
Abstract
Multi-marker association tests can be more powerful than single-locus analyses because they aggregate the variant information within a gene/region. However, combining the association signals of multiple markers within a gene/region may cause noise due to the inclusion of neutral variants, which usually compromises the power of a test. To reduce noise, the “adaptive combination of P-values” (ADA) method removes variants with larger P-values. However, when both rare and common variants are considered, it is not optimal to truncate variants according to their P-values. An alternative summary measure, the Bayes factor (BF), is defined as the ratio of the probability of the data under the alternative hypothesis to that under the null hypothesis. The BF quantifies the “relative” evidence supporting the alternative hypothesis. Here, we propose an “adaptive combination of Bayes factors” (ADABF) method that can be directly applied to variants with a wide spectrum of minor allele frequencies. The simulations show that ADABF is more powerful than single-nucleotide polymorphism (SNP)-set kernel association tests and burden tests. We also analyzed 1,109 case-parent trios from the Schizophrenia Trio Genomic Research in Taiwan. Three genes on chromosome 19p13.2 were found to be associated with schizophrenia at the suggestive significance level of 5 × 10−5.
Similar content being viewed by others


Introduction
Multi-marker association tests can be more powerful than single-locus analyses because these tests combine variant information within a gene/region. Moreover, the multiple-testing penalty is moderate compared with that encountered in single-locus analyses. However, combining the association signals of multiple markers within a gene/region may cause noise due to the inclusion of neutral variants, which usually compromises the power of a multi-marker association test. To eliminate noise from neutral variants, the “adaptive combination of P-values” (ADA) method was proposed for the analyses of unrelated subjects1,2 and family data3.
The ADA method was originally proposed for rare-variant association testing2. While “rare” is frequently defined arbitrarily, here, according to Ionita-Laza et al.4, we defined variants with a minor allele frequency (MAF) \( < 1/\sqrt{2n}\) as rare, where n is the number of individuals in the study. The per-site P-values were first calculated for each individual variant site, and the ADA method was used to truncate larger per-site P-values that were more likely to be attributed to neutral variants. The P-value is the probability of obtaining a statistic as extreme as or more extreme than the observed statistic under the null hypothesis (H 0) of no association. However, a P-value provides no information regarding the alternative hypothesis (H 1). For example, a P-value of 10−9 may appear to provide strong evidence against H 0; however, if the test is low-powered, it may be almost as unlikely under H 1 as under H 0 5,6,7. In genome-wide association studies (GWAS), the power to detect disease-associated single-nucleotide polymorphisms (SNPs) varies with MAFs. In this work, we show that truncating variants according to P-values is not optimal, when both rare and common variants are considered (see the subsection “Ranking by Bayes factor vs. P-value”).
Zhou and Wang8 have extended the ADA method to address both rare and common variants (namely, RC-ADA, or “rare and common variants by adaptive combination of P-values”). However, the RC-ADA method also truncates neutral variants according to their P-values. In RC-ADA8, rare variants and common variants are weighted according to Beta(MAF;1,25) and Beta(MAF;0.5,0.5)4, respectively, where MAF is the MAF of the considered SNP. Compared with the commonly used weight function Beta(MAF;1,25), Beta(MAF;0.5,0.5) decreases slowly as the MAF increases. RC-ADA preserves the associations of common variants by assigning them this weight function.
An alternative summary measure to the P-value is the Bayes factor (BF)9,10, which is the ratio of the probability of the data under the alternative hypothesis to that under the null hypothesis, as follows:
where H 1 and H 0 are the alternative hypothesis and the null hypothesis, respectively. In this work, we show that truncating variants according to BFs is superior to truncating variants according to P-values, because BFs quantify the “relative” evidence supporting H 1. Here, we propose an adaptive combination of BFs (ADABF) method by extending our previous ADA method2 and the “adaptive rank truncated product” (ARTP) method11,12. As described in the “Methods” section, the highest k BFs in favor of H 1 are combined, in the observed sample and in each of the resamples, respectively. The optimal k that achieves the strongest signal is allowed to vary in the observed sample and in each of the resamples. Then, the significance of the gene/region is assessed by comparing the strongest signal in the observed sample with its counterparts in the resampling replicates.
The logic underlying this work can be traced back to the “variable-threshold (VT)” approach13. In the VT approach, Price et al. assume that a certain unknown MAF threshold, T, exists, and variants with MAFs lower than T are more likely to be disease-associated. Therefore, they compute the statistic for each MAF threshold and then search for the optimal MAF threshold with permutations. However, the MAF has little relevance to the association signals14,15. Disease-associated variants can be either rare or common. Here, we propose the ADABF method, which is based on the concept of VT, but we assume that a certain unknown BF threshold exists, and variants with BFs larger than this threshold are more likely to be disease-associated.
By performing extensive simulations with case-parent trios and unrelated case-control data, we find that our ADABF test is valid because the type I error rates match the nominal significance levels. Moreover, the ADABF test is more powerful than the other gene-based tests16,17,18,19. Various multi-marker methods and the single-locus transmission disequilibrium test (TDT)20,21 were then applied to the empirical data from the Schizophrenia Trio Genomic Research in Taiwan (S-TOGET)22.
Results
Simulation Results
Table 1 provides the type I error rates observed in 1,000,000 simulation replications (10,000 replications performed for each of the 100 Cosi data sets). All five tests are valid because their type I error rates match the nominal significance levels. Figures 1 (for case-parent trios) and 2 (for unrelated case-control data) present the power given the genome-wide significance level of 2.5 × 10−6 (\(=0.05/20000\), corresponding to a Bonferroni correction for testing 20,000 independent genes23,24). The power for each scenario was evaluated using 10,000 simulation replicates (100 replicates for each Cosi data set). The ADABF method outperformed the other multi-marker tests because it excluded the variants with smaller BFs.

Simulation results of the case-parent trios. Top row: OR = 1.5 for a deleterious allele and OR = 0.67 for a protective allele; bottom row: OR = 1.25 for a deleterious allele and OR = 0.8 for a protective allele. Left column: all causal variants were deleterious; right column: ~50% of the causal variants were deleterious and the other ~50% were protective. The x-axis shows the number of causal variants, whereas the y-axis shows the power (given a significance level of 2.5 × 10−6).
As described in the “Methods” section, we created the “ADABF1” test representing our ADABF method coupled with another prior distribution. While the prior used in ADABF was chosen according to the WTCCC GWAS6, the purpose of adding ADABF1 was to evaluate the sensitivity of the results to the prior setting. ADABF1 (standard deviation of the prior distribution = 0.1) performed similarly to ADABF (standard deviation of the prior distribution = 0.2, following the WTCCC GWAS6). ADA also performed well because it truncated the variants with larger P-values. The popular kernel test (denoted by “TK”) was more powerful than the burden test (or linear combination test, denoted by “TLC”). Because the percentage of causal variants (\(2/150\) or \(4/150\), as described in “Simulation Study”) was not large, TK was generally more powerful than TLC. This result is consistent with the finding observed in rare-variant association testing for unrelated case-control data25.
Table 2 provides the average computation time (in seconds) for each test in our simulations, which was measured on a Linux platform with an Intel Xeon E5-2690 2.9 GHz processor and 8 GB memory. As described in the “Methods” section, we used the sequential resampling approach26 to compute the P-values of ADABF, ADABF1, and ADA. The minimum and maximum resampling numbers were set as 102 and 107, respectively. A longer time would be required to obtain a more significant result. Therefore, the average computation time increased as the power and the number of causal variants increased.
For unrelated case-control data, we also evaluated the “Variable Weight Test for testing the effect of an Optimally Weighted combination of variants” (VW-TOW)18. Because this test requires permutations to compute the P-values, we could not afford the computation time to evaluate it under the genome-wide significance level of 2.5 × 10−6 (\(=0.05/20000\)). Instead, we performed VW-TOW with 10,000 permutations and evaluated its power under the significance level of 0.01 (as shown in Figure S1). We found that its power performance was similar to that of TK. Zhou and Wang showed that ADA2 was more powerful than VW-TOW18 in testing the effects of both rare and common variants and rare variants alone8. Because ADABF is based on a similar concept in which the neutral variants are removed, it is not surprising that this method can outperform VW-TOW18.
When the haplotypes were generated according to the linkage disequilibrium (LD) patterns in Asians, the simulation results were similar to the abovementioned findings (Figures S2-S4 in our supplementary information).
Application of Tests to the Genetic Analysis Workshop 17 Simulated Data
We further applied these multi-marker association tests to the Genetic Analysis Workshop 17 (GAW 17) simulated exome data27. Here, we analyzed two quantitative traits, i.e., Q4 and Q1. Q4 was not associated with any variants, whereas Q1 was influenced by 39 variants located in nine genes27. Conditional on the genotype data, the trait simulations were performed 200 times to generate 200 replicates for the 697 unrelated individuals.
TLC and TK were performed using the “SKAT” R package (version 1.2.1)4,19,28. The “Davies” method was used to compute the P-values29. VW-TOW was implemented using the R code downloaded from the authors’ website, i.e., http://www.math.mtu.edu/~shuzhang/software.html, and the number of permutations was set as 106. The rare variant threshold (RVT) used in VW-TOW was set as \(1/\sqrt{2n}\), where n is the sample size (697). Age and smoking status served as covariates adjusted in TLC, TK, and VW-TOW.
To perform ADABF, ADABF1, and ADA2, we first considered the linear regression for each locus as follows:
where Y is the quantitative trait (Q4 or Q1) and G l is the genotype score (0, 1, or 2) of the l th variant (l = 1, …, 24487). We obtained the maximum likelihood estimate (MLE) of β l (l = 1, …, 24487) and the corresponding variance by fitting the linear regression (Eq. 2). The prior distribution of the true effect sizes (β l ’s) was assumed to be N(0,W), where the prior variance was W = 0.22 = 0.04 for ADABF and W = 0.12 = 0.01 for ADABF1 (see Figure S5). The prior for ADABF was the prior setting from the WTCCC GWAS6, and was adopted for ADABF throughout this work. Although this prior was originally proposed for dichotomous traits6, we considered it suitable for standardized quantitative traits with a mean of 0 and a standard deviation of 1 (because this prior implied that 95% of the true effect sizes range from −0.4 to 0.4).
The P-values of ADABF, ADABF1, and ADA2 were all obtained using the sequential resampling approach26 in which the minimum and maximum numbers of resampling were set as 102 and 107, respectively.
To assess the type I error rates, for each replication, we sequentially tested the association of each gene with Q4. Summarizing 200 replications, we obtained 641,000 (= 200 × 3205) P-values for each multi-marker association method. Because Q4 did not depend on any variant, we assessed the type I error rates by calculating the percentages of the 641,000 P-values that were smaller than the significance level, i.e., \(0.05/3205=1.56\times {10}^{-5}\), where 3,205 was the number of genes in the GAW 17 data. The first row in Table 3 provides the type I error rates. VW-TOW, ADABF, and ADABF1 yielded type I error rates that were the closest to the significance level (\(0.05/3205=1.56\times {10}^{-5}\)).
To quantify the power, we analyzed the association of all the nine causal genes that influenced Q127. Among the nine genes, the power for six genes (ARNT, ELAVL4, FLT4, HIF3A, VEGFA, and VEGFC) was smaller than 0.1 for all the tests and it was impossible to compare the different methods using this very low power. The second to fourth rows shown in Table 3 provide the power for the remaining three causal genes, i.e., KDR, FLT 1, and HIF1A, respectively. For each gene and each method, we obtained 200 P-values after analyzing all the 200 replicates. We quantified the power by calculating the percentage of the 200 P-values that were smaller than the significance level (\(0.05/3205=1.56\times {10}^{-5}\), where 3,205 was the number of genes in the GAW 17 exome data).
Overall, ADABF was the most powerful test. It provided the largest summation of power for detecting the three genes. Different from the above simulation results, TLC was not the least powerful test for the following two reasons:
-
(1)
According to the simulation model of the GAW 17 data, for all causal variants, the minor allele was associated with a higher mean Q127. Therefore, the power of TLC would not be compromised due to the coexistence of trait-increasing and trait-decreasing variants.
-
(2)
As described in the above simulation results, TLC is vulnerable to a small causal percentage (i.e., the percentage of causal variants among all variants in the gene). In contrast to the small causal percentage in the abovementioned simulations (\(2/150\) or \(4/150\)), the causal percentages of the three genes were all larger than 30% here (shown in Table 3).
Application to the Schizophrenia Trio Genomic Research in Taiwan (S-TOGET)
Schizophrenia is a highly heritable disease30. Previous studies have suggested that 1/3 to 1/2 of the genetic variants responsible for schizophrenia are common31,32, and these variants are genotyped using GWAS arrays. Therefore, GWAS is an important tool for exploring the genetic architecture of schizophrenia.
A portion of the Taiwanese case-parent trios obtained from the S-TOGET from 2009 to 2014 were subjected to GWAS genotyping22, approved by the Research Ethics Committee of the National Taiwan University Hospital (NTUH-REC no. 200810016 R). We confirmed that all experiments were performed in accordance with the relevant guidelines and regulations.
Totally 3,374 subjects were genotyped using the PsychChip array, which was developed by the Psychiatric Genomics Consortium (PGC) and Illumina (Illumina, San Diego, CA). After removing individuals with call rates < 98%, Mendelian errors, or sex inconsistency, 1,109 case-parent trios were used for analysis.
The PsychChip array (PsychChip_15048346_B) included ~580,000 markers in total. After removing invariant markers, markers with call rates < 98%, and markers that were significant for the Hardy-Weinberg equilibrium test (P-value < 10−6 in controls), 325,994 autosomal markers were retained for analysis.
The PsychChip array is a genotyping chip customized for psychiatric phenotypes. Unlike most commercial GWAS arrays, the PsychChip array allows investigators to simultaneously examine multiple genetic variants, including SNPs and rare variants. Of the 325,994 autosomal variants, 65,658 variants had MAFs < 1%, and 21,989 variants had MAFs ranging from 1% to 5%, where the MAFs were calculated according to the parents of the 1,109 trios.
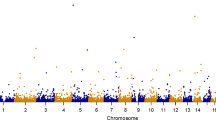
We first used the single-variant TDT20,21 to analyze the 1,109 case-parent trios. As shown in the bottom-right plot of Fig. 3, no variant was found to be associated with schizophrenia at the genome-wide significance level of 5 × 10−8 (\(0.05/1,000,000\)) or at the suggestive significance level of 10−6 (\(1/1,000,000\))33,34.

Simulation results of the unrelated cases and controls. Top row: OR = 1.5 for a deleterious allele and OR = 0.67 for a protective allele; bottom row: OR = 1.25 for a deleterious allele and OR = 0.8 for a protective allele. Left column: all causal variants were deleterious; right column: ~50% of the causal variants were deleterious and the other ~50% were protective. The x-axis shows the number of causal variants, whereas the y-axis shows the power (given a significance level of 2.5 × 10−6).

Manhattan plots of the Schizophrenia Trio Genomic Research in Taiwan (S-TOGET) data. Red lines indicate the genome-wide significance levels, i.e., 2.5 × 10−6 for the gene-based analyses and 5 × 10−8 for the single-locus analysis, respectively. Blue lines mark the suggestive significance levels, i.e., 5 × 10−5 for the gene-based analyses and 10−6 for the single-locus analysis, respectively. The three points surpassing the suggestive significance threshold represent the signals of the three genes (EVI5L, PRR36, and LYPLA2P2), although only the most significant gene (PRR36) is labeled.
We then resorted to multi-marker analyses. Because the SNP positions of the S-TOGET data were based on the human genome GRCh37/hg19 assembly, we mapped variants into genes according to the same assembly in the UCSC Genome Bioinformatics database (http://www.genome.ucsc.edu). We also included the 5’ and 3′ flanking regions of each gene. The 5′ flanking region may contain regulatory sequences such as promoters that control gene transcription. The 3′ flanking region may contain sequences that terminate transcription. Multi-marker analyses may include ±5 kb35, ±10 kb36, ±20 kb37, or ±30 kb38 flanking regions of a gene. Because incorporating additional flanking sequences increases the coverage of more distant regulatory elements, we grouped the variants within ± 30 kb flanking regions of a gene into a multi-marker analysis according to Song et al.38. In total, there were 24,769 autosomal genes.
TLC and TK were performed using the “rvTDT” R package (version 1.0)16. ADABF, ADABF1, and ADA2 were performed using the sequential resampling approach26, in which the minimum and maximum numbers of resampling were set as 102 and 107, respectively. The genome-wide significance level for the gene-based analyses is usually determined at 2.5 × 10−6 (\(0.05/20,000\))23,24, and the suggestive significance level is set at 5 × 10−5 (\(1/20,000\)), respectively.
As shown in Fig. 3 and Table 4, no gene was found to be associated with schizophrenia at the genome-wide significance level of 2.5 × 10−6. Three genes on chromosome 19p13.2, including EVI5L (ecotropic viral integration site 5 like), PRR36 (proline rich 36), and LYPLA2P2 (lysophospholipase II pseudogene 2), were detected to be associated with schizophrenia at the suggestive significance level of 5 × 10−5. This is a consistent result across all the five gene-based association tests except TLC.
The 13 SNPs in the EVI5L-PRR36-LYPLA2P2 region are described in Table 5. Some of the odds ratios (ORs) of the minor alleles compared with the major alleles were greater than 1, whereas others were less than 1. The TLC test could suffer from a power loss in this situation. Hence, it was not surprising that TLC could not identify the association signal of this region.
Discussion
In this work, we proposed the “adaptive combination of Bayes factors” (ADABF) method, which is applicable to a mixture of common and rare variants and can be applied to GWAS or next-generation sequencing (NGS) data.
Chromosome 19p13.2 has been found to be associated with panic disorder39. Based on our analysis for the S-TOGET trio data, three genes in this region, including EVI5L, PRR36, and LYPLA2P2, were detected to be associated with schizophrenia at the suggestive significance level of 5 × 10−5. Four multi-marker tests including ADABF, ADABF1, ADA, and TK all suggest that PRR36 is the most significant gene. This gene encodes a large protein - Proline Rich Protein 36 (PRP36)40. The second significant gene identified by the four multi-marker tests is EVI5L. It is also a protein-coding gene, but its function remains unknown41. The third significant gene identified by the four tests is LYPLA2P2, which is a pseudo gene42.
The gene next to the EVI5L-PRR36-LYPLA2P2 region (7865161–7975117 base pair) is MAP2K7 (mitogen-activated protein kinase kinase 7, also known as the “MKK7” gene, 7968665–7979363 base pair). Knocking out MAP2K7 results in schizophrenia-like behavioral deficits in mice43,44,45. A substantial effect size was observed for common variants in a case-control sample from the Glasgow area and a replication sample of Northern European descent46,47.
In our analysis of the EVI5L gene, the most prominent signal was achieved by combining the top four significant SNPs (see Table 5, i.e., rs525420, rs1651016, rs652260, and rs555609). This was a consistent prioritization of SNPs across ADABF, ADABF1, and ADA. These four SNPs have not been reported to be associated with schizophrenia. As shown in these four SNPs, the ORs of the minor alleles compared with the major alleles are larger than 1.25 or smaller than 0.8, corresponding to one of our simulation scenarios. To detect variants with smaller effect sizes, the number of case-parent trios must be increased.
In this work, we used the prior in the WTCCC GWAS6 [β~N(0,W), with a variance of W = 0.22 = 0.04] as the prior for ADABF. To evaluate the sensitivity of our results to this choice, we also considered another prior variance, i.e., W = 0.12 = 0.01. We found that our simulation and the S-TOGET results were very stable across these two settings. As noted by Stephens and Balding5, W can be chosen dependently on the MAF according to prior settings that are believed to best fit the underlying genetic architecture of a disease. Therefore, theoretically, we can develop better ways to prioritize SNPs.
With the advent of NGS technology, there has been a great interest in rare-variant association testing. However, both rare and common variants contribute to the etiology of complex diseases such as the Hirschsprung disease48, schizophrenia49, and type 2 diabetes50. Certain specialized arrays such as PsychChip were designed for the detection of both common and rare variants. There is a need to develop a powerful method for the joint analysis of rare and common variants. Compared with ADA2 and RC-ADA8, our ADABF method is recommended for its applicability to variants with a wide spectrum of MAFs. Compared with other multi-marker association tests such as TLC16,51, TK4,16,19,28, and VW-TOW18, our ADABF method is recommended for its robustness to the inclusion of neutral variants.
Methods
Here, we describe the method to analyze case-parent trios, but it can be generalized to unrelated case-control analyses. For a variant with two alleles, i.e., M 1 (the allele of interest) and M 2, the TDT tests whether the M 1 allele is transmitted to an affected child more often than the M 2 allele from heterozygous parents20.
Let OR be the odds ratio of allele M 1 compared with allele M 2. We denote \(\hat{\beta }\) as the MLE of log(OR). According to the asymptotic normality of MLE, \(\hat{\beta } \sim N(\beta ,V)\). Let b be the number of transmissions of M 1 from heterozygous parents to the affected offspring, and let c be the number of such transmissions of M 2 . We then obtain \(\hat{\beta }=\,\mathrm{log}(b/c)\) and \(\hat{V}=\frac{b+c}{bc}\). The prior distribution of the true effect sizes is assumed to be a normal distribution, i.e., β~N(0,W). Throughout this work, we follow the WTCCC GWAS6 to specify the prior variance, i.e., W = 0.22 = 0.04. The prior distribution is presented in the left column of Figure S5 of our supplementary information. This method is designated by “ADABF.”
To evaluate the performance sensitivity of ADABF using this prior, we also specify another prior variance, i.e., W = 0.12 = 0.01 (right column of Figure S5). This method is designated by “ADABF1.”
According to Wakefield7,52, the BF is as follows:
where \({\hat{\beta }}^{2}/\hat{V}\) is the Wald statistic. For unrelated subjects, \(\hat{\beta }\) and \(\hat{V}\) are the MLE and its corresponding variance from the linear regression (continuous traits) or logistic regression (dichotomous traits) for a particular variant. The greater the significance of an association, the larger the \({\hat{\beta }}^{2}/\hat{V}\) and BF. Given a fixed \(\hat{\beta }=\,\mathrm{log}(1.2)=0.18\), a larger \(\hat{V}\) corresponds to a lower power and a decrease in the BF, because the power is not sufficient for providing strong evidence supporting H 1 (Figure S6). Given a fixed P-value = 0.02 (i.e., a fixed \({\hat{\beta }}^{2}/\hat{V}\)), the BF is small when \(\hat{V}\) is extremely small (Figure S7). An extremely small \(\hat{V}\) implies an extremely small \({\hat{\beta }}^{2}\) given a P-value of 0.02, and thus, the data are unlikely under H 1. Moreover, a large \(\hat{V}\) represents a low power that is not sufficient for supporting H 1 (Figure S7).
Ranking by Bayes factor vs. P-value
In this subsection, we show that the BF ranking is superior to the P-value ranking, in a region with a mixture of rare and common variants. We performed 200,000 simulation replications to compare the rankings of a causal variant by the BF or P-value. In each replication, one of ~150 variants was specified as the causal variant. As described in the simulation study, a mixture of rare and common variants was observed in the region (see Figure S8). The disease status (Y = 1 denotes disease) was generated according to the following model:
where \(\alpha =\,\mathrm{log}(0.05/0.95)=-2.94\), implying a disease prevalence of 5%. G c was the genotype score (0, 1, or 2) of the causal variant, and the effect size was \(\beta =\,\mathrm{log}(1.5)\). In total, 200,000 replicates were performed to compare the ranking of a causal variant by the BF (x-axis in Fig. 4) to that by the P-value (y-axis in Fig. 4). The results shown in Fig. 4 and Table 6 are stratified according to the MAF of the causal variant.

Ranking by Bayes factor vs. P-value. We performed 200,000 simulations to compare the rankings of a causal variant using the Bayes factor (x-axis) and the P-value (y-axis). The chromosomal region included ~150 rare or common variants, and one of these variants was specified as the causal variant. The scatter plot was stratified according to the MAF of the causal variant. The black line in each plot represents x = y.
Let R B and R P be the rankings of the causal variant by BF and P-value, respectively. For a region containing 150 variants, 1 ≤ R B , R P ≤ 150. A smaller rank would be better, meaning that the causal variant would be ranked in priority order. The following three outcomes could be obtained: (1) the BF ranking was superior if R B < R P , (2) the BF ranking was identical to the P-value ranking if R B = R P , and (3) the BF ranking was inferior if R B > R P . According to Table 6, the mean rank of the causal variant by the BF was smaller than (or equal to) that by the P-value, across all ranges of causal-allele frequencies. As the MAF of the causal variant increased, the power to detect that causal variant also increased and both mean ranks improved. More replicates showed that the BF ranking outperformed the P-value ranking, across all ranges of causal-allele frequencies.
Compared with the P-value ranking, rare causal variants will benefit from the BF ranking (see the top-left plot of Fig. 4). This finding can be attributed to a rare causal variant generally having a larger P-value (say, P-value = 0.2) and a larger \(\hat{V}\) (say, \(\hat{V}\) = 0.1). As shown in Figure S9(c), its BF will be larger than that of a common neutral variant with the same P-value but a smaller \(\hat{V}\) (say, \(\hat{V}\) = 0.005). That is, a common variant with a P-value = 0.2 may actually be a neutral variant, because this large P-value is obtained from reliable information (smaller \(\hat{V}\)). However, a rare variant with a P-value = 0.2 may still be causal, because this large P-value is obtained from less reliable information (larger \(\hat{V}\)). Rare variants seldom have small P-values, and therefore, our previous ADA method2 prioritizes the rare variants with P-values smaller than 0.2. However, in a region with a mixture of rare and common variants, a P-value threshold of 0.2 is too liberal for common variants. In this situation, it will be better to consider the “relative” evidence in favor of H 1 (i.e., BF), instead of P-values.
Summarizing the BFs in a Chromosome Region
Let BF l be the BF of the l th variant. Denote the ordered BFs by BF (1) ≤ BF (2) ≤ … ≤ BF (L) for a region containing L variants. The summary score aggregating the highest k BFs is as follows:
where I(BF l ≥ BF (k)) is 1 if the l th variant is among the top k most significant variants according to BF, and is 0 otherwise. Because the natural logarithm of the BF is linked to log-likelihoods, log(BF) is considered the “weight of evidence”10. Therefore, in Eq. (5), we summarize the association evidence provided by L variants in the region of interest. Because log(BF) represents the “weight of evidence”10, we do not impose any additional weight according to the MAF. As previously mentioned, the MAF has little relevance to association signals14,15. Disease-associated variants can be either rare or common. If we believe that rare variants are more likely to be non-neutral, the Beta(MAF;1,25) function can be used to weight the contribution of individual BFs.
Based on Eq. (5), we obtain S 1, …, S L for a region containing L variants. Then, we use the efficient sequential resampling approach proposed by Liu et al.26 to assess the significance of the association between the region and a disease. The procedure is performed as follows:
-
(i)
We first draw B = 100 sets of \({\hat{{\beta }}}_{0}\) (the L × 1 vector of point estimates under the null hypothesis) from the multivariate normal distribution N(0 L×1,V L×L ), where the (i, j)th element of V L × L is \({R}_{i,j}\sqrt{{\hat{V}}_{i}{\hat{V}}_{j}}\). \({\hat{V}}_{i}\) and \({\hat{V}}_{j}\) are the estimated variances of \({\hat{\beta }}_{i}\) and \({\hat{\beta }}_{j}\), respectively [i, j = 1, …, L. Recall that \({\hat{\beta }}_{i}\) and \({\hat{V}}_{i}\) are obtained from a regression model of the i th variant, such as Eq. (2)]. Yang et al.53 have shown that the correlation among the association statistics in a region can be well approximated by the correlation among the genotypes. Therefore, R i,j is estimated from the correlation of the genotypes at the i th and j th loci. When analyzing case-parent trios, only the founder genotypes are used to calculate R i,j .
-
(ii)
For the b th set of \({\hat{{\boldsymbol{\beta }}}}_{0}\), we calculate the BFs using Eq. (3) and the summary scores using Eq. (5). Given k (k = 1, …, L), we compare S k with S k (b) (b = 1, …, B) and obtain the P-value of S k by \([{\sum }_{b=1}^{B}I({S}_{k}^{(b)}\ge {S}_{k})]/B\). In the observed sample, we find the minimum P-value across k (k = 1, …, L), which is denoted by MinP. The minimum P-value of the b th resample is calculated similarly and denoted by MinP (b), b = 1, …, B. Finally, the adjusted P-value is \([{\sum }_{b=1}^{B}I(Min{P}^{(b)}\le MinP)]/B\).
-
(iii)
If the adjusted P-value based on 100 sets of \({\hat{{\boldsymbol{\beta }}}}_{0}\) is smaller than 0.1, we draw 10 times more sets (i.e., B = 1,000) to increase the precision of the P-value. This procedure is repeated until the P-value is larger than \(10/B\) or a desired precision level is reached.
The R code of our ADABF method can be downloaded from http://homepage.ntu.edu.tw/~linwy/ADABF.html.
Competitor Methods
The ADABF test (prior variance W = 0.22 = 0.04) was compared with the ADABF1 (prior variance W = 0.12 = 0.01) and ADA tests2. To make a fair comparison, these three tests were all performed using the “adaptive rank truncated product” (ARTP) method11,12. Therefore, the highest k BFs (or the smallest k P-values) are combined, in the observed sample and in each of the resamples, respectively. The abovementioned sequential resampling approach26 was used to assess the significance of the association between the region of interest and the disease, and the minimum and maximum numbers of resampling were set as 102 and 107, respectively.
Furthermore, the TLC and TK tests were performed for comparison. These two tests were performed using the “rvTDT” R package (version 1.0)16 and the “SKAT” R package (version 1.2.1)4,19,28, to analyze the case-parent trios and unrelated subjects, respectively. To make a fair comparison, we did not assign any MAF-weighting function to the TLC, TK, ADABF, ADABF1, or ADA tests.
For the analysis of unrelated subjects, we also compared ADABF with VW-TOW18. VW-TOW is a test used to detect associations of rare and common variants, and was proposed by Sha et al.18. These authors divided the variants into rare (if MAF < RVT) and common (if MAF ≥ RVT), and then searched for the optimal weights for the two groups of variants, separately. The statistics from the two parts of the variants were then combined, and the P-value was calculated with permutations. This test was performed using the R code, which was downloaded from the authors’ website at http://www.math.mtu.edu/~shuzhang/software.html, and the number of permutations was set as 10,000. The RVT was set as \(1/\sqrt{2n}\), where n was the sample size4. Because VW-TOW was proposed for the analyses of unrelated individuals18, it was not evaluated for case-parent trio data.
Simulation Study
With the Cosi program54, we generated 100 data sets following the LD patterns in Europeans. Each of the 100 Cosi data sets contained 10,000 chromosomes from a 20 kilo base (kb) pairs region. That is, totally 100 20-kb regions were considered. On average, ~150 variants could be observed in a 20-kb region. The distribution of the MAFs of the variants is shown in Figure S8 in the supplementary information, which presents as an L-shaped distribution that is typical of allele frequencies55.
To evaluate the type I error rates, the disease status (Y = 1 denotes disease) was generated according to the following model:
where α = −2.94. To study the power, the disease status was generated according to the following model:
where α = −2.94, d was the number of causal variants (d = 2 or 4), G k c was the genotype score (0, 1, or 2) of the k th causal variant, and the effect sizes were \(\beta \text{'}s=\pm \mathrm{log}(1.5)\) or \(\pm \mathrm{log}(1.25)\). β was positive or negative depending on whether the causal variant was deleterious or protective, respectively. When \(\beta =\pm \mathrm{log}(1.5)\), the odds ratio (OR) was 1.5 for a deleterious allele and \(1/1.5=0.67\) for a protective allele. When \(\beta =\pm \mathrm{log}(1.25)\), the OR was 1.25 for a deleterious allele and \(1/1.25=0.8\) for a protective allele. The following two scenarios were evaluated:
-
(1)
All causal variants were deleterious and
-
(2)
In total, 50% of the causal variants were deleterious, and 50% of the causal variants were protective.
The following two data structures were simulated: (1) 2,000 case-parent trios and (2) 2,000 unrelated subjects, of which 1,000 were cases, and 1,000 were controls.
References
Lin, W. Y. Beyond Rare-Variant Association Testing: Pinpointing Rare Causal Variants in Case-Control Sequencing Study. Sci Rep 6, 21824, https://doi.org/10.1038/srep21824 (2016).
Lin, W. Y., Lou, X. Y., Gao, G. & Liu, N. Rare Variant Association Testing by Adaptive Combination of P-values. Plos One 9, e85728, https://doi.org/10.1371/journal.pone.0085728 (2014).
Lin, W. Y. & Liang, Y. C. Conditioning adaptive combination of P-values method to analyze case-parent trios with or without population controls. Sci Rep 6, 28389, https://doi.org/10.1038/srep28389 (2016).
Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J. D. & Lin, X. Sequence kernel association tests for the combined effect of rare and common variants. American journal of human genetics 92, 841–853, https://doi.org/10.1016/j.ajhg.2013.04.015 (2013).
Stephens, M. & Balding, D. J. Bayesian statistical methods for genetic association studies. Nat Rev Genet 10, 681–690, https://doi.org/10.1038/nrg2615 (2009).
WTCCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Wakefield, J. Bayes factors for genome-wide association studies: comparison with P-values. Genetic epidemiology 33, 79–86, https://doi.org/10.1002/gepi.20359 (2009).
Zhou, Y. & Wang, Y. Detecting association of rare and common variants by adaptive combination of P-values. Genetics research 97, e20, https://doi.org/10.1017/S0016672315000208 (2015).
Jeffreys, H. Theory of probability (3rd ed.). Oxford, U.K.: Oxford University Press (1961).
Kass, R. E. & Raftery, A. E. Bayes factors. Journal of the American Statistical Association 90, 773–795 (1995).
Yu, K. et al. Pathway analysis by adaptive combination of P-values. Genetic epidemiology 33, 700–709, https://doi.org/10.1002/gepi.20422 (2009).
Dudbridge, F. & Koeleman, B. P. Rank truncated product of P-values, with application to genomewide association scans. Genetic epidemiology 25, 360–366, https://doi.org/10.1002/gepi.10264 (2003).
Price, A. L. et al. Pooled association tests for rare variants in exon-resequencing studies. American journal of human genetics 86, 832–838 (2010).
Li, C., Li, M., Lange, E. M. & Watanabe, R. M. Prioritized subset analysis: improving power in genome-wide association studies. Hum Hered 65, 129–141 (2008).
Lin, W. Y. & Lee, W. C. Incorporating prior knowledge to facilitate discoveries in a genome-wide association study on age-related macular degeneration. BMC Res Notes 3, 26 (2010).
Jiang, Y. et al. Utilizing population controls in rare-variant case-parent association tests. American journal of human genetics 94, 845–853, https://doi.org/10.1016/j.ajhg.2014.04.014 (2014).
Lee, S., Lin, X. & Wu, M. C. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13, 762–775 (2012).
Sha, Q., Wang, X. & Zhang, S. Detecting association of rare and common variants by testing an optimally weighted combination of variants. Genetic epidemiology 36, 561–571, https://doi.org/10.1002/gepi.21649 (2012).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. American journal of human genetics 89, 82–93, https://doi.org/10.1016/j.ajhg.2011.05.029 (2011).
Spielman, R. S., McGinnis, R. E. & Ewens, W. J. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). American journal of human genetics 52, 506–516 (1993).
Terwilliger, J. D. & Ott, J. A haplotype-based ‘haplotype relative risk’ approach to detecting allelic associations. Hum Hered 42, 337–346 (1992).
Wang, S. H. et al. Polygenic risk for schizophrenia and neurocognitive performance in patients with schizophrenia. Genes Brain Behav. https://doi.org/10.1111/gbb.12401 (2017).
Epstein, M. P. et al. A statistical approach for rare-variant association testing in affected sibships. American journal of human genetics 96, 543–554, https://doi.org/10.1016/j.ajhg.2015.01.020 (2015).
Li, C. et al. Genome-Wide Association Study Meta-Analysis of Long-Term Average Blood Pressure in East Asians. Circ Cardiovasc Genet 10, e001527, https://doi.org/10.1161/CIRCGENETICS.116.001527 (2017).
Basu, S. & Pan, W. Comparison of statistical tests for disease association with rare variants. Genetic epidemiology 35, 606–619, https://doi.org/10.1002/gepi.20609 (2011).
Liu, Q., Chen, L. S., Nicolae, D. L. & Pierce, B. L. A unified set-based test with adaptive filtering for gene-environment interaction analyses. Biometrics 72, 629–638, https://doi.org/10.1111/biom.12428 (2016).
Almasy, L. et al. Genetic Analysis Workshop 17 mini-exome simulation. BMC Proc 5(Suppl 9), S2, https://doi.org/10.1186/1753-6561-5-S9-S2 (2011).
Lee, S., Wu, M. C. & Lin, X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13, 762–775, https://doi.org/10.1093/biostatistics/kxs014 (2012).
Davies, R. B. Algorithm AS 155: the distribution of a linear combination of χ2 random variables. Journal of the Royal Statistical Society. Series C (Applied Statistics) 29, 323–333 (1980).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427, https://doi.org/10.1038/nature13595 (2014).
International Schizophrenia Consortium. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752, https://doi.org/10.1038/nature08185 (2009).
Ripke, S. et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 45, 1150–1159, https://doi.org/10.1038/ng.2742 (2013).
He, Y. et al. Multi-breed genome-wide association study reveals novel loci associated with the weight of internal organs. Genet Sel Evol 47, 87, https://doi.org/10.1186/s12711-015-0168-7 (2015).
Zhang, F. et al. Genome-wide association studies for hematological traits in Chinese Sutai pigs. BMC Genet 15, 41, https://doi.org/10.1186/1471-2156-15-41 (2014).
Yan, Q. et al. Kernel-Machine Testing Coupled with a Rank-Truncation Method for Genetic Pathway Analysis. Genetic epidemiology. https://doi.org/10.1002/gepi.21813 (2014).
Muchero, W. et al. High-resolution genetic mapping of allelic variants associated with cell wall chemistry in Populus. BMC Genomics 16, 24, https://doi.org/10.1186/s12864-015-1215-z (2015).
Huang, H., Chanda, P., Alonso, A., Bader, J. S. & Arking, D. E. Gene-based tests of association. PLoS Genet 7, e1002177, https://doi.org/10.1371/journal.pgen.1002177 (2011).
Song, F. et al. Exonuclease 1 (EXO1) gene variation and melanoma risk. DNA Repair (Amst) 11, 304–309, https://doi.org/10.1016/j.dnarep.2011.12.005 (2012).
Gregersen, N. O. et al. Association between genes on chromosome 19p13.2 and panic disorder. Psychiat Genet 26, 287–292, https://doi.org/10.1097/Ypg.0000000000000147 (2016).
Proline rich 36 [Homo sapiens (human)]. National Center for Biotechnology Information (NCBI). https://www.ncbi.nlm.nih.gov/gene/80164, Retrieved 24 August 2017.
Ecotropic viral integration site 5 like [Homo sapiens (human)]. National Center for Biotechnology Information (NCBI). https://www.ncbi.nlm.nih.gov/gene/115704, Retrieved24 August 2017.
Lysophospholipase II pseudogene 2 [Homo sapiens (human)]. National Center for Biotechnology Information (NCBI). https://www.ncbi.nlm.nih.gov/gene/?term=LYPLA2P2, Retrieved24 August 2017.
Lipina, T. V. & Roder, J. C. Drug Discovery for Schizophrenia. Royal Society of Chemistry (2015).
Openshaw, R. L., Thomson, D. M., Penninger, J. M., Pratt, J. A. & Morris, B. J. Mice haploinsufficient for Map2k7, a gene involved in neurodevelopment and risk for schizophrenia, show impaired attention, a vigilance decrement deficit and unstable cognitive processing in an attentional task: impact of minocycline. Psychopharmacology (Berl). https://doi.org/10.1007/s00213-016-4463-y (2016).
Yamasaki, T. et al. Stress-activated protein kinase MKK7 regulates axon elongation in the developing cerebral cortex. J Neurosci 31, 16872–16883, https://doi.org/10.1523/JNEUROSCI.1111-11.2011 (2011).
Winchester, C. L. et al. Converging evidence that sequence variations in the novel candidate gene MAP2K7 (MKK7) are functionally associated with schizophrenia. Hum Mol Genet 21, 4910–4921, https://doi.org/10.1093/hmg/dds331 (2012).
Thompson, R. Genetic and functional investigation of FXYD6 and MAP2K7 as risk factors in schizophrenia. PhD thesis, University of Glasgow (2013).
Alves, M. M. et al. Contribution of rare and common variants determine complex diseases-Hirschsprung disease as a model. Dev Biol 382, 320–329, https://doi.org/10.1016/j.ydbio.2013.05.019 (2013).
Gratten, J. Rare variants are common in schizophrenia. Nat Neurosci 19, 1426–1428, https://doi.org/10.1038/nn.4422 (2016).
Bonnefond, A. & Froguel, P. Rare and common genetic events in type 2 diabetes: what should biologists know? Cell Metab 21, 357–368, https://doi.org/10.1016/j.cmet.2014.12.020 (2015).
Morris, A. P. & Zeggini, E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genetic epidemiology 34, 188–193 (2010).
Wakefield, J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. American journal of human genetics 81, 208–227, https://doi.org/10.1086/519024 (2007).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44(369-375), S361–363, https://doi.org/10.1038/ng.2213 (2012).
Schaffner, S. F. et al. Calibrating a coalescent simulation of human genome sequence variation. Genome Res 15, 1576–1583 (2005).
Luikart, G. & Cornuet, J. M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12, 228–237 (1998).
Acknowledgements
The authors would like to thank the Editorial Board Member and the anonymous reviewers for their insightful and constructive comments. This work was supported by grants 105–2314-B-002–030 and 106–2314-B-002-040 from the Ministry of Science and Technology of Taiwan (to W.-Y.L.), NIH/NHGRI grant U54HG003067, NIMH grants R01 MH085521, R01 MH085560, the Gerber Foundation, the Sidney R. Baer, Jr. Foundation, NARSAD: The Brain and Behavior Research Foundation, and the Stanley Center for Psychiatric Research. The authors would like to thank the GAW 17 workshop organizers for the permission to use their data in this study. Preparation of the Genetic Analysis Workshop 17 Simulated Exome Data Set was supported by the GAW grant, R01 GM031575, and in part by NIH R01 MH059490. The workshop used sequencing data from the 1000 Genomes Project (http://www.1000genomes.org). The authors would like to thank Drs. Benjamin Neale at the Broad Institute, Stephen V. Faraone at SUNY Upstate Medical University, and Nan Laird at Harvard University for the S-TOGET data generation. The authors would also like to thank Dr. Benjamin Neale for helpful comments and suggestions.
Author information
Authors and Affiliations
Contributions
W.-Y.L. conceived the idea of this study, developed the statistical methodology and the analysis tools, performed the simulations and real data analysis, and drafted the manuscript. W.J.C. contributed to the design of this study and the writing of the manuscript, and provided the case-parent trio data. C.-M.L., H.-G.H., S.A.M., S.J.G., and M.T.T. participated in the manuscript writing and the data collection. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, WY., Chen, W.J., Liu, CM. et al. Adaptive combination of Bayes factors as a powerful method for the joint analysis of rare and common variants. Sci Rep 7, 13858 (2017). https://doi.org/10.1038/s41598-017-13177-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13177-7
This article is cited by
-
Sex differences in the genetic architecture of depression
Scientific Reports (2020)
-
Rare variant association testing in the non-coding genome
Human Genetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
