
Abstract
Pachydermoperiostosis (PDP) is a rare inherited multisystem disease characterized with digital clubbing, pachydermia and periostosis. Variants in either HPGD or SLCO2A1 that interrupt the prostaglandin E2 (PGE2) pathway have been shown to be involved in PDP. Here, in addition to six confirmed variants in HPGD or SLCO2A1, we identified four novel SLCO2A1 variants in eight PDP patients from seven Chinese Han families. In addition, gastric mucosa hyperplasia was observed in all affected individuals and interleukin-6 (IL-6), tumor necrosis factor-alpha (TNFα) and receptor activator of nuclear factor kappa ligand (RANKL) expression were elevated in hypertrophic gastric mucosa. Two of eight patients who had severe arthralgia were treated with celecoxib. After three months, their arthralgia was partly relieved and IL-6, TNFα and RANKL expression were decreased in accordance with their relieved hypertrophic gastric mucosa. Our study broadens the variation spectrum of SLCO2A1 and suggests that the gastric mucosa hyperplasia might be a common characteristic of PDP. Moreover, celecoxib would be a considerable choice for PDP patients. We also revealed that IL-6, TNFα and RANKL may play important roles in the molecular mechanisms of gastric mucosa hyperplasia in PDP for the first time.
Similar content being viewed by others


Introduction
Pachydermoperiostosis (PDP), also named primary hypertrophic osteoarthropathy (HOA), is a rare genetic disease, whose major manifestations include digital clubbing, periostosis and pachydermia. Other clinical characteristics, such as arthralgia, arthritis, seborrhea, acne, hyperhidrosis, cutis verticis gyrate (CVG), acroosteolysis and ptosis were also reported1, 2. So far, variants in two genes involved in prostaglandin catabolic pathway, HPGD (MIM 259100)3 and SLCO2A1 (MIM 614441)4, have been found to cause PDP. HPGD encodes the degradative enzyme 15-hydroxyprostaglandin dehydrogenase (15-PGDH), a critical enzyme of metabolizing prostaglandins5. And SLCO2A1 encodes the prostaglandin transporter (PGT) that uptakes prostaglandins into cellular matrix for further degradation6, 7.
Most of the previous studies focused on bone and skin changes of PDP patients, while their gastric conditions were seldom reported. In fact, most PDP patients did not receive endoscopy or other relative examinations so that their gastric lesions might be ignored. Still, relative symptoms have been repeatedly reported by different groups independently. Lakshmi TS et al. first put forward that a PDP patient was associated with Menetrier’s disease8, a rare disease characterized by massive overgrowth of mucous cells (foveola) in the mucous membrane lining the gastric, resulting in large gastric folds. M.BHARATH et al. also reported a PDP patient associated Menetrier’s disease in 20169. Three independent reports showed hypertrophic gastric mucosa in PDP patients and gastric biopsy revealed gastric mucosa hyperplasia of the surface epithelium, cystic changes, stromal edema with inflammatory cells infiltration10,11,12. Other three PDP patients with hypertrophic gastritis were also respectively reported but they didn’t receive histological examination13,14,15. In addition, digestive system disease, such as ulcer, juvenile polyps, gastric cancer, Crohn’s disease and protein-losing enteropathy, were also reported occurring as occasional associations of PDP16,17,18. Early in 2001, Zrinka Jajic et al. performed gastroscopy examination in 21 of 76 PDP patients (27.6%) and they found all of these patients had ulcer or hypertrophic gastritis19. In 2011, according to Fortes BC’s summary, more than 20% PDP patients present gastric system abnormities20.
The specific pathogenesis of PDP is still unclear. Besides the PGE2 metabolism, inflammation and unbalance between the bone resorption and osteogenesis also play important roles in PDP3, 4, 7. Several cytokines and cell receptors, such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNFα), receptor activator of nuclear factor Kappa ligand (RANKL), were reported increasing in serum and synovial fluid of PDP patients21,22,23. We detected the IL-6, TNFα and RANKL expression in hypertrophic gastric mucosa of PDP patients for the first time.
Patients and Methods
Patients and Ethics
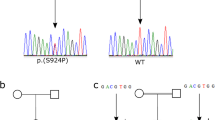
This study was approved by the Ethics Committee of The Second Xiangya Hospital of Central South University and all methods were in accordance with the Declaration of Helsinki. All participants have provided informed written consent to genetic and biochemical analysis and to publication of all identifying images. All PDP patients were of Han ethnicity and were diagnosed using established clinical and radiological criteria as a standard1. The pedigrees of these seven families are shown in Fig. 1. All patients were born from healthy non-consanguineous couples except patient1 and patient2, who were born from second-cousin unaffected parents. (Fig. 1a) Parental material of patient5 and patient6 and maternal material of patient4 were not available, as well as father’s material of patient8.

Pedigrees and variants of seven families affected by pachydermoperiostosis. The arrows in pedigrees show probands in the families. Black symbols mean PDP patients. Gray symbols mean unaffected homozygous or compound heterozygous carriers. Red arrows show novel variants on SLCO2A1 and black arrows show reported variants on SLCO2A1 and HPGD in chromatograms. Het, heterozygous. Hom, homozygous. Wt, wild type.
Variant analysis
Genomic DNA was extracted from peripheral white blood cells using the QIAamp DNA Blood Maxi Kit (QIAGEN KK, Tokyo, Japan). The whole DNA sequence of HPGD and SLCO2A1 were obtained from the available online database (UCSC: NM_00860.5, NM_005630.2). PCR was performed to amplify all exons and exon-intron boundaries of SLCO2A1 and HPGD followed by direct sequencing (Primers seen in Table S1). Online software, Mutationtaster (http://www.mutationtaster.org)24, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)25, and SIFT (http://sift.jcvi.org/)26 were used to predict the damaging effect of missense variants, and the HSF(http://www.umd.be/HSF3/)27 was used to analyze the damaging effect of splicing site variants. The pathogenicity of variants was interpreted according to ACMG guidelines28.
Immunohistochemical staining
8 PDP patients and 4 relative normal controls’ paraffin embedded gastric mucosa tissues were performed immunohistochemical staining. Specimens were grilled for 2 hours at 60° C oven at first. Then the specimens were dewaxed with xylene and rehydrated with gradient alcohol. Antigen retrieval was performed by using 0.01 mmol citrate buffer to make the samples infiltrate in for 15 minutes after steaming in pressure-cooker. After retrieval, the tissues were dealt with rabbit antibody against human IL-6 (abcam, ab9324) and mouse antibody against human RANKL (abcam, ab45039) and TNFα (abcam, ab6671) overnight at 4 °C. Biotinylated secondary antibody was adopted in SP kit at room temperature for 10 minutes followed with incubating of an appropriate amount of HRP-conjugated secondary antibody at room temperature for 30 minutes. Immunostaining was carried out through using DAB for chromogen and hematoxylin for nuclear staining. The specimens were cleared in xylene after dehydrated with gradient alcohol. Finally the neutral beams were applied to seal the specimens. All immunostained specimens were examined with the microscope (BA210T, Motic) and assessed by two pathologists blinding to clinical features. Under the Microscopy, the images and IOD (integrated optical density) were acquired with the Image-Pro Plus software 6.0.
Results
Clinical manifestations
All eight patients are males and began to show symptoms of PDP before their age of 20. Their major clinical manifestations were summarized in Table 1 and all of them were diagnosed as major PDP. Patient4, the propositus in family3, developed swollen knee joints gradually which finally lead to severe motion limitation at the age of 24 (Fig. S1c). Patient8 does not have diaphyseal periostosis but shows distinctive features clubbing of fingers and toes and acroosteolysis (Fig. S1d,e). Other members of these seven families are normal except patient5’s sister who received subtotal gastrectomy because of peptic ulcer when she was 24 years old.
Endoscopy and endoscopic ultrasonography (EUS) were also performed on all patients. A patient with mild chronic superficial gastritis (Fig. 2 C) was also tested as a control. According to WE Bolch, the thickness of normal gastric mucosa is about 1.0–1.6 mm29. The control had a gastric mucosa of 1.8 mm. Comparing with the control, all PDP patients had similarly or more thickened gastric mucosa (Fig. 2): Patient1, patient2 and patient7, in particular, presented severely rough, thickened and gyrus-like gastric mucosa (9.1 mm, 4.6 mm and 5.6 mm respectively); Patient3, patient4, and patient8 showed moderately rough and thickened gastric mucosa (3.9mm, 3.8 mm, and 4.2 mm respectively); Patient5 and patient6 (2.3 mm and 2.4 mm respectively) had mildly thickened gastric mucosa comparable to that of the control. Furthermore, histological examination was employed to detect the pathologic changes in patients. Consistent with the results of endoscopy, all patients had different degrees of gastric mucosa hyperplasia. The patients with most thickened gastric mucosa showed most severe gastric mucous glands hyperplasia, intracellular mucus increase and inflammation (Fig. 2 P1 a–d). The inflammation and mucous glandular proliferation were reduced in moderately thickened gastric mucosa patients (Fig. 2 P4 e–h) and least severe in mildly thickened patients (Fig. 2 P6 i–l).

Representative clinical and histological manifestations of gastric mucosa hyperplasia in PDP patients. EUS, endoscopic ultrasonography. P, patient. C, control, a patient with mild chronic superficial gastritis. HE, hematoxylin eosin staining. AB-PAS, alcian blue-periodic acid stiff staining. Endoscopy and EUS results as well as HE staining and AB-PAS staining of gastric mucosa of patitent1 (a–d), patient4 (e–h), patient6 (i–l) and control (m–p). Patient1 (a, b), patient4 (e, f) and patient6 (i, j) present severely, moderately and mildly thickened gastric mucosa respectively. In consistence, gastric mucous glands hyperplasia, intracellular mucus increase and inflammation from patient1 (c, d), patient4 (g, h) and patient6 (k, l) reduced gradually. All the images and materials were originated from gastric antrum. Pathological diagnosis was provided by professional pathologist.
Variants identification in HPGD and SLCO2A1
Employing PCR followed by direct sequence analysis, we detected variants in both HPGD and SLCO2A1 in all patients. All detected variants were shown in Fig. 1 and Table S2. Five confirmed variants, four novel variants in SLCO2A1 (Fig. 1a–f) and one reported variant in HPGD (Fig. 1g) were found. Among the four novel variants, there were two missense variants c.1069 T > C (Fig. 1f) and c.1406 C > T (Fig. 1d), a nonsense variant c.1771C > T (Fig. 1c) and an adenine-to-cytosine transition at the invariant + 4 position of the acceptor site of intron1 (c.96 + 4 A > C) (Fig. 1f). Since parents’ materials of patient5, patient6 and maternal materials of patient4 and paternal materials of patient8 were not available, we would not be able to get their genetic information. From our current data, all the variants found in patients were inherited from their normal parents. According to the ACMG guidelines28, all these variants are classified to be either pathogenic or likely pathogenic except c.96 + 4 A > C, which is of uncertain significance. Noticeably, patient5 and his little sister (Fig. 1d II-2) share the same compound heterozygous variants, she received subtotal gastrectomy at 24 years old because of peptic ulcer which was also reported as an association of PDP16. But she did not show any signs of skin and bone changes of PDP so far. The incomplete penetrance of PDP symptoms in females is observed repeatedly30, 31 and appeared to be significant. Currently, the underlying mechanism is not so clear. And previous researches suggested that it might be the crosstalk between estrogen and prostaglandin signal pathway32, 33. Other individuals in these families were heterozygous carriers or had no variants.
IL-6, TNFα and RANKL expression were increased in hypertrophic gastric mucosa of PDP patients
The expression of IL-6, TNFα and RANKL was detected in eight PDP patients and four normal controls. As Fig. 3 shows, IL-6, TNFα and RANKL expression were elevated in hypertrophic gastric mucosa of PDP patients (Fig. 3, P1, P4, P6) compared with normal control (Fig. 3C). And the differences in IOD between patients and controls were statistically significant (Fig. S2). In addition, in accordance with the severity of the hyperplasic gastric mucosa of eight PDP patients, IL-6, TNFα and RANKL expression were strongest in serious hypertrophic gastric mucosa (Fig. 3 P1 a e i), then gradually reduced in moderately (Fig. 3 P4 b f j) and mildly (Fig. 3 P6 c g k) thickened gastric mucosa.

IL-6, TNFα and RANKL expression were increased in the hypertrophic gastric mucosa of PDP patients. P1, patient1; P4, patient4; P6, patient6; C, normal control. a–d, e–h and i–l showed immunohistochemistry staining of IL-6, TNFα and RANKL respectively. IL-6, TNFα and RANKL expression were stronger in hypertrophic gastric mucosa of PDP patients (P1 a,e,i, P4 b,f,j, P6 c,g,k) than that of healthy control (C d,h,l). IL-6, TNFα and RANKL expression were strongest in patient1 and gradually decreased in patient4 and patient6. Immunostained specimens were assessed by pathologists blinding to clinical features.
Celecoxib treatment was considered to be effective for PDP patients
For patient2 and Patient3 with severe arthralgia, we treated them with celecoxib, a nonsteroidal anti-inflammatory drug used to treat pain or inflammation through specifically inhibiting COX-2 and reducing PGE2 generation, for three months34, 35. According to the patients’ response, their arthralgia was partly relieved. Moreover, endoscopy as well as histological examination (Fig. 4) showed that although the proliferation of mucous glands still existed, both the inflammation of the gastric mucosa and the intracellular mucus were reduced as expected (Fig. 4 Patient2 g h, Patient3 o p). Moreover, IL-6, TNFα and RANKL expression in gastric mucosa of patient2 and patient3 were also decreased after them receiving celecoxib treatment for three months (Fig. 5 Patient2 d-f, Patient3 j-l). And these declines were obvious in IOD (Table S2).

Changes of endoscopy and pathological features of patient2 and patient3 after them accepting celecoxib treatment. EUS, endoscopic ultrasonography. HE, hematoxylin eosin staining. AB-PAS, alcian blue-periodic acid stiff staining. P2, patient2. P3, patient3. Patient2 and patient3 were treated with celecoxib for 3 months. The effect of the treatment was shown by endoscopy (a,e,i,m) and endoscopic ultrasonography (b,f,j,n) as well as HE staining (c,g,k,o) and AB-PAS staining (d,h,l,p). The thickness of gastric mucosa declined after treatment (e,f,m,n). Consistently, patients showed inflammation, mucous glandular proliferation and intracellular mucus increase before treatment (c,d,k,l) and all these symptoms were reduced by treatment, but glandular proliferation still exist (g,h,o,p). Pathological diagnosis was provided by professional pathologist.

IL-6, TNFα and RANKL expression decreased in gastric mucosa of patient2 and patient3 after them receiving celecoxib treatment. P2, patient2; P3, patient3. (a,d,g and j) showed immunohistochemical staining of IL-6. (b,e,h and k) showed immunohistochemical staining of TNFα. (c,f,i and l) showed immunohhistochemical staining of RANKL. Patient2 and patient3 received celecoxib treatment for three months. IL-6, TNFα and RANKL expression in their gastric mucosa decreased after the treatment (d–f, j–l). Immunostained specimens were assessed by pathologists blinding to clinical features.
Discussion
Homozygous variants and compound heterozygous variants in HPGD and SLCO2A1 were reported to cause PDP3, 4. In this study, we identified four novel variants in SLCO2A1, among which c.96 + 4 A > C is predicted to cause an abnormal splicing event by in silico analysis. Unfortunately, we have not been able to provide corresponding data to prove it because of the unavailability of the patient’s RNA. Further studies are needed to assess the effects of this splice site variant. Localized within highly homologues regions, both residue Tyr357 and Pro469 are highly conserved across species. In silico analysis indicated that c.1069T > C and c.1406C > T have high probability to affect the protein function (Table S3). Correspondently, the MAF (Minor Allele Frequency) are low in Exome Aggregation Consortium (EXAC: http://exac.broadinstitute.org/) and 1000 Genomes Project (1000 G: http://browser.1000genomes.org). According to the ACMG guideline28, we classified these two novel variants as likely pathogenic. The fourth novel variant c.1771C > T in SLCO2A1, which wasn’t recorded in either EXAC or 1000 G, replaces the Arg591 with a stop codon. Early in 2013, a SLCO2A1 null variant c.1807C > T (p.R603X), resulting in a loss of the last transmembrane domain of PGT, was found in a patient with PDP36. Comparing with it, c.1771C > T causes a loss of an even bigger fragment, including an important structure for PGT function as a transporter37. Consequently, we assumed that the c.1771C > T variant would affect its function as well. These variants combined with another SLCO2A variant were considered as the cause of PDP for patient4 (Fig. 1c II-1), patient5 (Fig. 1d II-1) and his little sister (Fig. 1d II-2) and patient7 (Fig. 1f II-4).
Gastric system condition in PDP patients was rarely concerned. But early in 1983, Lam et al. described two PDP brothers who had hypertrophic gastritis16. In 2001, Zrinka Jajic et al. showed gastroscopy test was positive (ulcer and hypertrophic gastritis) in all 21 PDP patients (100%). But this report did not attract the attention of researchers. About 261 PDP patients were reported between 2001 and 2017. Gastroscopy was performed only in 25 patients (9.58%) and 16 patients were showed to be positive (64%): 7 patients had gastric mucosa hyperplasia and 4 patients received histological examination, 3 patients had gastritis, 4 patients had gastric polyps and 2 patients had gastric carcinoma. So a large proportion of PDP patients did not receive the digestive system examination. In our study, we systematically revealed the gastric mucosa hyperplasia in all of eight PDP patients. We believe that the incidence of digestive system abnormities in PDP, especially the gastric mucosa hyperplasia, is much higher than it have been reported20. So gastric mucosa hyperplasia could be regarded as a common manifestation of PDP and related examinations were advised in every PDP patient.
Although the pathogenesis of PDP is currently unknown, causal variants of HPGD and SLCO2A1 increase PGE2 level3, 4, 7, 38. PGE2 has a conflicting effect on gastric mucosa. As a protection mechanism, physiological PGE2 contributes to the recovery of gastric mucosa injury through promoting gland cell regeneration and other effects39. But in pathological condition, elevated PGE2 could accelerate gastric mucosa cell proliferation and plays an important role in the development of tumors through various mechanisms40, 41. Consistently, we observed in our study that all eight PDP patients had different degree of gastric mucosa hyperplasia from combined results of endoscopy and histological examination. What’s more, this would suggest an increased risk of gastric cancer in PDP patients. And PDP patients do tend to develop gastric tumor at a relatively young age17, 20, 39, 40. Therefore, it is possible that high PGE2 level in PDP patients might result in gastric mucosa hyperplasia even hyperplasic tumor in a similar way. And the majority of PGE2 formed in vivo is derived from COX-2 and COX-2 also contributes significantly to the overproduction of PGE2 42. Thus, we treated patient2 and patient3 who had severe arthralgia with celecoxib. After three months, their arthralgia and gastric mucosa hyperplasia were partly relieved and no side-effects were complained. Celecoxib is a NSAID which specifically inhibit COX-2 and reduce PGE2 generation. Our results support published individual case reports that specific COX-2 inhibitor NSAIDs treatment, such as celecoxib, etoricoxib etc, could relief PDP patients’ suffer and could not worry about its side-effects on digestive system13, 43, 44.
Other publications described inflammation and bone metabolism were also involved in PDP23, 45. IL-6, TNFα and RANKL are three of the most important factors. IL-6 is a pro-inflammatory cytokine that produces multifunctional effects, such as proliferation and differentiation and act as a key player during inflammation46, 47. TNFα is also a pro-inflammatory cytokine associated with pleiotropic effects on different cell-types. It is produced by lots of cell types involved in inflammation. TNFα can stimulate PGE2 synthesis and can also increase RANKL expression23, 48. RANKL is a member of the TNF superfamilies. The main function of RANKL is to stimulate bone resorption through osteoclastogenesis and the activation of multinucleated mature osteoclasts49, 50. RANKL expression can be induced by PGE2, IL-6 and TNFα51. High level of IL-6, TNFα and RANKL in serum and synovial fluid was observed in PDP patients21,22,23. IL-6, TNFα and RANKL were regarded as bone turnover markers and bone resorption and arthritis in PDP were probably mediated by them21, 52. Our results showed IL-6, TNF α and RANKL expression were elevated in hypertrophic gastric mucosa of PDP patients (Fig. 3, Fig. S2). Moreover, their expressions were in keeping with the severity of gastric mucosa hyperplasia (Fig. 3) and decreased after the patients received specific COX-2 inhibitor (celecoxib) treatment (Fig. 5, Table S2). We speculate that PGE2, IL-6, TNFα and RANKL are involved in the molecular mechanisms of gastric mucosa hyperplasia in PDP.
Further researches are needed to more completely elucidate the association between gastric mucosa hyperplasia, PGE2, IL-6, TNFα and RANKL increase in PDP.
Conclusion
Here, we identified four novel variants in the SLCO2A1 gene and demonstrated that gastric mucosa hyperplasia might be a common clinical manifestation of PDP and celecoxib would be a considerable treatment for PDP patients. We also revealed that IL-6, TNFα and RANKL may play important roles in the molecular mechanisms of gastric mucosa hyperplasia in PDP for the first time.
References
Castori, M. et al. Pachydermoperiostosis: an update. Clinical genetics 68, 477–486, doi:10.1111/j.1399-0004.2005.00533.x (2005).
Zhang, Z., Zhang, C. & Zhang, Z. Primary hypertrophic osteoarthropathy: an update. Frontiers of medicine 7, 60–64, doi:10.1007/s11684-013-0246-6 (2013).
Uppal, S. et al. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nature genetics 40, 789–793, doi:10.1038/ng.153 (2008).
Zhang, Z. et al. Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. American journal of human genetics 90, 125–132, doi:10.1016/j.ajhg.2011.11.019 (2012).
Pichaud, F., Delage-Mourroux, R., Pidoux, E., Jullienne, A. & Rousseau-Merck, M.-F. Chromosomal localization of the type-I 15-PGDH gene to 4q34–q35. Hum Genet 99, 279–281 (1997).
Naoaki Kanai, Run Lu, Joseph A. Satriano, Yi Bao, Allan W. Wolkoff, Victor L. Sch-uster. Identification and Characterization of a Prostaglandin Transporter. SCIENCE 4, 1453–1467 (1995).
Seifert, W. et al. Mutations in the prostaglandin transporter encoding gene SLCO2A1 cause primary hypertrophic osteoarthropathy and isolated digital clubbing. Human mutation 33, 660–664, doi:10.1002/humu.22042 (2012).
T SS Lakshmi, P Narasimha Rao, Motilal Nagaria. Primary pachydermoperiostosis associated with Menetrier’s disease..pdf. Indian J Dermatol Venereol Leprol. 67, 256-258 (2001).
BHARATH KUMARAN, M. & Pachydermoperiostosis, K. M. and Menetrier’ s Disease in a Young Male. Indian Journal of Applied Research 6, 158–159 (2016).
Ukinc, K. et al. Pachydermoperiostosis with gynecomastia and osteoporosis: a rare case with a rare presentation. International journal of clinical practice 61, 1939–1940, doi:10.1111/j.1742-1241.2005.00748.x (2007).
Narayanan, S., Mohamed Gani, V. M. & Sundararaju, V. Primary hypertrophic osteoarthropathy with hypertrophic gastropathy. Journal of clinical rheumatology: practical reports on rheumatic & musculoskeletal diseases 16, 190–192, doi:10.1097/RHU.0b013e3181e04d80 (2010).
Sun, X. F. et al. Primary hypertrophic osteoarthropathy with gastric hypertrophy. J Rheumatol 38, 959–960, doi:10.3899/jrheum.101077 (2011).
Gomez Rodriguez, N., Ibanez Ruan, J. & Gonzalez Perez, M. Primary hypertrophic osteoarthropathy (pachydermoperiostosis). Report of two familial cases and literature review. Reumatologia clinica 5, 259–263, doi:10.1016/j.reuma.2009.01.007 (2009).
Karnan, S., Krishnamoorthy, V., Ethiraj, P. & Sathyanathan, B. P. Touraine-Solente-Gole syndrome: The complete form needs to be recognized. Indian journal of nuclear medicine: IJNM: the official journal of the Society of Nuclear Medicine, India 27, 201–204, doi:10.4103/0972-3919.112743 (2012).
Madruga Dias, J. A. et al. Pachydermoperiostosis in an African patient caused by a Chinese/Japanese SLCO2A1 mutation-case report and review of literature. Seminars in arthritis and rheumatism 43, 566–569, doi:10.1016/j.semarthrit.2013.07.015 (2014).
Lam, S. K. U. M. et al. Pachydermoperiostosis, hypertrophic gastropathy, and peptic ulcer. GASTROENTEROLOGY 84, 834–839 (1983).
Ikeda, F. et al. Pachydermoperiostosis associated with juvenile polyps of the stomach and gastric adenocarcinoma. Journal of gastroenterology 39, 370–374, doi:10.1007/s00535-003-1304-7 (2004).
De Mestier, L. et al. Gastric juvenile polyposis with high-grade dysplasia in pachydermoperiostosis. Case reports in gastroenterology 5, 508–515, doi:10.1159/000326955 (2011).
Jajic, Z., Jajic, I. & Nemcic, T. Primary hypertrophic osteoarthropathy: Clinical, radiologic, and scintigraphic characteristics. Archives of Medical Research 32, 136–142, doi:10.1016/S0188-4409(01)00251-X (2001).
Fortes, B. C. et al. Pachydermoperiostosis associated with gastric neoplasia. Rev Assoc Med Bras 57, 128–130 (2011).
Rendina, D. et al. Interleukin (IL)-6 and receptor activator of nuclear factor (NF)-κB ligand (RANKL) are increased in the serum of a patient with primary pachydermoperiostosis. Scandinavian Journal of Rheumatology 37, doi:10.1080/03009740701772457 (2008).
Matsumoto, T., Tsurumoto, T. & Shindo, H. A case of pachydermoperiostosis associated with arthritis. Modern Rheumatology 13, 371–373, doi:10.3109/s10165-003-0240-y (2003).
da Costa, F. V. et al. Infliximab Treatment in Pachydermoperiostosis. JCR: Journal of Clinical Rheumatology 16, 183–184, doi:10.1097/RHU.0b013e3181df91c6 (2010).
Schwarz, J. M., Rodelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods 7, 575–576, doi:10.1038/nmeth0810-575 (2010).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nature methods 7, 248–249, doi:10.1038/nmeth0410-248 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols 4, 1073–1081, doi:10.1038/nprot.2009.86 (2009).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research 37, e67, doi:10.1093/nar/gkp215 (2009).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine: official journal of the American College of Medical Genetics 17, 405–424, doi:10.1038/gim.2015.30 (2015).
Huh, C. H., Bhutani, M. S., Farfan, E. B. & Bolch, W. E. Individual variations in mucosa and total wall thickness in the stomach and rectum assessed via endoscopic ultrasound. Physiol Meas. 24, 15–22 (2003).
Niizeki, H. et al. The novel SLCO2A1 heterozygous missense mutation p.E427K and nonsense mutation p.R603* in a female patient with pachydermoperiostosis with an atypical phenotype. The British journal of dermatology 170, 1187–1189, doi:10.1111/bjd.12790 (2014).
Diggle, C. P. et al. Prostaglandin transporter mutations cause pachydermoperiostosis with myelofibrosis. Human mutation 33, 1175–1181 (2012).
Hatano, R. et al. Sex hormones induce a gender-related difference in renal expression of a novel prostaglandin transporter, OAT-PG, influencing basal PGE2 concentration. Am J Physiol Renal Physiol 302, 342–349, doi:10.1152/ajprenal.00366.2011.-Based (2012).
Ospina, J. A., Brevig, H. N., Krause, D. N. & SP, D. Estrogen suppresses IL-1beta-mediated induction of COX-2 pathway in rat cerebral blood vessels. Am J Physiol Heart Circ Physiol 286, 2010–2019 (2004).
Penning, T. D. et al. Synthesis and biological evaluation of the 5-Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of 4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J Med Chem 40, 1347–1365 (1997).
Guda, K. et al. Inactivating mutation in the prostaglandin transporter gene, SLCO2A1, associated with familial digital clubbing, colon neoplasia, and NSAID resistance. Cancer prevention research 7, 805–812, doi:10.1158/1940-6207.CAPR-14-0108 (2014).
Cheng, R. et al. Three novel mutations in the SLCO2A1 gene in two Chinese families with primary hypertrophic osteoarthropathy. Eur J Dermatol 23, 636–639, doi:10.1684/ejd.2013.2154 (2013).
Chan, B. S., Satriano, J. A. & VL, S. Mapping the substrate binding site of the prostaglandin transporter PGT by cysteine scanning mutagenesis. J Biol Chem. 274, 25564–25570 (1999).
Busch, J. et al. Mutations in the prostaglandin transporter SLCO2A1 cause primary hypertrophic osteoarthropathy with digital clubbing. The Journal of investigative dermatology 132, 2473–2476, doi:10.1038/jid.2012.146 (2012).
Brzozowski, T., Konturek, P. C., Konturek, S. J., Brzozowska, I. & T, P. Role of prostaglandins in gastroprotection and gastric adaptation. J Physiol Pharmacol 56, 33–55 (2005).
Fujimura, T., Ohta, T., Oyama, K., Miyashita, T. & K, M. Role of cyclooxygenase-2 in the carcinogenesis of gastrointestinal tract cancers: a review and report of personal experience. World J Gastroenterol 12, 1336–1345 (2006).
Imai, K. et al. [In vitro cell proliferation assay method using rat gastric cultured cells and effect of anti-ulcer drugs on the proliferation of cultured cells]. Yakugaku zasshi: Journal of the Pharmaceutical Society of Japan 114, 316–324 (1994).
Murphey, L. J. et al. Quantification of the major urinary metabolite of PGE2 by a liquid chromatographic/mass spectrometric assay: determination of cyclooxygenase-specific PGE2 synthesis in healthy humans and those with lung cancer. Analytical biochemistry 334, 266–275, doi:10.1016/j.ab.2004.08.019 (2004).
Li, Z. T., Wang, D. & Wang, S. Successful treatment of pachydermoperiostosis with etoricoxib in a patient with a homozygous splice-site mutation in the SLCO2A1 gene. The British journal of dermatology. doi:10.1111/bjd.14480 (2016).
Bernardo, S. G., Emer, J. J., Burnett, M. E. & Gordon, M. Hypertrophic osteoarthropathy presenting as unilateral cellulitis with successful treatment using pamidronate disodium. The Journal of clinical and aesthetic dermatology 5, 37–46 (2012).
Tinoco-Fragoso, F. & Domínguez-Cherit, M.-F. S. J1. Pachydermoperiostosis, a unique entity with distinctive clinical features. Dermatology Online Journal 21, 12–12 (2015).
Dittrich, A., Hessenkemper, W. & Schaper, F. Systems biology of IL-6, IL-12 family cytokines. Cytokine and Growth Factor Reviews 26, 595–602, doi:10.1016/j.cytogfr.2015.07.002 (2015).
Ataie-Kachoie, P., Pourgholami, M. H. & Morris, D. L. Inhibition of the IL-6 signaling pathway: A strategy to combat chronic inflammatory diseases and cancer. Cytokine and Growth Factor Reviews 24, 163–173, doi:10.1016/j.cytogfr.2012.09.001 (2013).
Köhm, M., Burkhardt, H. & Behrens, F. Anti-TNFα-therapy as an evidence-based treatment option for different clinical manifestations of psoriatic arthritis. Clinical and experimental rheumatology 33, S109–114 (2015).
Liu, W. & Zhang, X. Receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (Review). Molecular Medicine Reports 11, 3212–3218, doi:10.3892/mmr.2015.3152 (2015).
Silva, I. & Branco, J. C. Rank/RANKL/OPG: Literature review. Acta Reumatologica Portuguesa 36, 209–218 (2011).
Hanada, R., Hanada, T. & Penninger, J. M. Physiology and pathophysiology of the RANKL/RANK system. Biological Chemistry 391, 1365–1370, doi:10.1515/BC.2010.149 (2010).
Steeve, K. T., Marc, P., Sandrine, T., Dominique, H. & Yannick, F. IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine & Growth Factor Reviews 15, 49–60, doi:10.1016/j.cytogfr.2003.10.005 (2004).
Acknowledgements
We thank the patients and their family for participating in this study. Research supported by China Hunan Provincial Science&Technology Department and Development and Reform Comission of Hunan Province.
Author information
Authors and Affiliations
Contributions
Hui Huang and Yongjun Wang wrote the main manuscript text. Yong Cao, Boda Wu, Yonggui Li and Jianguang Tang collected the samples. Hui Huang, Liangliang Fan, Jianzhong Hu and Xiaoliu Shi prepared table 1, Figure 1, figure S1 figure S2 and tables S1–3. Hui Huang, Yongjun Wang, Yi Jiang, Jianzhong Hu and Xiaoliu Shi prepared Figures 2–5. Jianzhong Hu and Xiaoliu Shi guided the research and the writing. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, H., Wang, Y., Cao, Y. et al. Interleukin-6, tumor necrosis factor-alpha and receptor activator of nuclear factor kappa ligand are elevated in hypertrophic gastric mucosa of pachydermoperiostosis. Sci Rep 7, 9686 (2017). https://doi.org/10.1038/s41598-017-09671-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09671-7
This article is cited by
-
Four Variants of SLCO2A1 Identified in Three Chinese Patients with Chronic Enteropathy Associated with the SLCO2A1 Gene
Digestive Diseases and Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
