
Abstract
Antibiotic resistance is a major threat to public health. The development of chemo-informatic tools to guide medicinal chemistry campaigns in the efficint design of antibacterial libraries is urgently needed. We present AB-DB, an open database of all-atom force-field parameters, molecular dynamics trajectories, quantum-mechanical properties, and curated physico-chemical descriptors of antimicrobial compounds. We considered more than 300 molecules belonging to 25 families that include the most relevant antibiotic classes in clinical use, such as β-lactams and (fluoro)quinolones, as well as inhibitors of key bacterial proteins. We provide traditional descriptors together with properties obtained with Density Functional Theory calculations. Noteworthy, AB-DB contains less conventional descriptors extracted from μs-long molecular dynamics simulations in explicit solvent. In addition, for each compound we make available force-field parameters for the major micro-species at physiological pH. With the rise of multi-drug-resistant pathogens and the consequent need for novel antibiotics, inhibitors, and drug re-purposing strategies, curated databases containing reliable and not straightforward properties facilitate the integration of data mining and statistics into the discovery of new antimicrobials.
Measurement(s) | molecular physical property analysis objective |
Technology Type(s) | Computer Modeling |
Similar content being viewed by others
Background & Summary
The increasing spread of antibiotic resistance in clinics is causing a global health crisis. Mobile genetic element encoding for resistance genes can be transferred among bacterial populations, leading to the need to make more efficient the discovery of new antibiotics and molecules able to improve their efficacy1,2. Gram-negative bacteria, such as Escherichia coli, Pseudomonas aeruginosa and Acinetobacter baumannii, are particularly challenging due to the presence of an outer membrane which reduces the permeability of antimicrobials and therefore their efficacy3,4,5. A major obstacle is represented by efflux pumps that act in synergy with the outer membrane ejecting a plethora of compounds with various chemical-physical properties, among which are different classes of antibiotics6,7,8. In addition, inactivating enzymes such as β-lactamases9 contribute to exacerbate the problem. To date, both academia and industry struggle to identify new antibiotic classes and optimize available compounds10,11,12,13,14,15. Although several strategies have been adopted (e.g., drug repurposing16 or systematic exploitation of natural compounds17), holistic approaches able to take into account multiple factors contributing to antimicrobial resistance are lacking18. Previous works focused primarily on the role of chemical-physical properties of antimicrobial molecules in their accumulation profile, searching for general “rules”19,20. For example, O’Shea and Moser19 found that antibiotics effective towards Gram-negative bacteria are generally characterized by high molecular weight (MW, around 600 Da) and high polarity (as expressed by cLogD7.4 below 0). More recently, Richter et al.21 identified the presence of a primary amine, flexible bond number (5 or less) and globularity (describing molecular shape), as key features to predict the accumulation of antibiotics in Gram-negative bacteria22. Predictive rules of efflux inhibition and avoidance were also identified23. This latter study combined standard molecular descriptors to properties derived from structure-based analyses (e.g., interaction descriptors extracted from molecular docking), allowing for a more complete and multi-factorial view. Definition of general rules able to predict both the permeability and the activity of antimicrobial compounds can greatly benefit from the application of machine learning approaches able to speed up the drug discovery process24,25. The application of these methods requires collection of data for the learning phase26,27, that highlights the need for curated molecular databases providing ready-to-use features.
In this regards, over the years, several molecular databases containing standard descriptors (e.g., PubChem28, DrugBank29, ChEMBL30, ZINC1531) or quantum-mechanical (QM) properties32,33,34,35,36 have been reported. These data have been extensively used in quantitative-structure-activity-relationships (QSAR) studies37,38, and recent works exploited additional information coming from molecular dynamics (MD) simulations39,40,41,42,43, that represent an effective tool to address key structural and kinetic features of biological systems44,45,46,47. However, although freely available servers for the automatic generation of force-field (FF) parameters are available48,49,50,51,52, small molecule parameterization remains often a non-trivial task53.
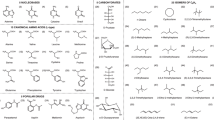
Following a previous work54, we present a homogeneous database of accurate all-atom FF parameters of more than 300 antimicrobial compounds, together with μs-long MD trajectories and QM-related data (e.g., ground-state optimized geometries). We additionally provide molecular descriptors of different nature: i) classical parameters usually considered in QSAR studies (e.g., MW, atom/ring counts, LogP, …); ii) MD-derived properties (e.g., root-mean-square fluctuations, statistics of intra- and inter-molecular H-bonds, hydration-shells structure and dynamics, …); iii) QM-based parameters (e.g., energies of frontier molecular orbitals, electronic gap, electric dipole moment, …). The computational protocol adopted is schematically depicted in Fig. 1. The molecules considered, ranging in size from cycloserine (13 atoms, MW = 102.09 Da) to rifalazil (132 atoms, MW = 941.09 Da), cover 24 classes of antimicrobial compounds with different mechanisms of action, plus miscellaneous compounds (e.g., fluorescent dyes such as rhodamine 6G and HT33342). In particular, about 30% of compounds in the whole dataset are β-lactams, and 10% are inhibitors of key bacterial proteins. High MW compounds (>~1000 Da) such as polymyxin, glycol- and lipo-peptides were omitted from the selection, due to the high computational costs/convergence issues associated to the QM calculation. A schematic depiction of representative compounds showing the overall chemical variability of the sample is given in Fig. 2. The complete set of antimicrobial families and compounds is reported in Table 1.

Schematic view of the computational protocol adopted to generate AB-DB. The different steps reporting some representative molecular descriptors are highlighted: molecular characterization, QM calculations, FF parameters generation, MD simulations. For further technical details see Methods section.

Schematic representation of four representative antimicrobials belonging to different classes, namely (a) penicillins, (b) (fluoro)quinolones, (c) aminoglycosides, and (d) aminocoumarins. Graphics rendered with DiscoveryStudio101.
To the best of our knowledge AB-DB is unique in supplying homogeneously-derived properties of antimicrobial compounds. The accurate FF parameters can be reused for further MD simulations of compounds either alone or interacting with their macromolecular target(s). The MD trajectories can be exploited for ligand- or structure-based studies, in particular for molecular docking. The successful application of this technique requires the knowledge of the bio-active conformation of ligands55,56,57,58, that is not always found by classical searching algorithms59,60. Our curated, homogeneous and not straightforward properties can feed machine learning models towards the discovery of new antimicrobials61,62. Input/output files are also supplied to ensure data reproducibility. In the near future we plan to update AB-DB including more compounds, covering additional antimicrobial classes.
Methods
For each antimicrobial compound we obtained the 3D structure data file (.sdf format) from the PubChem database, except for 13 compounds for which the 3D conformation is not available. In those cases, marked in italic in Table 1, the starting structure was taken from the ChEMBL database, or from available X-ray structures. We then used the ChemAxon’s Marvin suite of programs63 to calculate the dominant protonation states at physiological pH. For known uncertain cases (e.g., tetracyclines), for which several micro-species with similar population were predicted, the choice has been driven by available experimental data on pKa values. The comparison between the experimental and calculated pKa values of the ionizable groups of a representative set of challenging molecules is reported in Table 2. The large relative errors (even exceeding 100%) were expected for these classes of antimicrobials since pKa determination in Marvin ChemAxon is based on molecular charge distribution, and these molecules are characterized by a complex electronic structure (e.g., with several possible resonance states). For each molecule we then proceeded with QM calculations and FF generation followed by all-atom MD simulations. Properties generated in each step will be referred to as QSAR, MD, and QM descriptors, respectively. The three types of analysis performed in this study are graphically exemplified in Fig. 3 for a test-case molecule and described in details below.

Graphical exemplification of the three types of analysis performed to generate AB-DB. Puromycin molecule has been selected as a test-case. QSAR: number of rotatable bonds (left), QM and FF: atomic partial charges (center), from negative (red) to positive (blue) values, MD: conformations extracted from MD trajectories (right). Graphics rendered with PyMOL102.
Quantum-mechanical calculations and force-field generation
The 3D structures of the molecules downloaded from the PubChem or ChEMBL databases (see above) underwent quantum-chemical calculations at the Density Functional Theory (DFT) level64, using the Gaussian16 package65. We employed the hybrid B3LYP functional66, in conjunction with the split-valence 6–31G** Gaussian basis-set67. The combination B3LYP/6–31G** represents a good compromise between accuracy and computational cost and is widely used for small molecules68,69,70. In all cases we disabled molecular symmetry (Symmetry = None), adopted restrictive convergence criteria for self-consistent-field iterations (10−8 Ha, SCF(Conver = 8)), and used a pruned (99,590) grid (Int = UltraFine) for numerical integration. For the few cases for which convergence criteria were not reached, geometry optimization was first performed with a smaller basis-set (6–31G) and converged geometry and molecular orbitals were used as a starting point for the subsequent B3LYP/6–31G** step. For each compound we optimized the ground-state structure employing the Polarizable Continuum Model71 to mimic the effect of water solvent (SCRF = (PCM,Solvent = Water)), particularly to avoid formation of strong intra-molecular H-bonds. We then performed full vibrational analyses obtaining real frequencies in all cases, thus confirming the geometries obtained to be global minima. We processed the output of Gaussian16 with GaussSum72 to extract molecular orbital data. On the optimized geometry we then performed B3LYP/6–31G** single-point energy calculations in vacuum to generate the atomic partial charges fitting the molecular electrostatic potential. We used the Merz-Kollman scheme73 to construct a grid of points around the molecule under the constraint of reproducing the overall electric dipole moment of the molecule (Pop = (ESP,Dipole,Regular)). The two-step restrained electrostatic potential (RESP) method74 implemented in the Antechamber package75 was used to generate atomic partial charges at the DFT level, instead of the automatic AM1-BCC charges76. This step enabled the generation of the FF files using the General Amber Force Field 2 (GAFF2)77. In a single case, namely the siderophore enterobactin loaded with Fe(III)78, FF files were obtained using the metal center parameter builder module79 of the Amber18 package80, slightly modified accordingly to the QM settings described above.
Molecular dynamics simulations
All-atom MD simulations were performed in explicit water solution using Amber18. Systems were solvated within a box of TIP3P water model81 and K+/Cl− counter ions82, to reach an ionic concentration of 0.1 M, using the program tleap of Amber1880. GAFF2 parameters obtained as described above were adopted for antimicrobial compounds. All systems underwent an energy minimization, a heating followed by a cooling phase, and a short productive dynamics to relax the simulation box. Finally the production 1 μs-long MD simulation was performed, under the NPT ensemble (1 Atm and 310 K) using the isotropic Berendsen barostat83 and the Langevin thermostat84. Further details on MD settings can be found in ref. 54.
Descriptors generation
From the output of QM and MD simulations we extracted all molecular descriptors (~80 in total for each compound, see list in Table S1). Most QSAR descriptors were computed on the QM optimzed geometries using the calculator plugin of the Marvin ChemAxon program63. Given the importance of octanol/water partition coefficient in drug design85, we provide an additional estimate of this parameter by means of the XLOGP3 program86. Furthermore, for each compound we derived the molecular properties associated with the “entry rules”, a series of guidelines that have been recently proposed to increase small-molecule accumulation in Gram-negative bacteria21. QM-based properties were obtained from the Gaussian16 output files of the implicit-solvent geometry optimization. Isotropic and anisotropic polarizabilities were derived from the polarizability tensor according to ref. 87. We additionally provided the molecular dipole moment in vacuum consistent with the atomic partial charges of the FF files, computed as described above. From the all-atom MD simulations we obtained structural and dynamical features by means of the CPPTRAJ program88. First and second water shells were extracted using a lower (upper) cutoff of 3.4 (5.0) Å. For the analysis of intra- and inter-molecular H-bonds we adopted angle and distance cutoffs of 135° (donor-hydrogen-acceptor angle) and 3.5 Å (donor-acceptor), respectively89. The number and population of structural clusters were determined using a hierarchical agglomerative algorithm90 and the molecule root-mean-squaredeviation (RMSD) value as a metric. To evaluate atomic root-mean-square fluctuations (RMSF) we used the utility g_rmsf of the GROMACS package91. During the MD runs we also monitored three morphology descriptors related to the gyration tensor, i.e., asphericity, acylindricity, and kappa2, as implemented in the PLUMED plugin92. Asphericity and acylindricity give a measure of the deviation of the mass distribution from spherical and cylindrical symmetry, respectively; the relative shape anisotropy kappa2 is limited between 0 and 1 and reflects both symmetry and dimensionality93. The minimal projection area (MPA) is the minimum of the circular areas projected perpendicularly to the principal axes of inertia of the molecule, calculated based on the atomic van der Waals radii (Å). The dynamical evolution of the MPA have been determined with the combined use of Open Babel94 and ChemAxon’s calculator plugin63.
Data Records
AB-DB is available on figshare95. The computed molecular descriptors are given in the comma separated file all-descriptors.csv. A compressed TAR archive for each family is provided (e.g., carbapenems.tgz). In turn, every archive contains sub-folders named after the compound and the net charge considered in the calculations (e.g., carbapenems/ertapenem_-1/). For 34 molecules the two protonation states most populated at pH = 7.4 were considered (see Methods section). In these cases two folders per compound are reported, with different values of the net charge (e.g., quinolones/ciprofloxacin_0/ where the compound is considered as zwitterionic, and quinolones/ciprofloxacin_-1/ where the nitrogen atom of the piperazine ring is considered in its neutral form). Each compound folder contains a 2D sketch of the molecule (2d.png), and a total of 20 files distributed into three sub-directories reporting QM (QM/), FF (FF/), and MD (MD/) data. Figure 4 shows a schematic representation of the database structure describing the path of all files provided.

Schematic representation of the AB-DB structure reporting filenames of each sub-directory.
Quantum-mechanical data
QM/ folders contain files derived from QM calculations (see Quantum-mechanical calculations and force-field generation section). In details, the opt-freq.com and opt-freq.log are the input and output files of the Gaussian16 geometry optimization and frequency analysis in implicit solvent. The minimization steps are collected in the optimization.xyz file and the final optimized structure is given in structure data file format as optimized.sdf. This file, generated with Open Babel94 from the corresponding .xyz file and carefully checked manually, is also provided for reproducibility purposes since it has been used to compute QSAR descriptors. We also collected the electronic structure and the harmonic vibrational frequencies into electronic.dat and vibrational.dat files, respectively. The elec-pot.com and elec-pot.log are respectively the input and output files of the Gaussian16 single-point energy calculation in vacuum, performed to derive atomic partial charges. The resulting electrostatic potential file is elec-pot.dat.
Force-field data
For each compound we supply in the corresponding FF/ folder the mol.mol2 and Amber mol.prep files, containing the optimized structure of the molecule with RESP partial charges. The Amber force-field modification file mol.frcmod with all parameters not included in the GAFF2 is also provided. For reproducibility purposes we make available the Amber parameter/topology mol_solv.parm7, and coordinate/restart mol_solv.rst7 files used to perform the MD simulation in explicit solvent. The corresponding mol_solv.pdb file generated using the ambpdb program80 is also provided.
Molecular dynamics data
MD/ folders store the μs-long MD trajectories performed in explicit water solution (see Molecular dynamics simulations section) in the file trajectory.pdb (100000 frames). The representatives of the ten most populated clusters extracted from the trajectory are given in clusters.pdb, and their corresponding fraction in clusters.dat. The statistics of intra- and inter-molecular H-bonds are collected in hbonds-intra.dat and hbonds-inter.dat, respectively.
Technical Validation
AB-DB is built making use of the different computational steps detailed above: molecular characterization, QM calculations, FF generation, MD simulations, and extraction of physico-chemical descriptors. Concerning the starting configurations used for the subsequent steps, we carefully checked the protonation state of all compounds at physiological pH, paying particular attention to uncertain cases with two major populated species (see Table 2 and Methods section for details). In details, for these ambiguous cases, we searched the literature for experimental values of pKa, that were consistently used as reference throughout the class. As for the QM calculations, the DFT level of theory adopted is routinely used and has proven to be reliable for small organic molecules, providing FF parameters compatible with available FFs for macromolecules50. B3LYP/6–31G** calculations represent a good compromise between accuracy and computational cost96. Therefore, no further validation is here provided for QM calculations. In the following we present a thorough justification of the reliability of our data through the comparison with experiments.
Descriptors validation
Calculation of classical parameters reporting the topological properties of molecules, such as the number of atoms or the count of aliphatic bonds, is quite straightforward. Most popular databases (e.g. PubChem, DrugBank) indeed employ ChemAxon’s tools to automatically compute these properties. In AB-DB, we likewise used the same programs to obtain the QSAR descriptors. However, for LogP, which is known to be a key feature for antimicrobial penetration kinetics61,97,98, we exploited another widely used method (XLOGP3, see Methods section). Note that accurate prediction of LogP is a well-known challenge in computational chemistry, and is also common to find severe disagreement through experimental results obtained for the same compound99,100. In order to assess the quality of our predictions, we collected available experimental LogP values for a subset of molecules. Table 3 compares the experimental data, falling in the range [−1.69, 5.15], with the computed ones, highlighting the differences between the two methods. In most cases the two predicted values are similar and agree with the experimental LogP. However, as expected, ambiguous situations were also found. Methicillin, for instance, was well predicted by XLOGP3 (computed 1.96 vs. experimental ~1.90) while cxcalc yielded a poor estimation (computed 0.79). On the contrary, the latter program agrees with experiments for lomefloxacin (computed −0.43 vs. experimental −0.47), whereas the former failed (computed 0.27).
Validation of force-field parameters and molecular dynamics trajectories
To assess the reliability of the FF generated for all compounds we computed the RMSD between the QM B3LYP/6–31G** optimized geometry and the molecular mechanics minimum-energy structure obtained with the GAFF2 parameters of the database. Table 4 shows the good agreement between the two sets of structures, differing on average by less than 1 Å, with an overall mean value of 0.5 ± 0.1 Å. The registered low RMSDs prove the accuracy of the FF parameters presented in AB-DB and used for the MD simulations.
To give a measure of the quality of MD simulations, we compared the representative conformations extracted from MD trajectories (cluster representatives) of selected compounds with their 3D experimental structure available on the Protein Data Bank, in complex with biological targets. When multiple experimental structures were available for the same compound, we considered the one with the highest resolution. The total number of 85 experimental structures collected are listed in Table S2, reporting the corresponding PDB code and the weighted average RMSDs (<RMDS>w), obtained using cluster populations as weights. The average values associated to selected families are also given. The mean value of <RMDS>w considering all families is 1.8 ± 0.8 Å, with the highest and smallest value reached by aminocoumarins (3.6 ± 0.4 Å) and tetracyclines (0.9 ± 0.2 Å), respectively. As expected, bigger and more flexible molecules give rise to higher <RMDS>w, whereas smaller and more rigid compounds show lower values. Overall, the performed MD simulations based on GAFF2 parameters appear to be able to sample molecular conformations found in available experimental structures.
As emphasized in previous works54,60, MD simulations enable to go beyond a static picture of molecules, providing ranges of properties accounting for their dynamical nature and their impact on biological activity. Prominent examples are represented by MD simulations performed to differentiate the most active inhibitors of ERK2 kinase39 and Ptch1 multidrug efflux transporter58.
Code availability
QSAR calculations were performed using the ChemAxon’s Marvin suite of programs, version 21.1463. For QM calculations we used the Gaussian16 package, revision A.0365. The Amber18 package80 was used for MD simulations and FF generation. We used simple bash scripts to iteratively extract descriptors from outputs and generate AB-DB data-files.
References
Partridge, S. R., Kwong, S. M., Firth, N. & Jensen, S. O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31, e00088–17, https://doi.org/10.1128/CMR.00088-17 (2018).
Eichenberger, E. M. & Thaden, J. T. Epidemiology and mechanisms of resistance of extensively drug resistant Gram-negative bacteria. Antibiotics 8, https://doi.org/10.3390/antibiotics8020037 (2019).
Breijyeh, Z., Jubeh, B. & Karaman, R. Resistance of Gram-negative bacteria to current antibacterial agents and approaches to resolve it. Molecules 25, 1340, https://doi.org/10.3390/molecules25061340 (2020).
Im, W. & Khalid, S. Molecular simulations of Gram-negative bacterial membranes come of age. Annu. Rev. Phys. Chem. 71, 171–188, https://doi.org/10.1146/annurev-physchem-103019-033434 (2020).
Klobucar, K. & Brown, E. D. New potentiators of ineffective antibiotics: Targeting the Gram-negative outer membrane to overcome intrinsic resistance. Curr. Opin. Chem. Biol. 66, 102099, https://doi.org/10.1016/j.cbpa.2021.102099 (2022).
Zgurskaya, H. I., Walker, J. K., Parks, J. M. & Rybenkov, V. V. Multidrug efflux pumps and the two-faced Janus of substrates and inhibitors. Acc. Chem. Res. 54, 930–939, https://doi.org/10.1021/acs.accounts.0c00843 (2021).
Zgurskaya, H. I., Malloci, G., Chandar, B., Vargiu, A. V. & Ruggerone, P. Bacterial efflux transporters’ polyspecificity - a gift and a curse? Curr. Opin. Microbiol. 61, 115–123, https://doi.org/10.1016/j.mib.2021.03.009 (2021).
Alav, I. et al. Structure, assembly, and function of tripartite efflux and type 1 secretion systems in Gram-negative bacteria. Chem. Rev. 121, 5479–5596, https://doi.org/10.1021/acs.chemrev.1c00055 (2021).
Tooke, C. L. et al. β-lactamases and β-lactamase inhibitors in the 21st century. J. Mol. Biol. 431, 3472–3500, https://doi.org/10.1016/j.jmb.2019.04.002 (2019).
Blaskovich, M. A. T., Zuegg, J., Elliott, A. G. & Cooper, M. A. Helping chemists discover new antibiotics. ACS Infect. Dis. 1, 285–287, https://doi.org/10.1021/acsinfecdis.5b00044 (2015).
Zuegg, J., Hansford, K. A., Elliott, A. G., Cooper, M. A. & Blaskovich, M. A. T. How to stimulate and facilitate early stage antibiotic discovery. ACS Infect. Dis. 6, 1302–1304, https://doi.org/10.1021/acsinfecdis.0c00163 (2020).
Lewis, K. The science of antibiotic discovery. Cell 181, 29–45, https://doi.org/10.1016/j.cell.2020.02.056 (2020).
Miethke, M. et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 5, 726–749, https://doi.org/10.1038/s41570-021-00313-1 (2021).
Klug, D. M. et al. There is no market for new antibiotics: this allows an open approach to research and development. Wellcome Open Res. 6, https://doi.org/10.12688/wellcomeopenres.16847.1 (2021).
Durand-Reville, T. F. et al. Rational design of a new antibiotic class for drug-resistant infections. Nature 597, 698–702, https://doi.org/10.1038/s41586-021-03899-0 (2021).
Farha, M. A. & Brown, E. D. Drug repurposing for antimicrobial discovery. Nat. Microbiol. 4, 565–577, https://doi.org/10.1038/s41564-019-0357-1 (2019).
Oselusi, S. O., Christoffels, A. & Egieyeh, S. A. Cheminformatic characterization of natural antimicrobial products for the development of new lead compounds. Molecules 26, 3970, https://doi.org/10.3390/molecules26133970 (2021).
Ropponen, H.-K., Richter, R., Hirsch, A. K. & Lehr, C.-M. Mastering the Gram-negative bacterial barrier - chemical approaches to increase bacterial bioavailability of antibiotics. Adv. Drug Deliv. Rev. 172, 339–360, https://doi.org/10.1016/j.addr.2021.02.014 (2021).
O’Shea, R. & Moser, H. E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 51, 2871–2878, https://doi.org/10.1021/jm700967e (2008).
Brown, D. G., May-Dracka, T. L., Gagnon, M. M. & Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 57, 10144–10161, https://doi.org/10.1021/jm501552x (2014).
Richter, M. F. et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304, https://doi.org/10.1038/nature22308 (2017).
Haloi, N. et al. Rationalizing the generation of broad spectrum antibiotics with the addition of a positive charge. Chem. Sci. 12, 15028–15044, https://doi.org/10.1039/D1SC04445A (2021).
Mehla, J. et al. Predictive rules of efflux inhibition and avoidance in Pseudomonas aeruginosa. mBio 12, e02785–20, https://doi.org/10.1128/mBio.02785-20 (2021).
Carracedo-Reboredo, P. et al. A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotechnol. J. 19, 4538–4558, https://doi.org/10.1016/j.csbj.2021.08.011 (2021).
Stokes, J. M. et al. A deep learning approach to antibiotic discovery. Cell 180, 688–702.e13, https://doi.org/10.1016/j.cell.2020.01.021 (2020).
Tkatchenko, A. Machine learning for chemical discovery. Nat. Commun. 11, 4125, https://doi.org/10.1038/s41467-020-17844-8 (2020).
Schneider, P. et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 19, 353–364, https://doi.org/10.1038/s41573-019-0050-3 (2020).
Kim, S. et al. Pubchem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 49, D1388–D1395, https://doi.org/10.1093/nar/gkaa971 (2021).
Wishart, D. S. et al. Drugbank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 34, D668–72, https://doi.org/10.1093/nar/gkj067 (2006).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954, https://doi.org/10.1093/nar/gkw1074 (2017).
Sterling, T. & Irwin, J. J. Zinc 15 - ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337, https://doi.org/10.1021/acs.jcim.5b00559 (2015).
Montavon, G. et al. Machine learning of molecular electronic properties in chemical compound space. New J. Phys. 15, https://doi.org/10.1088/1367-2630/15/9/095003 (2013).
Ramakrishnan, R., Dral, P. O., Rupp, M. & von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, https://doi.org/10.1038/sdata.2014.221 (2014).
Isert, C., Atz, K., Jiménez-Luna, J. & Schneider, G. Qmugs: Quantum mechanical properties of drug-like molecules. arxiv https://arxiv.org/abs/2107.00367 (2021).
NIST computational chemistry comparison and benchmark database. NIST Standard Reference Database Number 101 http://cccbdb.nist.gov/, https://doi.org/10.18434/T47C7Z (2020).
Ropo, M., Schneider, M., Baldauf, C. & Blum, V. First-principles data set of 45,892 isolated and cation-coordinated conformers of 20 proteinogenic amino acids. Sci. Data 3, https://doi.org/10.1038/sdata.2016.9 (2016).
Ahamad, S., Islam, A., Ahmad, F., Dwivedi, N. & Hassan, M. I. 2/3d-QSAR, molecular docking and md simulation studies of FtsZ protein targeting benzimidazoles derivatives. Comput. Biol. Chem. 78, 398–413, https://doi.org/10.1016/j.compbiolchem.2018.12.017 (2019).
Zin, P. P. K., Borrel, A. & Fourches, D. Benchmarking 2d/3d/MD-QSAR models for imatinib derivatives: How far can we predict? J. Chem. Inf. Model. 60, 3342–3360, https://doi.org/10.1021/acs.jcim.0c00200 (2020).
Ash, J. & Fourches, D. Characterizing the chemical space of ERK2 kinase inhibitors using descriptors computed from molecular dynamics trajectories. J. Chem. Inf. Model. 57, 1286–1299, https://doi.org/10.1021/acs.jcim.7b00048 (2017).
Fourches, D. & Ash, J. 4d-Quantitative structure-activity relationship modeling: making a comeback. Expert Opin. Drug Discov. 14, 1227–1235, https://doi.org/10.1080/17460441.2019.1664467 (2019).
Jamal, S., Grover, A. & Grover, S. Machine learning from molecular dynamics trajectories to predict caspase-8 inhibitors against Alzheimer’s disease. Front. Pharmacol. 10, 780, https://doi.org/10.3389/fphar.2019.00780 (2019).
Esposito, C., Wang, S., Lange, U. E. W., Oellien, F. & Riniker, S. Combining machine learning and molecular dynamics to predict p-glycoprotein substrates. J. Chem. Inf. Model. 60, 4730–4749, https://doi.org/10.1021/acs.jcim.0c00525 (2020).
Bak, A. Two decades of 4d-QSAR: A dying art or staging a comeback? Int. J. Mol. Sci. 22, 5212, https://doi.org/10.3390/ijms22105212 (2021).
Boehr, D. D., Nussinov, R. & Wright, P. E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796, https://doi.org/10.1038/nchembio.232 (2009).
Baron, R. & McCammon, A. J. Molecular recognition and ligand association. Annu. Rev. Phys. Chem. 64, 151–175, https://doi.org/10.1146/annurev-physchem-040412-110047 (2013).
Zivanovic, S. et al. Exploring the conformational landscape of bioactive small molecules. J. Chem. Theory Comput 16, 6575–6585, https://doi.org/10.1021/acs.jctc.0c00304 (2020).
Salo-Ahen, O. M. H. et al. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 9, https://doi.org/10.3390/pr9010071 (2021).
Jo, S., Kim, T., Iyer, V. & Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865, https://doi.org/10.1002/jcc.20945 (2008).
Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. J. Automation of the CHARMM general force field (CGenFF) ii: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 52, 3155–3168, https://doi.org/10.1021/ci3003649 (2012).
Dupradeau, F.-Y. et al. R.E.DD.B.: A database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 36, D360–D367, https://doi.org/10.1093/nar/gkm887 (2008).
Bryce, R. Amber parameter database. http://amber.manchester.ac.uk/.
Morado, J. et al. Paramol: A package for automatic parameterization of molecular mechanics force fields. J. Chem. Inf. Model. 61, 2026–2047, https://doi.org/10.1021/acs.jcim.0c01444 (2021).
Lin, F.-Y. & MacKerell, A. D. J. Force Fields for Small Molecules (Humana, New York, 2019).
Malloci, G. et al. A database of force-field parameters, dynamics, and properties of antimicrobial compounds. Molecules 20, 13997–14021, https://doi.org/10.3390/molecules200813997 (2015).
Auer, J. & Bajorath, J. Distinguishing between bioactive and modeled compound conformations through mining of emerging chemical patterns. J. Chem. Inf. Model. 48, 1747–1753, https://doi.org/10.1021/ci8001793 (2008).
Bai, F. et al. Bioactive conformational generation of small molecules: A comparative analysis between force-field and multiple empirical criteria based methods. BMC Bioinform. 11, 545, https://doi.org/10.1186/1471-2105-11-545 (2010).
Anighoro, A., de la Vega de León, A. & Bajorath, J. Predicting bioactive conformations and binding modes of macrocycles. J. Comput. Aided Mol. Des. 30, 841–849, https://doi.org/10.1007/s10822-016-9973-5 (2016).
Kovachka, S. et al. Molecular insights into the Patched1 drug efflux inhibitory activity of Panicein A Hydroquinone: a computational study. Phys. Chem. Chem. Phys. 23, 8013–8022, https://doi.org/10.1039/D0CP05719C (2021).
Ebejer, J.-P., Morris, G. M. & Deane, C. M. Freely available conformer generation methods: How good are they? J. Chem. Inf. Model. 52, 1146–1158, https://doi.org/10.1021/ci2004658 (2012).
Malloci, G., Serra, G., Bosin, A. & Vargiu, A. V. Extracting conformational ensembles of small molecules from molecular dynamics simulations: Ampicillin as a test case. Computation 4, https://doi.org/10.3390/computation4010005 (2016).
Cooper, S. J. et al. Molecular properties that define the activities of antibiotics in Escherichia coli and Pseudomonas aeruginosa. ACS Infect. Dis. 4, 1223–1234, https://doi.org/10.1021/acsinfecdis.8b00036 (2018).
Zhao, S. et al. Defining new chemical space for drug penetration into gram-negative bacteria. Nat. Chem. Biol. 16, 1293–1302, https://doi.org/10.1038/s41589-020-00674-6 (2020).
ChemAxon. Marvin version 21.14. http://www.chemaxon.com (2021).
Kohn, W. Nobel lecture: Electronic structure of matter - wave functions and density functionals. Rev. Mod. Phys. 71, 1253–1266, https://doi.org/10.1103/RevModPhys.71.1253 (1999).
Frisch, M. J. et al. Gaussian 16, revision a.03. Gaussian Inc. Wallingford CT (2016).
Becke, A. D. Density-functional thermochemistry. iii. The role of exact exchange. J. Chem. Phys. 98, 5648–5652, https://doi.org/10.1063/1.464913 (1993).
Pople, J. Quantum chemical models (Nobel Lecture). Angew. Chem. Int. Ed. 38, 1894–1902, 10.1002/(SICI)1521-3773(19990712)38:13/14<1894::AID-ANIE1894>3.0.CO;2-H (1999).
Tirado-Rives, J. & Jorgensen, W. L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 4, 297–306, https://doi.org/10.1021/ct700248k (2008).
Cleydson B. R. d. S. et al. Pinheiro Evaluation of Quantum Chemical Methods and Basis Sets Applied in the Molecular Modeling of Artemisinin. Comput. Mol. Biol. 3, 66–79, https://doi.org/10.4236/cmb.2013.33009 (2013).
Narbe Mardirossian and Martin Head-Gordon Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 115, 2315-2372, https://doi.org/10.1080/00268976.2017.1333644 (2017).
Tomasi, J., Mennucci, B. & Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094, https://doi.org/10.1021/cr9904009 (2005).
O’Boyle, N., Tenderholt, A. & Langner, K. cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845, https://doi.org/10.1002/jcc.20823 (2008).
Singh, U. C. & Kollman, P. A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 5, 129–145, https://doi.org/10.1002/jcc.540050204 (1984).
Bayly, C. I., Cieplak, P., Cornell, W. & Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280, https://doi.org/10.1021/j100142a004 (1993).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 25, 247–260, https://doi.org/10.1016/j.jmgm.2005.12.005 (2006).
Jakalian, A., Jack, D. B. & Christopher, I. Bayly Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641, https://doi.org/10.1002/jcc.10128 (2009).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174, https://doi.org/10.1002/jcc.20035 (2004).
Moynié, L. et al. The complex of ferric-enterobactin with its transporter from Pseudomonas aeruginosa suggests a two-site model. Nat. Commun. 10, https://doi.org/10.1038/s41467-019-11508-y (2019).
Li, P. & Merz, K. M. Jr. MCPB.py: A Python based metal center parameter builder. J. Chem. Inf. Model. 56, 599–604, https://doi.org/10.1021/acs.jcim.5b00674 (2016).
Case, D. et al. AMBER. University of California and San Francisco 2018.
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935, https://doi.org/10.1063/1.445869 (1983).
Joung, I. S. & Cheatham, T. E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 112, 9020–9041, https://doi.org/10.1021/jp8001614 (2008).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690, https://doi.org/10.1063/1.448118 (1984).
Loncharich, R. J., Brooks, B. R. & Pastor, R. W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of n-acetylalanyl-n′-methylamide. Biopolymers 32, 523–535, https://doi.org/10.1002/bip.360320508 (1992).
Isik, M. et al. Assessing the accuracy of octanol-water partition coefficient predictions in the SAMPL6 part II logP Challenge. J. Comput. Aided Mol. Des. 34, 335–370, https://doi.org/10.1007/s10822-020-00295-0 (2020).
Cheng, T. et al. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 47, 2140–2148, https://doi.org/10.1021/ci700257y (2007).
Sauer, S. P. A. Molecular Electromagnetism: A Computational Chemistry Approach (OUP Oxford, 2011).
Roe, D. R. & Cheatham, T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095, https://doi.org/10.1021/ct400341p (2013).
Nag, A., Chakraborty, D. & Chandra, A. Effects of ion concentration on the hydrogen bonded structure of water in the vicinity of ions in aqueous NaCl solutions. J. Chem. Sci. 120, 71–77, https://doi.org/10.1007/s12039-008-0009-0 (2008).
Shao, J., Tanner, S. W., Thompson, N. & Cheatham, T. E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 3, 2312–2334, https://doi.org/10.1021/ct700119m (2007).
van der Spoel, D. et al. Gromacs User Manual version 3.0. Nijenborgh 4, 9747 AG Groningen, The Netherlands (2001).
Bonomi, M. et al. Plumed: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Comm. 180, 1961–1972, https://doi.org/10.1016/j.cpc.2009.05.011 (2009).
Theodorou, D. N. & Suter, U. W. Shape of unperturbed linear polymers: polypropylene. Macromolecules 18, 1206–1214, https://doi.org/10.1021/ma00148a028 (1985).
O’Boyle, N. et al. Open Babel: An open chemical toolbox. J. Cheminf. 3, 33, https://doi.org/10.1186/1758-2946-3-33 (2011).
Gervasoni, S. AB-DB: Force-Field parameters, MD trajectories, QM-based data, and Descriptors of Antimicrobials, figshare, https://doi.org/10.6084/m9.figshare.c.5752703 (2022).
Rodrigues-Oliveira, A. F., Ribeiro, F. W. M., Cervi, G. & Correra, T. C. Evaluation of common theoretical methods for predicting infrared multiphotonic dissociation vibrational spectra of intramolecular hydrogen-bonded ions. ACS Omega 3, 9075–9085, https://doi.org/10.1021/acsomega.8b00815 (2018).
Davis, T. D., Gerry, C. J. & Tan, D. S. General platform for systematic quantitative evaluation of small-molecule permeability in bacteria. ACS Chem. Biol. 9, 2535–2544, https://doi.org/10.1021/cb5003015 (2014).
Blokhina, S. V., Sharapova, A. V., Ol’khovich, M. V., Volkova, T. V. & Perlovich, G. L. Solubility, lipophilicity and membrane permeability of some fluoroquinolone antimicrobials. Eur. J. Pharm. Sci. 93, 29–37, https://doi.org/10.1016/j.ejps.2016.07.016 (2016).
Stratton, C. F., Newman, D. J. & Tan, D. S. Cheminformatic comparison of approved drugs from natural product versus synthetic origins. Bioorg. Med. Chem. Lett. 25, 4802–4807, https://doi.org/10.1016/j.bmcl.2015.07.014 (2015).
Dorwald, F. Z. Lead Optimization for Medicinal Chemists (Wiley-VCH Verlag & Co. KGaA, Boschstr. 12, 69469 Weinheim, Germany, 2012).
Systémes, B. D. Biovia discovery studio 2021. San Diego: Dassault Systémes (2021).
The PyMOL molecular graphics system, version 1.3r1. The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC (2010).
Babic, S., Horvat, A. J., Mutavdžc-Pavlovic, D. & Kaštelan-Macan, M. Determination of pKa values of active pharmaceutical ingredients. Trends Analyt. Chem. 26, 1043–1061, https://doi.org/10.1016/j.trac.2007.09.004 (2007).
Hirano, T. et al. Mechanism of the inhibitory effect of zwitterionic drugs (levofloxacin and grepafloxacin) on carnitine transporter (OCTN2) in Caco-2 cells. Biochim. Biophys. Acta Biomembr. 1758, 1743–1750, https://doi.org/10.1016/j.bbamem.2006.07.002 (2006).
Alkhzem, A. H., Woodman, T. J. & Blagbrough, I. S. Individual pKa values of tobramycin, kanamycin b, amikacin, sisomicin, and netilmicin determined by multinuclear NMR spectroscopy. ACS Omega 5, 21094–21103, https://doi.org/10.1021/acsomega.0c02744 (2020).
Lesniakac, W., Larena, J. M., Harrisd, W. R., Pecoraroc, V. L. & Schachtab, J. An isocratic separation of underivatized gentamicin components, 1H NMR assignment and protonation pattern. Carbohydr. Res. 338, 2853–2862, https://doi.org/10.1016/j.carres.2003.08.005 (2003).
Topal, B. D., Golcu, A. & Ozkan, S. A. Electrochemical investigation and determination of the antibacterial loracarbef by voltammetric methods. Anal. Lett. 42, 689–705, https://doi.org/10.1080/00032710802678637 (2009).
Cocco, L. et al. Protonated state of methotrexate, trimethoprim, and pyrimethamine bound to dihydrofolate reductase. Arch. Biochem. Biophys. 226, 567–577, https://doi.org/10.1016/0003-9861(83)90326-0 (1983).
Rifampin. Tuberculosis 88, 151–154, https://doi.org/10.1016/S1472-9792(08)70024-6 (2008).
Song, J. et al. Discovery of prenyltransferase inhibitors with in vitro and in vivo antibacterial activity. ACS Infect. Dis. 6, 2979–2993, https://doi.org/10.1021/acsinfecdis.0c00472 (2020).
Ibrahim, T. S. et al. Novel 1,2,4-oxadiazole-chalcone/oxime hybrids as potential antibacterial DNA gyrase inhibitors: Design, synthesis, ADMET prediction and molecular docking study. Bioorg. Chem. 111, https://doi.org/10.1016/j.bioorg.2021.104885 (2021).
Sebastiano, M. R. et al. Impact of dynamically exposed polarity on permeability and solubility of chameleonic drugs beyond the rule of 5. J. Med. Chem. 61, 4189–4202, https://doi.org/10.1021/acs.jmedchem.8b00347 (2018).
Tsuji, A., Kubo, O., Miyamoto, E. & Yamana, T. Physicochemical properties of β-lactam antibiotics: Oil-water distribution. J. Pharm. Sci. 66, 1675–79, https://doi.org/10.1002/jps.2600661205 (1977).
Rashid, U. et al. Structure based medicinal chemistry-driven strategy to design substituted dihydropyrimidines as potential antileishmanial agents. Eur. J. Med. Chem. 115, 230–244, https://doi.org/10.1016/j.ejmech.2016.03.022 (2016).
Ito, T., Yano, I., Masuda, S. & Hashimoto, Y. & ichi Inu, K. Distribution characteristics of levofloxacin and grepafloxacin in rat kidney. Pharm. Res. 16, 534–539, https://doi.org/10.1023/A:1018871029244 (1999).
Rankovic, Z. CNS physicochemical property space shaped by a diverse set of molecules with experimentally determined exposure in the mouse brain. J. Med. Chem. 60, 5943–5954, https://doi.org/10.1021/acs.jmedchem.6b01469 (2017).
Klosinska-Szmurlo, E. et al. Physicochemical properties of lomefloxacin, levofloxacin, and moxifloxacin relevant to the biopharmaceutics classification system. Acta Chim. Slov. 61, 827–34 (2014).
Gunaydin, H. Probabilistic approach to generating MPOs and its application as a scoring function for CNS drugs. ACS Med. Chem. Lett. 7, 89–93, https://doi.org/10.1021/acsmedchemlett.5b00390 (2016).
Acknowledgements
This work was supported by National Institute of Allergy and Infectious Diseases/NIH grant no. R01AI136799 and “Progetti biennali d’Ateneo Finanziati dalla Fondazione di Sardegna - annualità 2020.” We thank Dr. Giovanni Serra (University of Cagliari) for technical assistance with computational resources.
Author information
Authors and Affiliations
Contributions
G.M., A.B., A.V.V., H.I.Z. and P.R. conceived the project, S.G. and G.M. performed the calculations, S.G., G.M. and A.B. analysed the results, curated the data and implemented the database. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gervasoni, S., Malloci, G., Bosin, A. et al. AB-DB: Force-Field parameters, MD trajectories, QM-based data, and Descriptors of Antimicrobials. Sci Data 9, 148 (2022). https://doi.org/10.1038/s41597-022-01261-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01261-1
This article is cited by
-
Predicting permeation of compounds across the outer membrane of P. aeruginosa using molecular descriptors
Communications Chemistry (2024)
