
Abstract
Phenotypically diverse species from recently evolved groups always share allele/haplotype due to insufficient differentiation in the early process. In this study, we performed population genetics analyses using sequences from the mitochondrial cytochrome b gene, and two nuclear genes to investigate the genetic differentiation of the closely related Schizothorax species complex, comprising a group of alpine fish living in the Nujiang River. The results from both mtDNA and nDNA markers revealed relatively low but pronounced genetic differentiation among the three Schizothorax species, i.e., Schizothorax gongshanensis, S. lissolabiatus, and S. nukiangensis. However, haplotype sharing was frequently occurred among the three species. Divergence time estimation suggested the last glaciation on the Tibetan Plateau (0.075–0.01 Ma) might drive the divergence of the species complex. Gene flow might contribute to the haplotype sharing between S. gongshanensis and S. lissolabiatus, and between S. gongshanensis and S. nukiangensis, whereas retention of ancestral polymorphisms seemed to be a better explanation of the haplotype sharing between S. lissolabiatus and S. nukiangensis. In addition, S. lissolabiatus populations should obtain more protection in the future because of their low genetic diversity and habitat fragmentation. In summary, our study assesses genetic differentiation among the three closely related Schizothorax species and explores the possible driving forces for their differentiation.
Similar content being viewed by others

Introduction
In many DNA-based analyses, the genes of different populations and species from recently evolved groups exhibit insufficient differentiation signature in this early process1, 2, resulting in allele sharing among phenotypically diverse species. The retention of ancestral polymorphisms and hybridization can both contribute to allele sharing. When an ancestral population harbors a genetic polymorphism, descendant lineages are expected to share polymorphic alleles, reflecting insufficient time to coalescence. However, when pre-mating barriers are not sufficient to hinder local gene flow among young species, these organisms also share genetic polymorphisms in some areas of the genome through hybridization3. Even low levels of gene flow can maintain the sharing of polymorphisms across porous species boundaries, and ancestrally polymorphic alleles can reach fixation over time due to natural selection and/or genetic drift4.
Schizothoracine (Cyprinidae) fishes, representing the largest and most diverse taxon of the highland ichthyofauna, dominate the torrential mountain streams and plateau lakes of Central Asia, the Himalayas, and peripheral regions of the Tibetan Plateau5, 6. The genus Schizothorax is the most diversified schizothoracine genus, possessing more than 100 species and subspecies (www.fishbase.org). However, in this genus, the discrepancies between classical taxonomy based on morphology and the molecular phylogeny were frequently observed7,8,9. For example, allele sharing is ubiquitous among different morphological species in the same drainage7, 9.
Nujiang River (upper Salween) is an important international river originating from the Tibetan Plateau and flowing through the southwest mountain regions of China. This region has retained a distinct fish fauna, reflecting its complex geological history and landscape diversity10. The Nujiang River basin harbors four recognized morphological species/subspecies of the genus Schizothorax, Schizothorax gongshanensis, S. lissolabiatus, S. nukiangensis and S. yunnanensis paoshanensis 6, 11, 12. S. gongshanensis and S. nukiangensis are endemic in Nujiang River. S. gongshanensis occurs only in a small area in the main stem between north Yunnan and east Tibet and in some tributaries in the middle and lower drainage (Figure S1), and S. nukiangensis is widespread throughout the main stem and large tributaries in Tibet6, 11,12,13. In contrast, S. lissolabiatus occupies many isolated drainages in Southwest China, e.g., Nujiang River, Lancang River, Red River, and upper Pearl river. However, Yang et al. (2012) found that S. lissolabiatus from the Nujiang River was phylogenetically distinct with S. lissolabiatus from other drainages9, which suggests independent evolutionary history of S. lissolabiatus in the Nujiang River and rules out the possibility of colonization from other drainages. In the Nujiang River, S. lissolabiatus only occurs in the tributary of the middle and lower drainage (Figure S1)6, 11,12,13. Schizothorax yunnanensis paoshanensis is exclusively observed in the Donghe River basin (Longwang spring and Beimiao reservoir) and Lanzha River in the middle Nujiang River drainage6, 11,12,13. Schizothorax yunnanensis paoshanensis and S. yunnanensis yunnanensis can be distinguished in the length of mouth palpus and distribution6. Schizothorax yunnanensis yunnanensis only distributes in the Lancang River6.
Previous phylogenetic analyses have demonstrated that S. gongshanensis, S. lissolabiatus, and S. nukiangensis cluster into a single lineage with low genetic difference and even share mitochondrial DNA (mtDNA) haplotypes9, 14. However, the level of genetic differentiation among the three closely related species remained unresolved. Furthermore, Schizothorax yunnanensis paoshanensis generated a sister group with a Schizothorax species complex in the Irrawaddy River basin rather than the Nujiang River species complex9, S. yunnanensis paoshanensis was excluded in the present study. Nearly all previous DNA-based studies of the Schizothorax species complex in the Nujiang River basin used only mtDNA fragments with limted sample size and did not include nuclear DNA (nDNA) information9, 13.
Herein, our aims were to assess genetic differentiation among the three closely related Schizothorax species complex and to explore the possible driving forces for their differentiation through dense sampling combining both mtDNA and nDNA markers. In addition, given that cyclical cooling-warming events during the Pleistocene could facilitate population divergence and consequent speciation15,16,17, we also examined whether the Pleistocene climatic oscillations influenced the genetic differentiation of the three closely related species.
Results
Sequence information
We used a total of 447 mitochondrial cytochrome b gene (Cytb) sequences, including 55 de novo sequences, 224 sequences (Genbank nos: KM070647–KM070729) from Chen et al.18, 151 sequences (Genbank nos: KP796151–KP796154, KP796156–KP796158 and KP796160–KP796168) from Yue et al.13, and 17 sequences downloaded from NCBI database. The overall Cytb sequences came from 26 locations in the Nujiang River basin (Table S1; Fig. 1). The 447 Cytb (1063 bp) sequences contained 49 variable sites and 33 parsimony-informative sites. A total of 40 haplotypes were defined from Cytb sequences (Table 1).

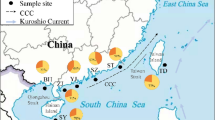
Map of the sampling sites for the Schizothorax species complex in the Nujiang River. The site numbers are presented in Table S1. 1–13, main stem sampling sites; 14–26, sampling sites for S. gongshanensis and S. lissolab1atus of the tributary in the Nujiang River. Populations are presented as pie-diagrams with slice-size proportional to the frequency of the three species (Red: S. gongshanensis; blue: S. lissolab1atus; and green: S. nukiangensis). Sample sites harbored two species are exhibited with larger pie-diagrams. Map was created in the ArcGIS version 10.1 and modified in Microsoft Office.
We obtained partial sequences for the recombinase-activating gene proteins 1 and 2 (RAG-1 and RAG-2) from a subset of all samples (Table S1). The dataset included 75 sequences (1466 bp) from RAG-1 and 61 sequences (1226 bp) from RAG-2. The longest non-recombining regions of RAG-1 (1466 bp) and RAG-2 (1226 bp) contained 16 and 15 variable sites, respectively.
Phylogenetic relationship, mtDNA haplotype and nDNA allele relationships
The phylogenetic trees of the in-group obtained using Cytb via the Bayesian inference (BI) and maximum parsimony (MP) approaches showed a marked consistency in topological congruence, differing only in the support values for certain nodes; thus, only the MP tree was presented (Figure S2). The trees showed that S. gongshanensis, S. lissolabiatus, and S. nukiangensis were clustered into a single lineage, and S. yunnanensis paoshanensis generated another lineage (Figure S2). Thus, S. yunnanensis paoshanensis was excluded in the subsequent analyses.
The grouping patterns of the Cytb median-joining network (MJN) clearly showed the haplotype relationships among the different species (Fig. 2). Only one haplotype (H1) was shared among the three species (including 32 S. gongshanensis specimens derived from the main stem, one S. gongshanensis specimen sampled from the tributary, 32 S. lissolabiatus specimens and one S. nukiangensis specimen). Four haplotypes (H1, H6, H8, H12) were shared between S. nukiangensis and S. gongshanensis (including 38 S. gongshanensis individuals and 115 S. nukiangensis individuals). Four haplotypes (H1, H32–H34) were shared by S. gongshanensis and S. lissolabiatus, and only one haplotype (H1) was shared by S. lissolabiatus and S. nukiangensis. From a total of 40 haplotypes, four haplotypes are private to S. gongshanensis, seven haplotypes are private to S. lissolabiatus, and twenty-two haplotypes are private to S. nukiangensis.

Median-joining network of Cytb for the Schizothorax species complex. Each colored circle represents different species, scaled according to its frequency in the entire sample. An empty circle indicates missing intermediate steps between observed haplotypes. The dotted rectangles indicate the populations of the three species with a shared Cytb haplotype. The location codes correspond to those in Table S1.
We built MJNs using only the longest non-recombining regions of RAG-1 and RAG-2. This strategy resulted in 13 and 12 alleles from RAG-1 and RAG-2, respectively (Table 1). The networks obtained for RAG-1 and RAG-2 exhibited slightly different results (Fig. 3). The RAG-1 MJN showed that S. gongshanensis from the main stem did share a particular allele with S. nukiangensis, and no shared alleles existed between the tributary populations (overall S. lissolabiatus specimens and S. gongshanensis from location 17) and the main stem populations (overall S. nukiangensis specimens and S. gongshanensis from the main stem) (Fig. 3a). In contrast, two mixed alleles were shared between the three species (S. gongshanensis, S. lissolabiatus and S. nukiangensis), and one mixed allele was shared between S. gongshanensis and S. nukiangensis, from the RAG-2 MJN (Fig. 3b). In the both MJNs, both S. gongshanensis and S. lissolabiatus from the tritutary were found to share an allele.

Median-joining network of the nuclear gene fragments for the Schizothorax species complex. Each colored circle represents different species, scaled according to its frequency in the entire sample. The dotted rectangles indicate the populations of the three species with a shared allele. An empty circle indicates missing intermediate steps between observed alleles. (a) RAG-1. (b) RAG-2.
Genetic diversity and genetic differentiation
We calculated the haplotype and nucleotide diversities of the three species using Cytb sequences (Table S2). The haplotype and nucleotide diversities were greatest in S. lissolabiatus (0.863 ± 0.010 and 0.0028 ± 0.0001, respectively) and lowest in S. gongshanensis (0.676 ± 0.084 and 0.0014 ± 0.0002, respectively).
Pairwise comparisons of the genetic differentiation (ɸST) revealed significant genetic differentiation between the species pairs (Table 2). Low but statistically significant differentiation (P < 0.001) was determined for the three species based on Cytb. The analysis of nDNA genes showed that most of the pairwise species comparisons exhibited significant genetic differentiation (except between S. gongshanensis and S. nukiangensis from RAG-2) (Table 2). Furthermore, in the hierarchical analysis of molecular variance (AMOVA) results, we examined a low but significant genetic differentiation among the three species (ɸCT = 0.05, P = 0.048) (Table 3).
We examined the pairwise genetic differentiation among populations with more than five specimens within each species (Tables 4, S3 and S4). For S. gongshanensis, low genetic differentiation was obtained between the two main stem populations, whereas moderate and significant differentiation was detected between the tributary population and the two main stem populations (Table S3). High and statistically significant differentiation (P < 0.05) was observed among most of populations within S. lissolabiatus (Table 4). With regard to S. nukiangensis, moderate and statistically significant differentiation was found between SJK population and other populations, whereas low genetic differentiation values were examined among the remaining populations (Table S4).
Gene flow
Simulation with IMa2 revealed statistically significant (P < 0.001) migration events among the three species (Fig. 4). Migration events occurred in both directions between S. gongshanensis and S. nukiangensis (2NM = 0.88 from S. gongshanensis to S. nukiangensis and 2NM = 1.4 from S. nukiangensis to S. gongshanensis), while migration events were unidirectional from S. lissolabiatus to S. gongshanensis (2NM = 1.3) and from S. lissolabiatus to S. nukiangensis (2NM = 0.27). The MDIV analyses showed that migration rate (M) was ranged from 2NM = 0.18 between S. lissolabiatus and S. nukiangensis to 2NM = 0.90 between S. gongshanensis and S. lissolabiatus (Table 5). Both analyses showed relatively low level of gene flow betweent S. lissolabiatus to S. nukiangensis.

Isolation with migration analyses for the three Schizothorax species based on Cytb. The arrows represent migration directions from the source population to the receiving population; the numbers next to arrows are 2NM values. Only statistically significant cases of gene flow are presented. *P < 0.05 and ***P < 0.001.
Divergence time estimation
Net average sequence distances between species varied from 0.0004 to 0.0005 and yielded estimates of divergence between S. gongshanensis, S. lissolabiatus and S. nukiangensis dating to 0.02–0.025 Ma (Table S5). The MDIV results indicated that the divergence time between S. gongshanensis and S. lissolabiatus (0.008 Ma) was more recent than the divergence time between S. gongshanensis and S. nukiangensis (0.033 Ma) and between S. lissolabiatus and S. nukiangensis (0.061 Ma) (Table 5).
Demographic history
Neutrality tests yielded statistically significant negative values for S. gongshanensis and S. nukiangensis (Table S2). The sum of square deviations and raggedness index suggested that the curves did not significantly deviate from the distributions expected under a model of sudden demographic expansion for S. gongshanensis (Table S2). The extended Bayesian skyline plot (EBSP) obtained similar results with the neutrality tests (Figure S3). EBSP suggested that population expansion for S. gongshanensis occurred between 0.06 Ma and 0.01 Ma. By contrast, S. nukiangensis appeared to experience population expansion between 0.04 Ma and 0.01 Ma. No signal of recent population expansion for S. lissolabiatus was detected in our analyses (Table S2; Figure S3).
Discussion
Genetic differentation among the three species
Genetic differentiation among the three closely related species was observed in the current study, although the haplotypes of the three taxa cluster into a single unresolved clade9, 19, and even the three species share mtDNA and nDNA haplotypes. Several lines of evidence for the genetic differentiation were comfirmed in our genetic analyses. First, only one Cytb haplotype, no RAG-1 allele, and two RAG-2 alleles shared between S. lissolabiatus and S. nukiangensis indicated remarkable genetic differentiation and low level of gene flow between them. Second, relatively low but statistically significant pairwise genetic differentiation (ɸST) was consistently obtained among the three species based on mtDNA and nDNA markers (Table 2). The genetic differentiation of nDNA markers larger than Cytb gene might be largely due to the difference in sample size and the number of populations used for nDNA genes and Cytb. We used the same sample to calculate ɸCT values and found that the genetic differentiation of Cytb was larger than the nuclear genes, except S. lissolabiatus vs. S. nukiangensis in RAG-1 (Table S6). The nuclear genes with degenerate base used to calculate ɸCT values were firstly resolved into two alleles, which could increase the number of alleles and variable sites. This factor also could influence the ɸCT calculations. Thirdly, a hierarchical AMOVA based on Cytb supported a low but significant genetic differentiation among the three species (ɸCT = 0.05, P = 0.048).
On the Tibetan Plateau, the maximum extent of glacier development occurred during the middle Pleistocene (0.5 Ma), while glacial retreat has occurred since 0.17 Ma20,21,22, in contrast to the European and North American ice sheets, with a maximum extent in the Last Glacial Maximum (0.023–0.018 Ma)22,23,24. The Tibetan Plateau entered the last glacial age at approximately 0.075 Ma, continuing until 0.01 Ma25, 26. Mutation rates estimated for the Schizothorax species complex in the upper Mekong River19 placed divergence times between the species from 0.02 to 0.025 Ma based on net average sequence distances and from 0.008 to 0.061 Ma based on MDIV. Both estimates of divergence time consistently pointed to their separation during the last glaciation on the Tibetan Plateau (0.075–0.01 Ma). Considering that estimated divergence times vary depending on the assumed substitution rate, we cautiously hypothesized that the Pleistocene glaciations on the Tibetan Plateau facilitated the differentiation of the Schizothorax species complex in the Nujiang River. A similar case was observed in the Gymnocypris chilianensis in the Hexi River system27. During the last glaciation (0.075–0.01 Ma)25, 26, the drier and cold weather might divide the ancestral population of the Schizothorax species complex and result in the three present species diverging in allopatry. Secondary contact between the three new species was established due to coming of the warmer (interstadial) episodes during the glaciations25. Each glacial period was composed of a series of alternating cooler (stadial) and warmer (interstadial) episodes21, 28. Population expansion of S. gongshanensis and S. nukiangensis during the late Pleistocene inferred from demographic analyses confirmed the potential events of secondary contact and effects of the late Pleistocene glaciations on the Schizothorax species complex. Furthermore, trophic alternation due to oligotrophic condition during the glaciations may also be a potential explanation for the divergence of the three species17. The three species are clearly morphologically distinct regarding the pattern of the lower lip and lower jaw and the number of gill rakers (Table S7), which are all strongly associated with the ability to process different food types in schizothoracine fish29, 30. The absence of food during the glaciations increased the possibility for food competition and facilitated forming the shape of trophically relevant structures. The significant morphological differences can be maintained by disruptive natural selection. A similar process was seen in the Gymnocypris species complex in Lake Sunmcuo31.
Haplotype shared among the three species
The sharing of haplotypes between/among different species can be attributed to one of two main processes, i.e., retention of ancestral polymorphisms due to recent divergence and gene flow. In our study, high frequency of haplotype shared was examined between S. gongshanensis and S. lissolabiatus, and between S. gongshanensis and S. nukiangensis (Figs 2 and 3). Because the hybrid zone where the two species-paris meet is broad (i.e., S. gongshanensis and S. nukiangensis are sympatric in some tributary and S. gongshanensis and S. nukiangensis occupy the same distribution between Fugong in Yunnan and southern section in Tibet in main stem11), recent gene flow seems a likely explanation. Furthermore, IM and MDIV results also detected substantial gene flow between the two species-pairs (Table 4, Fig. 4). In contrast, limited haplotypes shared and allopatric distribution between S. lissolabiatus and S. nukiangensis suggests rare gene flow between them, though relatively low level of gene flow was observed from the IM and MDIV calculations. However, the detected levels of gene flow might not be high enough to prevent differentiation; a 2NM greater than one would limit the divergence process in the absence of selection32. Consequently, shared ancestral polymorphisms is more suitable to interpret the haplotype sharing between the two species.
Low genetic diversity and fragmentation of S. lissolabiatus populations
Genetic analyses indicated that S. lissolabiatus populations (locations 14–26) displayed remarkably low mtDNA genetic diversity. Seven out of nine populations with more than five individuals were found to harbor a single Cytb haplotype, except locations 21 and 22, which contained three Cytb haplotypes (Table 1). In addition, higher pairwise population differentiation and limited Cytb haplotype sharing among different S. lissolabiatus populations suggested rare gene flow and fragmentation among the tributary populations (Table 1; Fig. 2). The tributary is pretty small and separated by numerous mountains. The isolated tributaries posed a natural barrier for gene exchange and might lead to inbreeding within population. The inbreding would make populations more and more homogeneous and lose much genetic diversity33. Considering that small and isolated populations are inherently more vulnerable to external environmental perturbations and chance fluctuations34, more attention should be paid to conserving these populations in the future.
Conclusions
Our study assembles mtDNA and nDNA loci to assess the genetic differentiation among the Schizothorax species complex in the Nujiang River basin and to explore the possible driving forces for their differentiation through dense sampling. Genetic analyses indicate relatively low but pronounced genetic differentiation between the three species-pairs and support the hypothesis that the last glaciation on the Tibetan Plateau (0.075–0.01 Ma) may drive the divergence of the species complex. Gene flow may be contribute to the haplotype sharing between S. gongshanensis and S. lissolabiatus, and between S. gongshanensis and S. nukiangensis, whereas retention of ancestral polymorphisms seems to be a better alternative for the explantion of the haplotype sharing between S. lissolabiatus and S. nukiangensis. In addition, the S. lissolabiatus populations with low genetic diversity and habitat fragmentation should be paid more attention for the future protection.
Methods and Materials
Ethics statement
All experimental protocols were approved by the Ethics Committee of the Institute of Hydrobiology, Chinese Academy of Sciences. The policies were enacted according to Chinese Association for Laboratory Animal Sciences, and coordinated with the Institutional Animal Care and Use Committee (IACUC) protocols (http://iacuc.usc.edu/).
Sample collection, laboratory techniques and molecular data
A total of 430 specimens of three Schizothorax species (S. gongshanensis, S. lissolabiatus and S. nukiangensis) were collected from the Nujiang River basin from 2007 to 2013 (Table S1). Fresh samples were assigned to species based on differential morphological characters in the field (Table S5). In addition, 17 published sequences of partial mitochondrial cytochrome b gene (Cytb) belonging to S. gongshanensis, S. lissolabiatus and S. nukiangensis were added to these analyses (Table S1). A small piece of white muscle tissue or fin was dissected from the right side of each specimen. All tissue samples used for genomic DNA extraction were preserved in 95% ethanol. A total of 26 sampling locations were considered in the present study (Fig. 1; Table S1). The sampling map was generated using the ArcGIS and modified in Microsoft Office.
Total genomic DNA was extracted from muscle or fin tissue samples by using a standard salt extraction method. A segment of the Cytb gene was amplified from all individuals using the universal primers L14724 and H1591535. The PCR conditions were identical for the partial Cytb gene, with an initial denaturation at 94 °C for 3 min, followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 59–64 °C for 1 min, extension at 72 °C for 1 min, and a final extension at 72 °C for 5 min. Gene fragments of recombinase-activating gene proteins 1 and 2 (RAG-1 and RAG-2) were obtained from a subset of samples (75 individuals for RAG-1 and 61 individuals for RAG-2) using previously published primer sequences36, 37. The amplification of genomic DNA was conducted with an initial denaturation at 94 °C for 3–5 min, followed by 30–35 cycles of denaturation at 94 °C for 30 s, annealing at 53–55 °C for 30 s, extension at 72 °C for 1.5 min and a final extension at 72 °C for 10 min. The PCR reaction contained approximately 100 ng of template DNA, 1 μl of each primer (10 pmol/μl), 3 μl of 10 × reaction buffer, 1.5 μl of dNTPs (2.5 mM each) and 2.0 U of Taq DNA polymerase in a total volume of 30 μl. The amplified fragments were purified by 1.0% low-melting agarose gel electrophoresis and sequenced with an ABI PRISM 3700 (Applied Biosystems, Foster City, California, USA) automatic DNA sequencer using the same primer pairs.
Sequence analyses
The nucleotide sequences were initially edited using DNASTAR multiple package (DNASTAR. Inc., Madison, WI, USA), aligned using Muscle38 and subsequently optimized in MEGA version 6.039. Nuclear gene sequences containing more than one ambiguous site were resolved using PHASE 2.1.140, 41, for which input files were prepared using SEQPHASE42. Recombination tests to detect the longest non-recombining region for each locus were conducted using IMGC43. Identical haplotypes of both mtDNA sequences and phased nuclear gene sequences were collapsed using DNASP 5.1044.
Phylogenetic analyses
Phylogentic relationships among the three Schizothorax species in the Nujiang River were reconstructed using Bayesian inference (BI) and maximum parsimony (MP) approaches for Cytb. Two species of genus Gymnocypris, G. eckloni and G. przewalakii, were selected as outgroups for Cytb sequences (Table S1). Four published Cytb sequences of S. yunnanensis paoshanensis from the Nujiang River basin were added to the phylogenetic analyses (Table S1). Nucleotide substitution models were selected using the Akaike information criterion in MRMODELTEST version 2.345. The best-fit model was GTR + I for Cytb. The BI analyses were performed in MrBayes 3.1.246. Four independent runs were performed for 20 million generations. The phylogenetic trees were sampled every 1000th generation, which resulted in 20 000 trees, and the first 25% were discarded as burn-ins. The MP analyses were implemented in MEGA version 6.0. Nodal support values were estimated from 1000 nonparametric bootstrap replicates.
MtDNA and nDNA network
We used NETWORK 4.647 to construct a median-joining network (MJN) for Cytb, RAG-1 and RAG-2, respectively. A network approach is the most appropriate method to examine intraspecific gene evolution in closely related species, particularly when few characters are available for phylogenetic analysis as a result of shallow levels of divergence48. For Cytb, we directly analyzed the datasets. For the two nuclear genes, we analyzed the longest non-recombining region generated using IMGC.
Genetic differentiation
Genetic variation, including haplotype diversity (h) and nucleotide diversity (π)49 with standard errors, was calculated for Cytb using DNASP 5.10. Pairwise genetic differentiation (ɸST) (i.e., ɸ-statistics)50,51,52 was calculated for the different species using Cytb and two nDNA loci in ARLEQUIN 3.553. Analysis of molecular variance (AMOVA) was performed in ARLEQUIN 3.5 using Cytb to evaluate genetic differentiation within and among species. A total of 1000 permutations were employed to estimate the corrected significance levels using ɸST analyses and AMOVA.
Divergence time estimation
Two approaches were used to estimate the divergence times among the three species using Cytb sequences. First, we used net avarage sequence distance between species to estimate the approximate divergence times among the species. Net avarage sequence distance was estimated with MEGA version 6.0 as dA = dXY − (dX + dY)/2, where dXY is the net average distance between species X and Y, and dX and dY are the mean intraspecific distances. Mutation rate for Cytb (2.04 × 10−8 substitutions per site per year19) was used to measure the approximate divergence times between species.
Second, we calculated divergence times among the three species with a nonequilibrium coalescence model that uses the variance in pairwise differences between Cytb sequences to generate estimates of divergence time independent of gene-migration rates between pairs of species54. We used Markov chain Monte Carlo simulations as implemented in the program MDIV54 to estimate θ = 2N eμ, where N e is the effective population size and μ is the mutation rate per sequence per generation. The time since divergence is t pop = t/N e, where t is the time since population divergence. M is the migration rate between populations, and T MRCA is the time to the most recent common ancestors between pair-wise species. MCMC simulations were run for 4 × 107 steps with the first 10% discarded as burn-in. The uniform prior distribution of maximum M and t pop was set to 10 and 30, respectively. Divergence times in generations before present (T pop) between pairs of species were estimated with T pop = [(t pop × θ)/2 K]/μ, where μ is the mutation rate per site per generation and K is sequence length. MDIV was run for three replicates with different random seeds using the HKY model55. The mutation rate of 2.04 × 10−8 substitutions per site per year was also used in MDIV.
Gene flow
To determine whether the haplotype sharing between the three species-pairs stems from recent gene flow or from the retention of ancestral polymorphisms, potential gene flow among the three species was estimated using the isolation with migration (IM) model with the program IMa232 and using the Markov chain Monte Carlo simulations with the program MDIV. IM analysis can address nonequilibrium scenarios where haplotype sharing may result from retention of ancestral polymorphism in recently diverged lineages and potential ongoing gene flow32. We used Cytb sequences for the IM and MDIV analyses. MDIV analysis was run using the aforementioned parameter settings. The method estimates the density functions and posterior-probability densities of the IM model parameters using a Markov chain (MCMC) method56. The functions of the model parameters were first estimated in M-mode with one million generations, and the first 10% were discarded as burn-in. The MCMC run was repeated three times to confirm convergence. Using these functions, the marginal posterior distribution and the maximum-likelihood estimates of the demographic parameters were then estimated in the L-mode. The HKY model of the DNA substitution was employed for Cytb and 40 heated metropolis-coupled Markov chains were employed to assure convergence.
Historical demography
We assessed demographic historical changes using three approaches. First, Tajima’s D 57 and Fu’s Fs 58 statistics were calculated using ARLEQUIN 3.5 to detect evidence of demographical expansions, with 1000 coalescent simulations. Second, mismatch distributions59 were calculated to infer the demographic history in ARLEQUIN 3.5. The two aforementioned methods used only Cytb sequences. Finally, an extended Bayesian skyline plot (EBSP) was implemented in BEAST v.1.660 to reveal demographic changes over time under neutral evolution61. EBSP facilitates the inclusion of mitochondrial and nuclear loci in the same analysis. The EBSP was performed for the three species independently, applying an evolutionary rate of 2.04% per million years. The evolutionary rates for two nuDNA genes were estimated as a function of the Cytb evolutionary rate. A strict clock model was set as prior, 100 million generations were run for S. gongshanensis and S. lissolabiatus, and 50 million generations were run for S. nukiangensis. Convergence was assessed with TRACER v.1.562.
Availability of supporting data
The data set supporting the results of this article is available in the GenBank under KT034083–KT034091, KT034105–KT034119, KT034121–KT034122, KT034131, KT034133–KT034149, KT034153–KT034175, KT034189–KT034199, KT034201, KT034213–KT034225, KT034229–KT034238, KT034259–KT034275, KT034322–KT034358, KU255547–KU255584, KY801703 and KY801706 and provided as supplementary information.
References
Via, S. & West, J. The genetic mosaic suggests a new role for hitchhiking in ecological speciation. Molecular ecology 17, 4334–4345 (2008).
Nosil, P., Funk, D. J. & Ortiz-Barrientos, D. Divergent selection and heterogeneous genomic divergence. Molecular ecology 18, 375–402 (2009).
Gow, J. L., Peichel, C. L. & Taylor, E. B. Contrasting hybridization rates between sympatric three-spined sticklebacks highlight the fragility of reproductive barriers between evolutionarily young species. Molecular ecology 15, 739–752 (2006).
Avise, J. C. Molecular Markers, Natural History and Evolution. Chapman & Hall, New York, NY (1994).
Cao, W. X., Chen, Y. Y., Wu, Y. F. & Zhu, S. Q. Origin and evolution of schizothoracine fishes in relation to the upheaval of the Xizang Plateau (ed. Tibetan Expedition of the Chinese Academy of Science) 118–130 (Science Press, 1981).
Chen, Y. F. & Cao, W. X. Schizothoracinae in Fauna Sinica, Osteichthyes, Cypriniformes III (ed. Yue, P.) 273–335. (Science Press, 2000).
Dimmick, W. W. & Edds, D. R. Evolutionary genetics of the endemic Schizorathicine (Cypriniformes: Cyprinidae) fishes of Lake Rara, Nepal. Biochemical Systematics and Ecology 30, 919–929 (2002).
He, D. K. & Chen, Y. F. Biogeography and molecular phylogeny of the genus Schizothorax (Teleostei: Cyprinidae) in China inferred from cytochrome b sequences. Journal of Biogeography 33, 1448–1460 (2006).
Yang, J., Yang, J. X. & Chen, X. Y. A re-examination of the molecular phylogeny and biogeography of the genus Schizothorax (Teleostei: Cyprinidae) through enhanced sampling, with emphasis on the species in the Yunnan-Guizhou Plateau, China. Journal of Zoological Systematics and Evolutionary Research 50, 184–191 (2012).
Chu, X. L. & Chen, Y. R. The Fishes of Yunnan, China. Cyprinidae. (Science Press, 1989).
Chen, X. Y. Checklist of Fishes of Yunnan. Zoological Research 34, 281–343 (2013).
Chen, Y. Y. The Fishes of the Hengduan Mountains Region. (Science Press, 1998).
Yue, X. J. et al. Population genetic structure of three schizothoracins from Nujiang River in Southwestern China. Acta Ecologica Sinica 30, 6418–6429 (2010).
Chen, W., Ma, X., Shen, Y., Mao, Y. & He, S. The fish diversity in the upper reaches of the Salween River, Nujiang River, revealed by DNA barcoding. Sci Rep-Uk 5 (2015).
Hewitt, G. M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58, 247–276 (1996).
Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913 (2000).
Hewitt, G. M. Genetic consequences of climatic oscillations in the Quaternary. Philosophical transactions of the Royal Society of London Series B, Biological sciences 359, 183–195; discussion 195 (2004).
Chen, W., Du, K. & He, S. Genetic structure and historical demography of Schizothorax nukiangensis (Cyprinidae) in continuous habitat. Ecology and evolution 5, 984–995 (2015).
Chen, W., Shen, Y., Gan, X., Wang, X. & He, S. Genetic diversity and evolutionary history of the Schizothorax species complex in the Lancang River (upper Mekong). Ecology and evolution 6, 6023–6036 (2016).
Shi, Y. F., Ren, B. H., Wang, J. T. & Derbyshire, E. Quaternary Glaciation in China. Quaternary Sci Rev 5, 503–507 (1986).
Shi, Y., Zheng, B. & Li, S. Last glaciation and maximum glaciation in Qinghai-Xizang (Tibet) Plateau. Journal of Glaciology and Geocryology 12, 1–15 (1990).
Zhou, S. Z., Wang, X. L., Wang, J. & Xu, L. B. A preliminary study on timing of the oldest Pleistocene glaciation in Qinghai-Tibetan Plateau. Quaternary International 154, 44–51 (2006).
Zhang, W., Cui, Z. J. & Li, Y. H. Review of the timing and extent of glaciers during the last glacial cycle in the bordering mountains of Tibet and in East Asia. Quaternary International 154, 32–43 (2006).
Shi, Y., Zheng, B. & Yao, T. Glaciers and environments during the last glacial maximum (LGM) on the Tibetan Plateau. Journal of Glaciology and Geocryology 19, 97–113 (1997).
Yi, C. L., Cui, Z. J. & Xiong, H. G. Numerical periods of Quaternary glaciations in China. Quaternary Sciences 25, 609–619 (2005).
Jing, M., Yang, G. & Sun, N. Study on the climatic changes between the last interglacial age and the last glacial age recorded by Ostracoda in eastern Qaidam Basin. Journal of Earth Sciences and Environment 26, 83–87 (2004).
Zhao, K. et al. Phylogeography of the endemic Gymnocypris chilianensis (Cyprinidae): sequential westward colonization followed by allopatric evolution in response to cyclical Pleistocene glaciations on the Tibetan Plateau. Molecular phylogenetics and evolution 59, 303–310 (2011).
Martrat, B. et al. Abrupt temperature changes in the Western Mediterranean over the past 250,000 years. Science 306, 1762–1765 (2004).
Tsao, W. S. & Wu, H. W. An investigation of the fish biology and fishery problems in Ganze-Apa region of western Szechwan province. Acta Hydrobiologica Sinica 2, 79–110 (1962).
Chen, Y. Y., Zhang, W. & Huang, S. Y. Speciation in schizothoracid fishes of Lake Lugu. Acta Zoologica sinica 28, 217–225 (1982).
Zhao, K. et al. The youngest split in sympatric schizothoracine fish (Cyprinidae) is shaped by ecological adaptations in a Tibetan Plateau glacier lake. Molecular ecology 18, 3616–3628 (2009).
Hey, J. Isolation with migration models for more than two populations. Molecular biology and evolution 27, 905–920 (2010).
Keller, L. F. & Waller, D. M. Inbreeding effects in wild populations. Trends in ecology & evolution 17, 230–241 (2002).
Whitlock, M. C., Ingvarsson, P. K. & Hatfield, T. Local drift load and the heterosis of interconnected populations. Heredity 84, 452–457 (2000).
Xiao, W., Zhang, Y. & Liu, H. Molecular systematics of Xenocyprinae (teleostei: cyprinidae): taxonomy, biogeography, and coevolution of a special group restricted in East Asia. Molecular phylogenetics and evolution 18, 163–173 (2001).
Lopez, J. A., Chen, W. J. & Orti, G. Esociform phylogeny. Copeia 449–464 (2004).
Lovejoy, N. R. & Collette, B. B. Phylogenetic relationships of new world needlefishes (Teleostei: Belonidae) and the biogeography of transitions between marine and freshwater habitats. Copeia 324–338 (2001).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–1797 (2004).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular biology and evolution 30, 2725–2729 (2013).
Stephens, M., Smith, N. J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. American journal of human genetics 68, 978–989 (2001).
Smith, S. A., Stephens, P. R. & Wiens, J. J. Replicate patterns of species richness, historical biogeography, and phylogeny in Holarctic treefrogs. Evolution; international journal of organic evolution 59, 2433–2450 (2005).
Flot, J. F. seqphase: a web tool for interconverting phase input/output files and fasta sequence alignments. Molecular ecology resources 10, 162–166 (2010).
Woerner, A. E., Cox, M. P. & Hammer, M. F. Recombination-filtered genomic datasets by information maximization. Bioinformatics 23, 1851–1853 (2007).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Nylander, J. MrModeltest v2. Program distributed by the author (ed. Nylander JAA). (Evolutionary Biology Centre, Uppsala University, Uppsala, 2004).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Bandelt, H. J., Forster, P. & Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Molecular biology and evolution 16, 37–48 (1999).
Posada, D. & Crandall, K. A. Intraspecific gene genealogies: trees grafting into networks. Trends in ecology & evolution 16, 37–45 (2001).
Nei, M. Molecular Evolutionary Genetics. (Columbia University Press, New York, NY, 1987).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution; international journal of organic evolution 38, 1358–1370 (1984).
Reynolds, J., Weir, B. S. & Cockerham, C. C. Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics 105, 767–779 (1983).
Slatkin, M. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139, 457–462 (1995).
Excoffier, L., Hofer, T. & Foll, M. Detecting loci under selection in a hierarchically structured population. Heredity 103, 285–298 (2009).
Nielsen, R. & Wakeley, J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics 158, 885–896 (2001).
Hasegawa, M., Kishino, H. & Yano, T. A. Dating of the Human Ape Splitting by a Molecular Clock of Mitochondrial-DNA. Journal of molecular evolution 22, 160–174 (1985).
Hey, J. & Nielsen, R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences of the United States of America 104, 2785–2790 (2007).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Molecular biology and evolution 9, 552–569 (1992).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC evolutionary biology 7, 214 (2007).
Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular biology and evolution 22, 1185–1192 (2005).
Rambaut, A. & Drummond, A. J. Tracer version 1.5. Available from http://beast.bio.ed.ac.uk/Tracer/ (2013).
Acknowledgements
We are very grateful to Yang Liandong, Mao Yuntao, Lu Suxiang, Du Kang, Shen Yanjun, Guan Lihong, Zhong Zaixuan, Xiong Huan and Zhou Chuanjiang for sample collection and experimental assistance. This work was supported by the Key Fund and NSFC-Yunnan mutual funds of the National Natural Science Foundation of China (Grant Nos 31130049 and U1036603).
Author information
Authors and Affiliations
Contributions
W.T.C. contributed to sampling, molecular experiment, data analyses and writing the manuscript, X.J.Y. contributed to molecular experiment and sampling. S.P.H. contributed to research design and writing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, W., Yue, X. & He, S. Genetic differentiation of the Schizothorax species complex (Cyprinidae) in the Nujiang River (upper Salween). Sci Rep 7, 5944 (2017). https://doi.org/10.1038/s41598-017-06172-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06172-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
