Fig. 2 Body weight (kg) by week.
Observed mean body weight (kg) over time for participants in the full analysis set during the in-trial observation period (from randomization to last contact with trial site, regardless of treatment discontinuation or rescue intervention). Error bars are standard error of the mean. Numbers below the panels are the number of participants contributing to the mean.
Extended Data Fig. 3 Cumulative distribution plot of change from baseline to week 104 in body weight.
(a, b) Cumulative distribution plot of observed percentage change from baseline over time in body weight for participants in the full analysis set during the in-trial observation period* (a) and on-treatment observation period† (b). *From randomization to last contact with trial site, regardless of treatment discontinuation or rescue intervention. †During treatment with trial product (any dose of trial medication administered within the previous 2 weeks (that is, any period of temporary treatment interruption with trial product was excluded)).
Extended Data Fig. 4 Comparison of change from baseline by week for selected cardiometabolic endpoints for semaglutide versus placebo.
(a-d) Observed mean percentage change from baseline over time for participants in the full analysis set during the in-trial observation period in waist circumference (a), systolic blood pressure (b), diastolic blood pressure (c), and HbA1c (d). Error bars are standard error of the mean; numbers below the panels are the number of participants contributing to the mean.
Extended Data Fig. 5 Shift from baseline to week 104 in glycemic status.
(a-d) Observed data for participants in the full analysis set treated with semaglutide 2.4 mg (a, c) or placebo (b, d) during the in-trial period. As illustrated by the gray shading, the week 104 bars present results at this time point among the subgroups of participants with baseline prediabetes (a and b) or baseline normoglycemia (c and d). Glycemic category was determined by investigators on the basis of available information (for example, medical records, concomitant medication, and blood glucose variables) and in accordance with American Diabetes Association criteria,30 which for prediabetes includes fasting plasma glucose levels of 100 mg/dL (5.6 mmol/L) to 125 mg/dL (6.9 mmol/L) or HbA1c levels of 5.7–6.4% (39–47 mmol/L), and for type 2 diabetes includes fasting plasma glucose levels of ≥126 mg/dL (7.0 mmol/L) or HbA1c levels ≥6.5% (48 mmol/L). *Number of participants in the full analysis set. †Number of participants with prediabetes (a and b) or normoglycemia (c and d) at baseline and evaluable data at week 104.
Extended Data Fig. 6 Comparison of body weight parameters for semaglutide versus placebo (trial product estimand).
(a) Observed mean percentage change from baseline in body weight over time for participants in the full analysis set during the on-treatment observation period (error bars are standard error of the mean; numbers below the panels are the number of participants contributing to the mean) and estimated treatment difference for the percentage change from baseline to week 104 in body weight based on the trial product estimand. (b) Observed proportions of participants and odds ratio for achieving weight loss of at least 5% from baseline at week 104 in the full analysis set during the on-treatment observation period, based on the trial product estimand. *Estimated means in percent. A time point is considered as on treatment if any dose of trial product has been administered within the previous 14 days. The trial product estimand assesses treatment effect assuming all participants adhered to treatment and did not receive rescue intervention. CI, confidence interval; ETD, estimated treatment difference.
Extended Data Fig. 7 Prevalence and duration of gastrointestinal events by severity.
(a-d) The proportion of participants receiving semaglutide or placebo who reported nausea (a), diarrhea (b), constipation (c), or vomiting (d) events classed as mild, moderate, or severe over the course of the treatment period. Data are from the on-treatment observation period (during treatment with trial product [any dose of trial medication administered within the previous 49 days (that is, any period of temporary treatment interruption with trial product was excluded)). Adverse events were classified by severity as mild (easily tolerated, causing minimal discomfort, and not interfering with everyday activities), moderate (causes sufficient discomfort and interferes with normal everyday activities), or severe (prevents normal everyday activities).
Supplementary information
Supplementary Information
List of investigators in the STEP 5 trial and Supplementary Tables 1–4.
Supplementary Data 1
Study protocol.
Supplementary Data 2
ICMJE forms.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garvey, W.T., Batterham, R.L., Bhatta, M. et al. Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial. Nat Med 28, 2083–2091 (2022). https://doi.org/10.1038/s41591-022-02026-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-022-02026-4
This article is cited by
-
Pharmacotherapy for obesity: moving towards efficacy improvement
Diabetology & Metabolic Syndrome (2024)
-
Structured lifestyle modification as an adjunct to obesity pharmacotherapy: there is much to learn
International Journal of Obesity (2024)
-
Targeting the incretin system in obesity and type 2 diabetes mellitus
Nature Reviews Endocrinology (2024)
-
Redefining peptide therapeutics with semaglutide
Nature Chemistry (2024)
-
Cost-effectiveness of weight-management pharmacotherapies in Canada: a societal perspective
International Journal of Obesity (2024)
