
Abstract
The WRKY family of transcription factors (TFs) is one of the ten largest families of TFs in higher plants and has been implicated in multiple biological processes. Here, we identified 121 WRKY TFs in moso bamboo, including five novel members that were not annotated in the Phyllostachys edulis genomic database. Estimation of the divergence time of paralogous gene pairs revealed an important role of the recent whole-genome duplication in the expansion of the WRKY family. Expression analysis based on quantitative reverse-transcription polymerase chain reaction (qRT-PCR) data revealed that a large number of PheWRKY genes varied significantly under cold or drought stress treatments, which could be defined as abiotic stress-responsive genes. The overexpression of PheWRKY72-2 in Arabidopsis resulted in a decreased sensitivity to drought stress during early seedling growth. PheWRKY72-2 may enhance plant tolerance to stress by functioning as a positive regulator of stoma closure. Our study provides a theoretical foundation and some experimental evidence for further functional verification of the PheWRKY family of TFs.
Similar content being viewed by others


Introduction
Moso bamboo (Phyllostachys edulis) is a large, woody bamboo with the highest ecological, economic, and cultural value of all bamboo types and accounts for 70% of the total cultivated bamboo worldwide1. Moso bamboo belongs to the monophyletic Bambusoideae, Ehrhartoideae, Pooideae (BEP) clade of the grass family (Poaceae). Moso bamboo is a perennial plant characterized by its woody culm and a prolonged vegetative phase that lasts for decades before flowering1. It is one of the most important non-timber forest resources in south-eastern China because of its striking shoot growth rate. Indeed, the shoot can grow as long as 1 m within 24 hrs and reaches a final height of 5–20 m in 45 to 60 days2. Interestingly, multiple moso bamboo plants often flower and die after flowering simultaneously1. Moreover, the plants rarely flower in a natural setting. Transcriptomic studies in P. edulis provide an excellent opportunity to investigate genes that could be useful for improving plant growth3. RNA sequencing studies suggest that drought stress may be related to flowering in bamboo4. P. edulis has a tetraploid origin that underwent whole-genome duplication approximately 7–12 million years ago (mya). Collinearity analysis of Oryza sativa and bamboo revealed that a large number of genes were lost at the same time4.
The WRKY family is one of the ten largest transcription factor (TF) families in higher plants5. The first cDNA encoding a WRKY protein to be cloned was SPF1 from Pachyrhizus erosus 6. Since then, several WRKY family member proteins have been identified from various plant species7,8,9,10,11, all of which play key roles in the response to ever-changing internal and external stimuli. The WRKY TFs are named after the WRKY domain, which consists of approximately 60 amino acids. These TFs play vital regulatory roles in developmental and physiological processes, such as seed dormancy12, embryo morphogenesis13, plant growth14, senescence15, 16, and metabolism17, 18. Furthermore, WRKY TFs are involved in responses to various abiotic and biotic stresses, such as bacteria19, 20, fungi15, 21, nematodes22, wounding23, heat, drought, salinity and cold24. Most WRKY family members contain highly conserved heptapeptides (WRKYGQK) at their N-terminals and a C2H2 or C2HC zinc-finger motif at their C-terminals7, 23, 25. However, some have altered motifs, including WRKYGEK and WRKYGKK21, 26. WRKY proteins are categorized into three distinct groups based on both the number of WRKY domains and the features of their zinc-finger-like motifs. The WRKY TFs with two WRKY domains containing C2H2 zinc-finger motifs belong to group 1. Group 2 TFs have a single WRKY domain that includes a C2H2 zinc-finger motif and are further divided into five subgroups based on their phylogenetic relationships. Group 3 TFs contain a single WRKY domain with a C2HC zinc-finger motif27.
Gao et al. (2013) identified a P. edulis WRKY family gene, PheWRKY67-2 (GenBank accession number: FP101056.1), involved in the defence of P. edulis against the pathogens Ceratosphaeria phyllostachydis and Arthrinium phaeospermum 28. However, to date, no studies have addressed the WRKY gene family and its function in the organ growth and development of P. edulis. Here, for the first time, we performed a detailed analysis of the subgroup classification, gene structure and conserved motif composition of 121 WRKY TFs in the P. edulis genome. We analysed the expression patterns of PheWRKYs from floral expression profiles and fast-growing shoot expression profiles to determine whether the expression patterns of PheWRKYs were related to fast-growing shoots and flower development in P. edulis. We further studied changes in expression of the PheWRKY genes in response to abiotic stress (drought or cold) in P. edulis seedlings. Furthermore, a drought stress and cold stress-inducible gene, PheWRKY72-2, was characterized in transgenic Arabidopsis to comprehensively understand its role in the response to abiotic stress. Our study provided a theoretical foundation and some experimental evidence for further functional verification of PheWRKYs.
Results
Identification of the WRKY family of TFs
We identified 121 WRKY members in the P. edulis genomic database (alternative splicing variants were not considered; Table 1), including five novel members that were not annotated in the P. edulis genomic database (Table S1). Because all the proteins and CDS sequences obtained from moso bamboo genome database were annotated by their putative orthologous sequences of O. sativa, so we renamed each moso bamboo WRKY sequences based on the individual similarity with rice WRKY proteins by blast method, and the different proteins of moso bamboo which corresponded to the same sequences in rice were suffixed with ‘−1’, ‘−2’, ‘−3’, ‘−4’ depending on levels of sequences similarity (Table S2)29. The polypeptides encoded by the PheWRKY genes ranged from 159 to 1,168 amino acids in length, with predicted pI values ranging from 4.7 to 10.1 and molecular weights ranging from 19.77 to 131.06 kD (Table S3).
Structure and classification of PheWRKYs
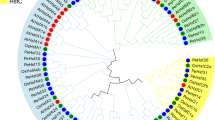
According to the zinc-finger structure (Table 1), sequence alignment (Figure S1), conserved motif (Fig. 1 and Figure S2) and gene structure (Figure S3), the PheWRKYs can be classified into three main groups. Twenty PheWRKY proteins that contained two WRKY domains were assigned to group 1. Fifty of these contained two intact WRKY domains. However, the other five members of group 1 (PheWRKY35-2, 85-1, 96-1, 96-2 and 96-3) had only one complete WRKY domain in their N-terminals, and their C-terminal domains lacked a zinc-finger. PheWRKY125, which should have been placed in group 3 because of its C2HC type zinc-finger, was assigned to group 1 because it contained two WRKY domains. Except for PheWRKY125, the zinc-finger structures of the PheWRKY proteins in group 1 were of C2H2 type with a C-X4-C-X22-23-H-X1-H motif (Table 1). Sixty-three PheWRKY proteins had a single WRKY domain, but in 62 members, the motif was C-X4-5-C-X23-H-X1-H; one member had the motif C-X5-C-X88-H-X1-H (PheWRKY9-1). All 62 members were assigned to group 2 and were further classified into five subgroups based on their conserved motif, their gene structure, and phylogenetic analysis. The subgroups included the following: group 2a (5 members), group 2b (11 members), group 2c (28 members), group 2d (7 members), and group 2e (12 members). The zinc-finger structure of group 3 was C2HC (27 members), with the motif C-X6-7-C-X23-30-H-X1-C (Table 1). Eleven proteins contained only partial WRKY domains and did not fit in any group. Most group 2c members shared motif 2 with most group 1 protein sequences, which also primarily harboured motifs 7 and 16. Motif 13 appeared only in group 2a, whereas motifs 8, 9 and 10 were found exclusively in group 2b. Group 2d harboured motifs 5, 6 and 11 (Fig. 1b).

Phylogenetic analysis of the WRKY family of proteins in P. edulis (a) and the distribution of conserved motifs within each group (b).
Structural analysis of the genes revealed that consisted with previous report30, gene family members within the same group shared similar gene structures in terms of their intron numbers and intron phases (Figure S3). The intron phases of most sequences in group 1 were 0 at the first two introns and 2 at the last two introns. Except for PheWRKY97-1, the intron phases of all other members from groups 2a and 2b were 0. For groups 2c, 2d, 2e and 3, most introns phases were 2. Group 2b had more introns (average: 4.375) than the other groups. In contrast, almost all members of groups 2a, 2c, 2d, and 2e had only two introns. Generally, the motif and gene structure analysis revealed that members from different species assigned to the same group always shared similar motifs, intron numbers, and phases (Fig. 1b, Figure S3).
Although the WRKYGQK motif was highly conserved, we found several sequence variations in 15 P. edulis WRKY proteins, most of which belonged to groups 3 and 2c (Table S4). We identified WRKYGKK as the most common variant in six domains, whereas WRKYGEK was common to five domains. The other four variants—WRKYGKK, CRKYGQA, WRKYGQQ, and WRKFGQK—were found in a single WRKY domain in PheWRKY7-3, PheWRKY80-1 (N-terminal), PheWRKY80-2 (N-terminal), and PheWRKY61 (C-terminal), respectively.
To confirm the specific zinc-finger structure of PheWRKY9-1 (C-X5-C-X88-H-X1-H), we designed a pair of primers at the 5′-UTR and 3′-UTR of PheWRKY9-1. Different RNA samples isolated from different tissues, including one-year-old leaves, two-year-old leaves, flowers, one-year-old culms, shoots, seeds, roots, and seedlings under various abiotic stress, were used as clone templates. Although we did not obtain the CDS sequence of PheWRKY9-1, which is annotated in the moso bamboo CDS database, from any of the tissues, we did identify a gene with a normal zinc-finger structure (C-X5-C-X23-H-X1-H). Compared with the annotated PheWRKY9-1 sequence from the moso bamboo CDS database, this gene lacked two exons (Fig. 2a and b), as revealed by sequencing studies. In addition, whether the specific zinc-finger exists or is merely a mis-annotation could not be determined.

(a) Structure of the PheWRKY9-1 gene. The sequence obtained from the P. edulis CDS database is indicated on top, and the sequence obtained by sequencing verification is shown below the sequence. (b) Sequence alignment of the PheWRKY9-1 sequence obtained from the P. edulis CDS database and the sequence obtained by sequencing verification.
Phylogenetic relationships
To examine the phylogenetic relationship among WRKY proteins in P. edulis, A. thaliana, O. sativa, and Brachypodium distachyon, we performed phylogenetic analyses of the WRKY domain sequences from all four species based on a neighbour-joining method using Mega 6 software. Because the N-terminal and C-terminal domains form distinct clusters, we designated the two domains as 1 N and 1 C, respectively (Fig. 3). The phylogenetic tree indicated a divergence between monocotyledons and dicotyledons. PheWRKY clearly shared more sequence similarity with OsWRKY and BdWRKY than with AtWRKY. We then used the bidirectional best hit (BBH) method, which is restricted to a 1:1 ratio of orthologues31, to arrange possible orthologues for PheWRKY from the three sequenced species mentioned above. Of the 121 sequences, we identified orthologues for 62 PheWRKY genes from at least one of the three plant species. The highest number of orthologues (52 members) was identified in O. sativa, whereas the lowest number (10 members) was detected in A. thaliana (Table S2).

Rooted phylogenetic tree of the WRKY TF families. A. thaliana, O. sativa, B. distachyon, and P. patens. ‘Group 1 N’ and ‘Group 1 C’ indicate N-terminal or C-terminal WRKY domains, respectively, in group 1. P. edulis, A. thaliana, O. sativa, B. distachyon, and P. patens proteins are indicated by black, green, purple, red, and blue dots, respectively.
These putative paralogous pairs accounted for 59.5%, 54.2%, 53.6% and 52.7% of the entire WRKY family in P. edulis, O. sativa, B. distachyon and A. thaliana, respectively. The median values of the divergence time for P. edulis, O. sativa, B. distachyon and A. thaliana were 13.9, 38.26, 42.84 and 23.25 mya, respectively. The divergence for most PheWRKY gene pairs (22 of 36) was approximately 6 to 15 mya, similar to the P. edulis whole-genome duplication event (7-12 mya)4 and much later than those of O. sativa, B. distachyon and A. thaliana (Table S5).
Expression patterns of PheWRKY genes in different seedling tissues
We detected the expression of 81 PheWRKY genes in different tissues of three-month-old seedlings by RT-PCR. These selected genes covered all subgroups and included both up-regulated genes and down-regulated genes in flower development and shoot growth. Among them, PheWRKY11-2, 22-2, 45-1, 77-2, 83-1 and 83-3 showed high expression levels in roots; 3-2, 13-3 and 19-1 showed high expression levels in stems; and 26, 67-1 and 71-2 were highly expressed in leaves (Figure S4).
Expression analysis of the PheWRKY genes’ roles in flower development and shoot growth
We divided the PheWRKY genes into two groups (Fig. 4b) based on their expression in shoot and one-year-old culm. The expression levels of genes from cluster 1 were more abundant in the culm than in the shoot. In contrast, the expression of cluster 2 was significantly elevated in the shoot growth stage than in the one-year-old culm (CK). The genes PheWRKY28, 29-3, and 62 were significantly up-regulated during the shoot growth stage, suggesting that they play roles in the rapid elongation of the bamboo shoot (Table S6).

Expression profiles of the PheWRKY genes based on transcriptome data. The colour scale represents log2-transformed reads per kilobase per million (RPKM) values. Green indicates low expression, and red indicates high expression. (a) Dynamic expression profiles of four floral stages. F1, F2, F3, and F4 represent four different flowering developmental stages: the floral bud formation, inflorescence growing, blooming and embryo formation stages, respectively. (b) Expression profiles of the fast-growing shoot and culm.
The expression profiles from the floral digital gene expression (DGE) data were divided into three cluster groups. Cluster 1 had genes with high expressions at stage F4. Cluster 2 genes exhibited steady expression with little fluctuation from stage F1 to F4. The genes from cluster 3 were initially up-regulated, reached their peak expression at stage F2 or F3, and declined thereafter. Compared with CK (leaf), PheWRKY48-1 appeared to be preferentially expressed throughout the flowering process (Fig. 4a and Table S6). To validate the reliability of RNA-Seq and DGE data, quantitative reverse-transcription polymerase chain reaction (qRT-PCR) assays were randomly performed on four selected genes (Figure S5). As expected, in most cases, the expression trends of the selected genes corresponded to the presented data, indicating that the DGE and RNA-seq data were highly reliable (Figure S5).
A co-expression network was constructed to analyse the PheWRKY interactions in different biological processes. Of the top ten hub genes (degree ≥ 9) in the network, six genes, including PheWRKY19-2, 39-1, 39-2, 62, 65-1and 114-1, were up-regulated in flower development (Figure S6).
Expression profiles of PheWRKY genes under abiotic stress conditions
We detected the expression of 81 PheWRKY genes under drought and cold stress by qRT-PCR. Of these, 32 genes showed different degrees of up-regulation with at least one stress treatment, including 28 genes responding to cold treatment and 22 genes responding to drought treatment (Fig. 5). Interestingly, the most up-regulated genes in drought treatment (17 of the 22) were always up-regulated in response to cold treatment. The expression levels of PheWRKY4, 26-2, 48-2, 49, 62, 69–1, 72–3 and 96–1 increased gradually and peaked at 12 h in response to cold treatment, whereas those of PheWRKY11–2, 111, 34–2, 45–1, 114–1, 69–2, 70–2 and 76 rapidly accumulated at 1 h and then decreased to low levels. For drought treatment, PheWRKY45–1, 62, 69–1, 88–1, 96–1, 96–2 peaked at 1 h, and PheWRKY4, 5–1, 26–2, 15–2, 19–2, 44, 49, 55, 65–1, 71–2, 72–3 peaked at 24 h. The expression profiles of PheWRKY1, 36, 69–2 and 74–2 under drought treatment displayed parabolic trends and peaked at 12 h (Fig. 5 and Table S6).

Expression profiles of WRKY genes under cold and drought stress. The expression results were obtained by qRT-PCR, and the relative expression levels of PheWRKY genes under various treatments compared to the controls were used for hierarchical cluster analysis with Cluster 3.0. qRT-PCR analysis was based on three biological replicates of each sample and three technical replicates of each biological replicate.
Our expression analysis revealed that 54 genes were significantly induced by at least one abiotic stress (cold stress or drought stress) or physiological process (flower development or shoot growth). Of these WRKY genes, 7, 4, 17 and 3 were regulated by only cold stress, drought stress, flower development, and shoot growth, respectively (Figure S7).
Overexpression of PheWRKY72–2 in Arabidopsis
Subcellular location analysis indicated that green fluorescent protein (GFP)-tagged PheWRKY72–2 was located in the nucleus, in accordance with its function as a TF (Fig. 6).

Subcellular localization analysis of PheWRKY72-2.
To further investigate the potential role of PheWRKY72–2, which was up-regulated under both drought and cold stress, PheWRKY72–2 was transformed into Arabidopsis (wild type [WT]). Following qRT-PCR analysis of PheWRKY72–2 in WT and transgenic lines confirmed that PheWRKY72–2 performed high expression level in transgenic lines but no expression could be detected in WT (Fig. 7). Under cold stress treatment, no significant differences could be found between the WT and transgenic plants (data not shown). For simulated drought stress, transgenic Arabidopsis seeds were surface sterilized and germinated on Murashige and Skoog (MS) agar medium containing 10% and 20% polyethylene glycol (PEG) (Fig. 7b and e). The overexpression of PheWRKY72–2 (T3 generation) resulted in significantly longer roots than those of WT plants on medium supplemented with 10% PEG (Fig. 7b and c). We investigated the root hairs further but found no visible difference between those of the WT and transgenic lines (Fig. 7d). However, when the PEG concentration reached 20%, the WT line began to exhibit etiolation symptoms, unlike the transgenic lines (Fig. 7e).

(a) Relative expression of PheWRKY72 in transgenic lines. The expression of internal control gene (β-tubuli) was arbitrarily set at 1.0. (b) Response of 35S-PheWRKY72-2 Arabidopsis transgenic lines to drought stress (10% PEG). (v) The primary root lengths of WT and transgenic seedlings under drought stress (10% PEG). (d) Observation of root hairs under drought stress (10% PEG). (e) Response of the 35S-PheWRKY72-2 Arabidopsis transgenic lines to drought stress (20% PEG).
We then compared the stomatal apertures under drought treatment because water loss mainly depends on stomatal regulation. To this end, we measured the stomatal apertures of WT and transgenic plants under control and drought treatments (3 h). The length and width of the stomata were determined, and the length/width ratio was used to reflect the degree of stomatal closure. Under normal growth conditions, no significant differences were detected (Fig. 8b). Under drought stress, the PheWRKY72-2ox lines showed a higher length/width ratio of stomata than the WT plants (Fig. 8a and b). These results suggest that stomatal closure in PheWRKY72-2ox plants is more sensitive to drought stress than that in WT plants. Under the drought treatment, OE-PheWRKY72-2 suppressed the expression of AtABI2, AtABI5 and AtABA2 and significantly increased the expression of AtABI4, AtAREB and AtLEA (Fig. 8c).

(a) Comparison of the stomatal aperture between transgenic and WT plants in response to drought stress. (b) Ratios of stomatal closure. The ratio of the length to the width was used as a measurement of stomatal closure. (c) Expression of ABA-signalling genes in WT and 35S-OsWRKY72 transgenic Arabidopsis under drought stress for 3 h. The expression of each gene in WT was arbitrarily set at 1.0. qRT-PCR analysis was based on three biological replicates of each sample and three technical replicates of each biological replicate.
Discussion
Characterization of the P. edulis WRKY gene family
To facilitate the analysis of P. edulis WRKY gene evolution in group 1, we further classified P. edulis group 1 WRKY genes into two subgroups based on their zinc-finger motifs32. Except for PheWRKY61, whose zinc-finger motif of C2HC-C2HC was assigned to group 1b, all the other members of group 1 could be allocated to group 1a. PheWRKY61 probably evolved through an intra-molecular duplication event of a group 3 WRKY domain that had already evolved to have the C2HC type zinc-finger. PheWRKY35, 85-1, 96-1, 96-2 and 96-3 all contained two WRKY domains but lacked a complete zinc-finger in their C-terminal domains. The closing genetic relationship of the N-terminal WRKY domains among PheWRKY85-1, 96-1, 96-2 and 96-3 determined based on the phylogenetic tree suggests that these sequences underwent some similar evolutionary events. Domain acquisition and domain loss events appear to have shaped the WRKY family of proteins33. Thus, these five genes might have arisen when a two-domain WRKY gene lost the zinc-finger motif on its C-terminal domain during evolution. Small variations in the WRKYGQK sequence are described for some OsWRKY proteins (accounting for 11 OsWRKY domains)32. These variations are also very common (11 domains) in B. distachyon, but only three A. thaliana members had variant heptapeptide sequences (WRKYGKK) (Table S4). In moso bamboo, we found that amino acid substitutions in the conserved heptapeptide signature (15 domains) were much more common than in the three model plants.
In previous reports, bamboo was considered to have a tetraploid origin. Approximately 7-12 mya, P. edulis experienced a long progression from tetraploidy to diploidy4. P. edulis carries two duplicates as that of O. sativa gene model sets4. For PheWRKY genes, a similar phenomenon was always observed. Although P. edulis suffered a large-scale gene loss event after the whole-genome duplication, 36 putative paralogous pairs of PheWRKY genes remained, far exceeding the numbers in A. thaliana, O. sativa and B. distachyon. Furthermore, most duplication events of WRKY paralogous pairs in this species occurred 6-15 mya, much later than those of the three model plants mentioned above. All of our results suggest that the recent whole-genome duplication, which was likely linked to polyploidy events, played an important role in the WRKY family expansion.
Diverse PheWRKYs are involved in various biological processes
WRKY TFs regulate a range of biological processes and plant responses to various abiotic stressors. At least 54 OsWRKY genes in O. sativa 34 and 23 CsWRKY genes in Cucumis sativus are differentially expressed under abiotic stresses35. Similarly, 55 VvWRKY genes differentially respond to at least one abiotic stress treatment36. We profiled the expression of 80 PheWRKY genes in plants subjected to cold and drought stress and detected 31 P. edulis WRKY genes with significantly higher expression in response to at least one abiotic stress using real-time PCR, indicating that, in P. edulis, the PheWRKY genes function in the abiotic stress response. The overexpression of PheWRKY72-2 enhanced plant tolerance to drought stress. In Arabidopsis, AtWRKY54 and AtWRKY70 enhance plant tolerance to osmotic stress by regulating the size of the stomatal aperture37. Various environmental stresses result in the rapid accumulation of ABA, leading to stomatal closure, which reduces water loss by transpiration38. Recent reports showed that AtWRKY46 participated in the feedforward inhibition of lateral root inhibition via regulation of ABA signaling and auxin homeostasis under osmotic/salt stress treatment39, 40. Besides, WRKY46 is regulated by light and modulates starch metabolism and ROS levels to inhibit stomatal closing under osmotic stress treatment by another independent way. Unlike the overexpression line of AtWRKY46 whose stomatal closing is impaired under osmotic stress treatment40, the transgenic lines of PheWRKY72-2 showed enhanced stomatal closure under drought stress by comparing with the WT line. ABA controls stomatal movement via a dual mechanism. Regulation can occur via the biochemical effects of ABA on guard cells38. Salt stress is often accompanied by drought stress, and both cause water deprivation through ABA-dependent and ABA-independent pathways41. Our results showed that OE-PheWRKY72-2 significantly promoted the expression of AtLEA, AtAREB and AtABI4 and repressed the expression of AtABI2, AtABI5 and AtABA2. Plants with up-regulated ABI1/2 expression exhibited decreased stress tolerance, whereas plants with down-regulated ABI1/2 expression exhibited increased stress tolerance42. Reactive oxygen species (ROS), such as superoxide, hydrogen peroxide and hydroxyl radicals, play a dual role in plants: They act as necessary signalling molecules but can cause damage to plant cells when overproduced under stress conditions43. The constitutive expression of AtAREB1 in Arabidopsis modulates ROS accumulation and endogenous ABA levels to improve drought tolerance43. In plants, the presence of late embryogenesis abundant (LEA) proteins has been associated with cellular tolerance to dehydration, which may be induced by freezing, saline conditions, or drying44. Thus, up-regulating AtLEA might improve stress tolerance in transgenic plants.
Compared with their expression in nonflowering moso bamboo leaves (CK), the significant up-regulation of PheWRKY19-1, 19-2, 35-2, 48-1 and 65-1 during flower development indicated their potential roles in morphogenesis and organ development. WRKY TFs associated with senescence45,46,47 and stress responses48 were significantly up-regulated in the panicles of P. edulis. Under normal circumstances, P. edulis rarely flowers, and the plants often flower concurrently and die collectively after flowering. However, collective death after flowering in a large area is usually associated with external environmental changes, such as climatic variation or the over-exploitation of bamboo1. The overexpression of AtWRKY75 or OsWRKY72 causes early flowering in Arabidopsis 49, and the overexpression of GsWRKY20 accelerates plant flowering by regulating flowering-related genes (i.e., flowering locus T [FT] and CONSTANS [CO]) and floral meristem identity genes (i.e., activator protein 1 [AP1], SEPALLATA 3 [SEP3] and AGAMOUS [AG])48. The involvement of WRKY factors in floral induction may be induced by signalling hormones, such as salicylic acid7, 50, jasmonic acid45, 46 or gibberellic acid10. We found that PheWRKY 28, 29-3, 49, 62, 80-3 and 111 have significant expression levels in growing shoots but not in culms. OsWRKY78 has positive effects on stem elongation and cell length, as determined by transgenic studies51. The up-regulation of several WRKY genes in shoot growth and development indicate that many WRKY genes function in the rapid growth and elongation of P. edulis shoots. In summary, PheWRKY genes play varied roles in different biological processes in P. edulis.
The functional conservative and divergence of orthologous genes
We further analysed the correlations of orthologous pairs under various processes. In Arabidopsis, the expression pattern of AtWRKY53 was identified as a leaf senescence signal44. Constitutively overexpressing AtWRKY53 under the control of the CaMV35S promoter accelerated the senescence symptoms, whereas WRKY53 RNAi and knock-out lines exhibited delayed development and senescence45. The orthologues, PheWRKY15-1 (4E-039) and PheWRKY74-1 (2E-31), were always found in high abundance in leaves collected from flowering plants (flowering implies senescence and death in P. edulis) compared to their abundance in leaves collected from other growth stages (Figure S8), indicating that there are conserved roles between WRKY homologues in leaf senescence. Moreover, we compared the expression trends of PheWRKY genes with those of their possible orthologues from A. thaliana, O. sativa 34, and B. distachyon 46 under cold and drought stress. Although the sample times and experimental conditions were different, our data revealed that some P. edulis homologues in other model plants showed an entirely opposite expression trend. PheWRKY85-1 shares 87% sequence similarity with OsWRKY85; however, OsWRKY85 is up-regulated under drought stress, whereas PheWRKY85-1 is down-regulated. The function of these segregated duplicated genes (PheWRKY85-1 and OsWRKY85) may have been altered during evolution34.
Conclusions
In this study, a total of 121 WRKY genes were identified in P. edulis. The expression profiles derived from DGE, RNA-seq and qRT-PCR analyses indicated that PheWRKY involved in various abiotic stress. The overexpression of PheWRKY72-2 in Arabidopsis increased drought tolerance by functioning as a positive regulator of stomatal closure. Better understanding this gene family and its potential involvement in growth, development and stress responses will facilitate further research on the evolutionary history and biological functions of the PheWRKY gene family.
Materials and Methods
Plant materials, growth conditions, and treatments
Samples of P. edulis culms were harvested in Huoshan County (116° 10′; 31° 12′), Anhui province from spring to autumn of 2014. Flowering plant samples from different developmental stages of the flowering process (F1-floral bud formation, F2-inflorescence development, F3-anthesis, and F4-embryo formation) were collected in Guilin city (110° 17′; 25° 048′), Guangxi Zhuang Autonomous Region, from April to August in 2014. Plants were sampled from sites suitable for bamboo growth that were free of insect pests and artificial destruction. Samples were collected according to previously described methods1, 3.
For the abiotic stress treatment, P. edulis seeds were germinated in culture dishes lined with soggy filter paper for 1 week and then transferred into four seed pots containing approximately 0.5 kg of vermiculite. The seedlings were grown in an artificial climate chamber with long-day conditions (16-hrs light, 8-hrs dark) at 26 °C in the light and 18 °C in the dark. Drought stress was created by irrigating the plants with media containing 18% (v/v) PEG. Cold stress was created by placing plants in a 4 °C lighted growth chamber; the other conditions were the same as described above. The leaves from three seedlings were harvested at 0, 0.5, 1, 3, 6, 12 and 24 hrs after abiotic stress treatment, rapidly frozen in liquid nitrogen and stored at −80 °C prior to use.
RNA isolation, reverse transcription and gene expression analysis
Total RNA was extracted using Trizol (Invitrogen, USA). First-strand cDNA synthesis was conducted with approximately 1 μg of RNA using the reverse transcriptase AMV (Promega, Madison, Wisconsin, USA). qRT-PCR was performed using the fluorescent intercalating dye Light Cycler 480 SYBR Green I Master Mix (Roche, Mannheim, Germany) on a Light Cycler 480 (Roche, Rotreuz, Switzerland). Primers were designed using Primer 3 software and Oligo dT7 (Table S7). According to the manufacturer’s instructions, the 20-μL reaction mixture contained 0.4 μL (10 μM) of each primer, 1.5 μL (30 ng) of cDNA and 10 μL of SYBR Green I Master Mix. TIP41 (tonoplast intrinsic protein 41)52 and β-tubulin were selected as internal controls. All reactions—technical and biological—were performed in triplicate. The ΔCT and ΔΔCT values were calculated by the formulas ΔCT = CT target − CT reference and ΔΔCT = ΔCT treated sample −ΔCT untreated sample, respectively.
Gene sequence verification
A full-length fragment of PheWRKY9-1 was cloned from P. edulis cDNA by RT-PCR using the following primer pair: 5′-ATGGAGGCGGTATCGGCGGT-3′ and 5′-TTAGATCGATCTGCGAGGTGCGGTC-3′.
Database searches
The annotated WRKY genes of A. thaliana, O. sativa, and B. distachyon were obtained from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/), the rice genome annotation project (http://rice.plantbiology.msu.edu/index.html) and the B. distachyon WRKY database (http://www.igece.org/WRKY/BrachyWRKY/BrachyWRKYIndex.html). All annotated WRKY genes of P. edulis were downloaded from the National Center for Gene Research (http://202.127.18.221/bamboo/down.php). To avoid missing potential members of the WRKY family, we created a local blast database using the CDS sequence that we obtained from the P. edulis CDS database and novel transcripts that we obtained from RNA-seq data. The novel transcripts were assembled in a GTF file (exported by Cufflink based on a series of transcriptome sequencing results), and the P. edulis genome data were analysed using Perl. Published AtWRKY sequences and OsWRKY were retrieved and used as queries in blastn searches against the CDS database, and sequences were selected as candidate genes if their E-value was ≤ −10. The Pfam (http://pfam.sanger.ac.uk/search) and Smart (http://smart.emblheidelberg.de/) databases were used to confirm each predicted WRKY sequence. The pI and molecular weight were estimated using the Compute pI/Mw tool from ExPASy (http://web.expasy.org/compute pi).
Multiple sequence alignment and phylogenetic tree construction
Multiple alignments of the amino acid sequences were performed using the ClustalX 2.1 program with default settings53. A phylogenetic tree based on sequence alignment was generated using MEGA 6.0 (http://www.megasoftware.net/) by the neighbour-joining method54. In addition, the BBH method31 was used to arrange possible orthologues and paralogues.
Gene structure and conserved motif analysis
The gene structure based on full-length CDS alignments with relevant genomic sequences was investigated using the online service of the Gene Structure Display Server55. MEME56 was used to identify motifs in the PheWRKY sequences.
Estimation of the duplication time in paralogous pairs
The Ka and Ks values of the paralogous genes were computed by the DNASP program. The synonymous substitution rate (Ks) was considered as a proxy for time to estimate the dates of the segmental duplication events. The formula T = Ks/2λ was used to o calculate the approximate date of the duplication event and the λ in formula represented for clock-like rates of synonymous substitution. The estimated clock-like rate for P. edulis, O. sativa and B. distachyon were 6.5 × 10−9 substitutions/synonymous site/year and that that for A. thaliana was 1.5 × 10−8 substitutions/synonymous site/year57, 58.
DGE data, transcriptome data analysis, and co-expression network generation
The DGE data of four different developmental stages of flowering (floral bud formation, inflorescence growing, blooming and embryo formation) and the transcriptomic data of fast-growing shoots at two developmental stages (shoots mixed by six unearthed heights [10, 50, 100, 300, 600, and 900 cm] and mature culm) were sequenced in our previous study1, 3 and used for PheWRKY gene expression analyses. For floral development, the DGE data of nonflowering, moso bamboo leaves (CK) were regarded as the reference gene database1. For shoot growth, the transcriptome data of one-year-old culms constituted the reference gene database3. The expression levels of PheWRKY genes at different growth stages were hierarchically clustered based on Euclidean distance with complete linkage found in Cluster 3.0. A gene was considered up-regulated if it had a false discovery rate (FDR) ≤0.05 and│log2 fold change│ ≥ 2.
The microarray data and RNA-seq data were used for coexpression analysis via the WGCNA package. The co-expression correlation was then calculated using Pearson’s correlation with R2 ≥ 0.95. The network picture was created using Cytoscape.
Overexpression of PheWRKY72-2
The complete open reading frame (ORF) of the PheWRKY72-2 (FP101056.1) gene cloned by Chen59 in our previous work was inserted into the pGEM-T Easy plasmid vector using the following primers: forward, 5′-TGCTCTAGAATGGAAGCCTACCCTATGCT-3′; reverse, 5′-CCGGAATTCTCAGTGGAACCGGCCAGAC-3′. TIANGEN DNA Polymerase (TIANGEN biological company, China) was used to amplify the PheWRKY72-2 gene. The PCR parameters were 94 °C for 5 min, followed by 28 cycles of 94 °C for 30 s, 62 °C for 1 min and 72 °C for 1 min. The PCR products was digested with BamH I and Xba I and then cloned into pGEM-T Easy (Promega, USA) after gel extraction. The coding region of the PheWRKY72-2 cDNA was cloned into the pCAMBIA 2300 vector under the control of the CaMV 35 S promoter and CAMV terminator. The constructed plasmid was named pCAMBIA2300-PheWRKY72-2 and confirmed by the chain-termination method on an ABI 3100 automated sequencer (USA).
The construct vector was introduced into WT Arabidopsis plants (Columbia-0) by Agrobacterium-mediated transformation. Transgenic plants were selected on kanamycin, and the first generation of transgenic plants was examined in terms of their phenotypes. At least 10 independent transgenic plants exhibiting severe phenotypes were selected, subjected to phenotypic characterization and screened for the transgene. Transgenic Arabidopsis seeds (T3 generation) and WT seeds were cultured with 10% and 20% PEG solution on MS agar medium. One-week-old seedlings were used to observe the phenotype. Three-week-old transgenic plants and WT which growth in normal growth condition were treated with PEG solution for 3 h and further used for RT-PCR experiment and stomatal closure observation.
Subcellular localization
The subcellular localization of PheWRKY72-2 was performed by transfecting GFP-tagged PheWRKY72-2 into Arabidopsis sheath protoplasts. The full-length cDNA of PheWRKY72-2 was fused in frame with the GFP cDNA and ligated between the CaMV 35 S promoter and the nopaline synthase terminator. The fluorescence signals in transfected protoplasts were examined by a confocal laser scanning microscope (Leica Microsystems).
Availability of supporting data
All sequencing data were deposited in the Short Read Archive at NCBI under accession number SRR961047, SRR1187864 and SRR1185317.
References
Gao, J. et al. Characterization of the floral transcriptome of P. edulis (Phyllostachys edulis) at different flowering developmental stages by RNA-seq and digital gene expression analysis. PLoS ONE 9, e98910 (2013).
Peng, X. H. et al. Genome-wide characterization of the biggest grass, bamboo, based on 10, 608 putative full length cDNA sequences. BMC Plant Biol. 10, 116 (2010).
Peng, Z. H. et al. Transcriptome sequencing and analysis of the fast growing shoots of P. edulis (Phyllostachys edulis). PLoS ONE 8, e78944 (2013).
Peng, Z. H. et al. The draft genome of the fast-growing non-timber forest species P. edulis (Phyllostachys heterocycla). Nat. Genet. 45, 456–461 (2013).
Ulker, B. & Somssich, I. E. WRKY transcription factors: from DNA binding towards biological function. Curr. Opin. Plant. Biol. 7, 491–498 (2004).
Ishiguro, S. & Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol. Gen. Genet. 244, 563–571 (1994).
Rushton, P. J., Macdonald, H., Huttly, A. K., Lazarus, C. M. & Hooley, R. Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes. Plant Mol. Biol. 29, 691–702 (1995).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends Plant Sci. 15, 247–258 (2010).
Liu, J. J. & Ekramoddoullah, A. K. M. Identification and characterization of the WRKY transcription factor family in Pinus monticola. Genome 52, 77–88 (2009).
Zhang, Y. J. & Wang, L. J. The WRKY transcription factor super family: its origin in eukaryotes and expansion in plants. BMC Evol. Biol. 5, 1 (2005).
Jiang, Y. Z. et al. Genome-wide identification and characterization of the Populus WRKY transcription factor family and analysis of their expression in response to biotic and abiotic stresses. J. EXP. BOT. 65, 6629–6644 (2014).
Pandey, S. P., Roccaro, M., Schön, M., Logemann, E. & Somssich, I. E. Transcriptional reprogramming regulated by WRKY18 and WRKY40 facilitates powdery mildew infection of Arabidopsis. Plant J. 64, 912–923 (2010).
Alexandrova, K. S. & Conger, B. V. Isolation of two somatic embryogenesis-related genes from orchardgrass (Dactylis glomerata). Plant Sci. 162, 301–307 (2002).
Chen, C. H. & Chen, Z. X. Potentiation of developmentally regulated plant defense response by AtWRKY18, a pathogen induced Arabidopsis transcription factor. Plant Physiol. 129, 706–716 (2002).
Maren, P., Silke, S., Stefan, B. & Ulrike, Z. Senescence Networking: WRKY18 is an Upstream Regulator, a Downstream Target Gene, and a Protein Interaction Partner of WRKY53. J. Plant Growth Regul. 33, 106–118 (2014).
Hinderhofer, K. & Zentgraf, U. Identification of a transcription factor specifically expressed at the onset of leaf senescence. Planta 213, 469–473 (2001).
Sun, C. et al. A novel WRKY transcription factor, SUSIBA2, participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell 15, 2076–2092 (2003).
Johnson, C. S., Kolevski, B. & Smyth, D. R. TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell 14, 1359–1375 (2002).
Deslandes, L. et al. Resistance to Ralstonia solanacearum in Arabidopsis thaliana is conferred by the recessive RRS1-R gene, a member of a novel family of resistance genes. PNAS 99, 2404–2409 (2002).
Dellagi, A. et al. A potato gene encoding a WRKY-like transcription factor is induced in interactions with Erwinia carotovora subsp atroseptica and Phytophthora infestans and is coregulated with class I endochitinase expression. Mol. Plant Microbe. Interact. 13, 1092–1101 (2002).
Chen, W. et al. Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 14, 559–574 (2002).
Higashi, K. et al. Modulation of defense signal transduction by flagellin-induced WRKY41 transcription factor in Arabidopsis thaliana. Mol. Gen. Genet. 279, 303–312 (2008).
Eulgem, T. & Somssich, I. E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 10, 366–371 (2007).
Ciolkowski, I., Wanke, D., Birkenbihl, R. P. & Somssich, I. E. Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol. Biol. 68, 81–92 (2008).
Dong, J. X., Chen, C. H. & Chen, Z. X. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 51, 21–37 (2003).
Zhou, Q. Y. et al. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant. Biotechnology J. 6, 486–503 (2008).
Eulgem, T., Rushton, P. J., Robatzek, S. & Somssich, I. E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206 (2000).
Cui, X. W. et al. Overexpression of a P. edulis (Phyllostachys edulis) transcription factor gene PheWRKY1 enhances disease resistance in transgenic Arabidopsis thaliana. Botany 91, 486–494 (2013).
Rice WRKY Working Group. Nomenclature report on rice WRKY’s – Conflict regarding gene names and its solution. Rice 5, 3 (2012).
Xie, Z. et al. Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol 137, 176–189 (2005).
Chen, F., Mackey, A. J., Vermunt, J. K. & Roos, D. S. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS ONE 2, e383 (2007).
Xie, Z. et al. Interactions of two abscisic-acid induced WRKY genes in repressing gibberellin signaling in aleurone cells. Plant J. 6, 231–242 (2006).
Rossberg, M. et al. Comparative sequence analysis reveals extensive microcolinearity in the lateral suppressor regions of the tomato, Arabidopsis, and Capsella genomes. Plant Cell 13, 979–988 (2001).
Ramamoorthy, R. et al. A comprehensive transcriptional profiling of the WRKY gene family in O. sativa under various abiotic and phytohormone treatments. Plant Cell Physiol. 49, 865–879 (2008).
Ling, J. et al. Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genomics 12, 471 (2011).
Guo, C. L. et al. Evolution and expression analysis of the grape (Vitis vinifera L.) WRKY gene family. J. EXP. BOT. 65, 1513–1528 (2014).
Zhu, J. K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 53, 247–273 (2002).
Krasensky, J. & Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 63, 1593–1608 (2012).
Ding, Z. J. et al. Transcription factor WRKY46 modulates the development of Arabidopsis lateral roots in osmotic/salt stress conditions via regulation of ABA signaling and auxin homeostasis. Plant J. 84, 56–69 (2015).
Ding, Z. J. et al. Transcription factor WRKY46 regulates osmotic stress responses and stomatal movement independently in Arabidopsis. Plant J. 79, 13–27 (2014).
Ramon, M., Rolland, F., Thevelein, J. M., Van, D. P. & Leyman, B. ABI4 mediates the effects of exogenous trehalose on Arabidopsis growth and starch breakdown. Plant. Mol. Biol. 63, 195–206 (2007).
Seo, Y. J. et al. Overexpression of the ethylene-responsive factor gene BrERF4 from Brassica rapa increases tolerance to salt and drought in Arabidopsis plants. Mol. Cells. 30, 271–277 (2010).
Li, X. Y. et al. Overexpression of Arachis hypogaea AREB1 gene enhances drought tolerance by modulating ROS scavenging and maintaining endogenous ABA content. Int. J. Mol. Sci. 14, 12827–42 (2013).
Michaela, H. & Dirk, K. H. LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC genomics 9, 118 (2008).
Miao, Y., Laun, T., Zimmermann, P. & Zentgraf, U. Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol. Biol. 55, 853–867 (2004).
Wen, F. et al. Genome-Wide Evolutionary characterization and expression analyses of WRKY Family genes in Brachypodium distachyon. DNA Research 13, 1–13 (2014).
Robatzek, S. & Somssich, I. E. A new member of the Arabidopsis WRKY transcription factor family, AtWRKY6, is associated with both senescence and defence related processes. Plant J. 28, 123–133 (2001).
Luo, X. et al. Ectopic expression of a WRKY homolog from Glycine soja alters flowering time in Arabidopsis. PloS ONE 8, e73295 (2013).
Yu, S., Li, G. C., Zhang, L. & Yu, D. Overexpression of OsWRKY72 gene interferes in the abscisic acid signal and auxin transport pathway of Arabidopsis. J. Biosci 35, 459–471 (2010).
Yang, P. Z., Chenm, C. H., Wang, Z. P., Fan, B. F. & Chen, Z. X. A pathogen- and salicylic acid-induced WRKY DNA-binding activity recognizes the elicitor response element of the tobacco class I chitinase genepromoter. Plant J. 18, 141–149 (1999).
Zhang, C. Q. et al. The WRKY transcription factor OsWRKY78 regulates stem elongation and seed development in O. sativa. Planta 234, 541–554 (2011).
Fan, C. J., Ma, J. M., Guo, Q. R., Li, X. T. & Wang, H. Selection of reference genes for quantitative real-time PCR in bamboo (Phyllostachys edulis). PLoS ONE 8, e56573 (2013).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2007).
Guo, A. Y., Zhu, Q. H., Chen, X. & Luo, J. C. GSDS: a gene structure display server. Yi Chuan 29, 1023–6 (2007).
Bailey, T. L., Williams, N., Misleh, C. & Li, W. W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–373 (2006).
Cao, J., Huang, J. L., Yang, Y. P. & Hu, X. Y. Analyses of the oligopeptide transporter gene family in poplar and grape. BMC Genomics 12, 465 (2011).
Zhang, Y. J., Ma, P. F. & Li, D. Z. High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS One 6, e20596 (2011).
Chen, Y. W. The physioligical response and research of PheWRKY2 from P. edulis under phosphorus stress. Dissertation, Chinese academy of forestry sciences (2012).
Acknowledgements
This project was supported by the National Keypoint Research and Invention Program in 13th Five-Year [grant No. 2016YFD060090102] and Fundamental Research Funds of ICBR [grant No.1632015009].
Author information
Authors and Affiliations
Contributions
L.L. performed the bioinformatics analyses, stress experiments, real-time PCR and drafted the manuscript. SHM helped in stress experiments and sample collection. Y.Z. contributed to RNA extraction and RT-PCR, ZCC assisted in gene cloning and plasmid construction. Y.M. and C.L.H. contributed to revisions of the manuscript. X.P.L. and J.G. designed the experiments and conceived the project, provided overall supervision of the study and revising the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Mu, S., Cheng, Z. et al. Characterization and expression analysis of the WRKY gene family in moso bamboo. Sci Rep 7, 6675 (2017). https://doi.org/10.1038/s41598-017-06701-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06701-2
This article is cited by
-
A wheat WRKY transcription factor TaWRKY17 enhances tolerance to salt stress in transgenic Arabidopsis and wheat plant
Plant Molecular Biology (2023)
-
WRKY transcription factors: evolution, regulation, and functional diversity in plants
Protoplasma (2023)
-
Biotechnological interventions in bamboo plants
Plant Cell, Tissue and Organ Culture (PCTOC) (2023)
-
Genome-wide identification, evolution and expression analysis of the aspartic protease gene family during rapid growth of moso bamboo (Phyllostachys edulis) shoots
BMC Genomics (2021)
-
The TCP transcription factor PeTCP10 modulates salt tolerance in transgenic Arabidopsis
Plant Cell Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
