
Abstract
Plant cell cultures are an attractive platform for the production of recombinant proteins. A major drawback, hindering the establishment of plant cell suspensions as an industrial platform, is the low product yield obtained thus far. Histone acetylation is associated with increased transcription levels, therefore it is expected that the use of histone deacetylase inhibitors would result in an increase in mRNA and protein levels. Here, this hypothesis was tested by adding a histone deacetylase inhibitor, suberanilohydroxamic acid (SAHA), to a cell line of the model legume Medicago truncatula expressing a recombinant human protein. Histone deacetylase inhibition by SAHA and histone acetylation levels were studied, and the effect of SAHA on gene expression and recombinant protein levels was assessed by digital PCR. SAHA addition effectively inhibited histone deacetylase activity resulting in increased histone acetylation. Higher levels of transgene expression and accumulation of the associated protein were observed. This is the first report describing histone deacetylase inhibitors as inducers of recombinant protein expression in plant cell suspensions as well as the use of digital PCR in these biological systems. This study paves the way for employing epigenetic strategies to improve the final yields of recombinant proteins produced by plant cell cultures.
Similar content being viewed by others

Introduction
Plant cell suspensions are coming to prominence in the Molecular Farming field as a platform for the production of high value molecules. They have several key advantages when compared to conventional production systems, such as the ability to perform post-translational modifications, the use of low cost culture medium and GMP compliance1. Unlike microbial cells, plant cells are able to perform the complex glycosylation needed for correct folding and activity of many proteins2. Due to cheaper growth conditions, plant cell suspension cultures offer a low-cost alternative to mammalian production systems. In addition, their controlled and confined growth using classical fermentation technology, which allows the implementation of GMP, overcomes regulatory issues concerning the use of transgenic plants (reviewed in3).
Molecular Farming has come a long way since 1989, when work by Hiatt and colleagues4 was featured on the cover of Nature. Many of the improvements since then were made by transferring knowledge obtained by researchers working on bacterial and mammalian platforms. Although plant cell cultures present many advantageous characteristics, the low final product yields that have been obtained to date remain the main drawback preventing this system from becoming a reliable production platform.
Efforts to reduce the overall cost of production in mammalian cell line systems have focused on increasing the final production yields5. One of the strategies used is to supplement media with protein expression inducers such as valproic acid6,7, sodium butyrate8,9,10 and hydroxamic acids11. These inducers are described to increase recombinant mRNA and protein expression levels by acting as histone deacetylase inhibitors (HDACi). Histone deacetylases (HDACs) remove acetyl groups from histones and their activity is regulated in the cell by the availability of acetyl-CoA (acetyl group donor) and by the HDAC/HAT (histone acetyltransferase) ratio12. HDACi are known to block the activity of HDAC enzymes, resulting in the hyperacetylation of histones. In turn, acetylated histones associate less tightly with DNA, facilitating access of the transcriptional machinery and potentially leading to higher mRNA synthesis and protein expression13. HDAC enzymes are present from prokaryotes to eukaryotes14 and it has been reported that their secondary structure, particularly around the enzyme’s DNA binding site, is highly conserved15. Some of these HDACi have recently been used as anti-cancer agents. Suberanilohydroxamic acid (SAHA) has been shown to arrest cancer cell growth, to induce autophagy and apoptosis, and to have an anti-proliferative activity16,17. The hydroxamic acid moiety of SAHA binds to the zinc ion located at the end of the catalytic tubular pocket18. The insertion of hydroxamic acids inside the catalytic pocket of a HDAC prevents binding of its natural substrates and thus results in inhibition of catalytic activity18,19. SAHA is described to inhibit HDAC classes I, II and IV from the RPD3-like superfamily17 and was the first HDACi approved by FDA for advanced cutaneous T-cell lymphoma cancer therapy20. In plants, these types of compounds have mainly been used for fundamental biology studies (Sodium Butyrate21,22, trichostatin A23,24,25 and SAHA, also known as vorinostat26) and to our knowledge there is no report of the use of these compounds as inducers of recombinant protein expression in plant cell cultures.
Medicago truncatula cell suspensions have been developed recently as a Molecular Farming platform and we have demonstrated a high yield of recombinant protein production for a fungal protein27. However, when human recombinant proteins (erythropoietin and lipocalin-type prostaglandin D2 synthase) were produced, the same high yields were not observed28,29. For this reason, a novel approach to improve the final yields of human recombinant proteins was designed, which involves the use of HDAC inhibitors. With this strategy, we intend to increase transgene expression levels by opening the chromatin through increased histone acetylation.
In this report, the potential of SAHA as a protein expression inducer in Medicago truncatula cell suspensions is addressed. Here, it is demonstrated, for the first time, that SAHA is able to increase the levels of expression of the human recombinant lipocalin-type prostaglandin D2 synthase (L-PGDS) produced by a stably transformed M. truncatula cell line. This protein has been chosen as a model since it is a glycosylated human protein that is involved in several physiological functions. It is localized in a variety of human tissues and is used as a molecular marker (reviewed in30). The effect of SAHA on histone acetylation and HDAC inhibition is also investigated.
Results and Discussion
Effect of adding SAHA on Medicago cell culture growth
The effect of SAHA on Medicago cell cultures expressing L-PGDS was initially screened by assessing its impact on cell viability. Cell viability was monitored by trypan blue dye exclusion. The addition of SAHA was tested on different days (0, 3 and 7) of growth as well as at different concentrations (2.5 and 5 µM). A cell culture without SAHA was also monitored as a control.
When SAHA was added at day 0, at 5 µM final concentration, cell viability dropped from around 90% to 60% on day 5 of growth. By day 14, cell viability decreased to 10%, suggesting that this concentration is unsuitable (Fig. 1). When SAHA was added at a lower concentration (2.5 µM), at day 0, cell viability decreased from 90% to 70% on day 5 of growth, with about 50% viable cells on day 14. Addition of SAHA at the beginning of the experiment appears to have a considerable effect on cell viability and cell division and the results are dose dependent. This might be due to lower cell density during early phases of growth, when compared to SAHA additions later on. The same effect is observed for mammalian cell cultures when this type of compound is used to induce protein expression, due to cell cycle arrest. Usually, inducers of protein expression are only added once a certain cell density has been attained31.

Cell viability tests performed using the trypan blue dye exclusion test throughout 14 days of culture. *p value ≤ 0.05; **p value ≤ 0.01; ***p value ≤ 0.001; statistical analysis represented by *follows the same color code as the graph. Error bars indicate S.E.M. for n = 6 (control) and n = 3 (SAHA).
Due to the toxic effects observed when using a concentration of 5 µM SAHA, further experiments were performed using a lower concentration (2.5 µM). In order to assess the effect of SAHA on recombinant L-PGDS protein production, two different time points for the addition of SAHA were tested (day 3 and day 7). When added on day 3, corresponding to the onset of exponential phase, SAHA did not have as severe a toxic effect as compared to that observed for addition on day 0 (Fig. 1). In fact, cell viability throughout growth was similar to that observed for the control without SAHA. Moreover, when SAHA was added on day 7 of growth (end of the exponential growth phase), the negative effect on cell viability was far greater than what was observed for addition on day 3. We hypothesize that an addition of SAHA at the end of the exponential growth phase might have a higher toxic effect due to a reduced rate of cell division and associated capacity to metabolize SAHA. Despite the decrease in cell viability following addition on day 7, the resulting protein production and gene expression levels were still assessed.
Analysis of L-PGDS gene expression
Two independent experiments were performed with SAHA added at different stages of growth; day 3 and day 7. The effect of SAHA on L-PGDS gene expression was analyzed by digital PCR. Digital PCR is a relatively new technique but there are already some reports describing its use in plant studies, namely for GMO quantification32 or in disease resistance33,34. In this report, digital PCR was chosen to study the expression of recombinant L-PGDS since the treatment with SAHA might affect the expression of any gene, even of the housekeeping genes that are usually used as an internal reference. The use of an internal reference would not be possible and digital PCR allows absolute quantification without the need for an internal control.
The QuantStudioTM 3D (QS3D) AnalysisSuiteTM software reads a digital PCR chip and assesses different parameters: the quality of the sample loading, the fluorescent signal from the positive PCR reactions and the background noise present in the chip reading. The chip quality is displayed by flags (green: good quality; yellow: medium quality–needs revision; red: poor quality) as seen on Figure S1. The loading quality is displayed by the continuous green color on the chip (see Figure S1B) and non-amplified and amplified reactions (yellow and blue, respectively) are also represented on chip (Figure S1A) and on scatter plots (Figure S1C).
In Figure S1, digital PCR data for three biological replicates of the same sampling point are shown and the scatter plot depicts the cluster of non-amplified and amplified reactions, which also allows for verification of the homogeneity of the results obtained. The QS3D AnalysisSuiteTM software takes into account the dilution factor of the sample in order to give a final value of copies per µL.
When SAHA was added, at either day 3 or day 7, the expression of the recombinant L-PGDS gene increased (Fig. 2). At day 11 there was no significant difference in transgene expression between the culture induced on day 3 and that induced on day 7. When compared to the control culture, transgene expression increased 1.9-fold (SAHA addition on day 3) and 2.2-fold (SAHA addition on day 7) for samples taken on day 11 of growth. The increase in gene expression is consistent with the observations made in mammalian cell cultures, where SAHA is considered a protein expression inducer35. The recombinant L-PGDS protein expression was thus assessed.

Gene expression analysis of Medicago cell lines expressing L-PGDS, with and without addition of 2.5 µM SAHA (at days 3 or 7 of growth). The calculated sample quantity (Copies/µL) represents the concentration of the cDNA sample in the PCR reaction mix. ***p value ≤ 0.001. Error bars indicate the theoretical confidence interval based on Poisson distribution calculated by the QS3D Analysis SuiteTM software for n = 6 (control) and n = 3 (SAHA).
Screening of recombinant protein levels
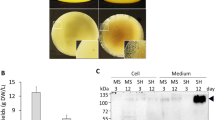
The production of recombinant L-PGDS, throughout growth, was assessed by western blotting. Addition of SAHA on day 3 resulted in higher accumulation of L-PGDS in the growth medium, compared with the control culture. This higher accumulation was observed two days after addition of the inducer of protein expression, with a 5-fold increase compared to the control (Fig. 3A). Accumulation of L-PGDS protein was also higher for these conditions on day 11, however by this point, the increase was only 2.4-fold when compared to the control. This might be due to degradation of proteins by proteases present in the cell medium (Santos et al. in preparation). On the other hand, when SAHA was added on day 7 of growth, a 2-fold decrease in L-PGDS accumulation in Medicago cell medium was observed three days after the addition. We hypothesized that this could be due to intracellular accumulation of L-PGDS protein. In order to assess this possibility, the levels of intracellular L-PGDS were investigated.

L-PGDS relative protein expression levels in cell medium. (A) Relative expression of L-PGDS with and without 2.5 µM SAHA throughout Medicago growth obtained from (B) western blot analysis of biological replicates of cell culture samples treated with SAHA on day 3 of growth and (C) on day 7 of growth with anti L-PGDS antibody. Control experiment lanes: 3, C5, C7 and C11. **p value ≤ 0.01; ***p value ≤ 0.001. Error bars indicate S.E.M. for n = 3. Full-length gels and immunoblots are shown in Supplementary Figure S2.
The amount of intracellular L-PGDS protein detected, when SAHA was added on day 3 of growth, was similar (day 5 and 11) or lower (day 7) than the control (see Fig. 4A and B). Despite a surge in protein production, the recombinant protein follows the secretory pathway, and is mostly released to the culture medium. However, when SAHA was added at day 7 of the growth, the surge in protein production led to the retention of L-PGDS protein inside the cell (see Fig. 4A and C), justifying the low levels of recombinant protein found in the cell medium. This accumulation was unforeseen given that the L-PGDS gene construct possesses a signal peptide that targets the protein to the secretory pathway. It has been described that under conditions of protein production peaks, the endoplasmic reticulum might become overloaded, which can result in greater intracellular accumulation of the recombinant protein36. Furthermore, stress or production of defective proteins might lead to unexpected subcellular protein sorting37. This phenomenon has already been described in mammalian cell cultures producing recombinant proteins. To overcome this, lower cell incubation temperatures are used to increase the expression of specific proteins. Lower temperatures could increase folding capacity and the expression of endoplasmic reticulum chaperones, which would ensure the recombinant proteins follows the secretory pathway and is exported to the medium38. Other studies suggest that the increase of recombinant protein production in CHO cells, following HDACi treatment at 33 °C (a lower temperature than the usually used), can be the result of elevated cellular secretory capacity39,40. The combination of these two strategies could be easily implemented in plant cell suspension cultures in order to further increase recombinant protein production.

L-PGDS relative protein expression levels in cell extracts. (A) Relative expression of L-PGDS with and without 2.5 µM SAHA throughout Medicago cell growth obtained from (B) western blot analysis of cell extract samples treated with SAHA on day 3 of growth and (C) on day 7 of growth, with anti L-PGDS antibody. Control experiment lanes: 3, C5, C7 and C11. **p value ≤ 0.01. Error bars indicate S.E.M. for n = 3. Full-length gels and immunoblots are shown in Supplementary Figure S3.
Histone deacetylase (HDAC) activity assay
Since transgene expression and recombinant protein levels increased in the presence of 2.5 µM SAHA, an in vitro assay was performed in order to confirm that this increase was correlated with HDAC inhibition by SAHA.
For this in vitro assay, nuclear extracts prepared from cells recovered at day 11 of M. truncatula culture were used. It is noteworthy that 2.5 µM SAHA was able to inhibit 74% (calculated using equation (1)) of the HDAC activity in Medicago samples. If HDACs are inhibited, fewer proteins become deacetylated. The amount of deacetylated histone was 2-fold lower in comparison to the control in the presence of SAHA (Fig. 5). This was important to evaluate since there are compounds that are able to inhibit mammalian HDACs but not plant HDACs. For instance, it has been described that sodium butyrate is a HDAC inhibitor in mammalian cells leading to histone hyperacetylation41. This compound was tested in alfalfa cells by Waterborg and colleagues, who concluded that it fails to inhibit alfalfa HDACs since it is readily metabolized into acetyl-CoA, which is then incorporated in acetylated histones42,43. The results we obtained from in vitro experiments confirmed that SAHA is able to inhibit the activity of M. truncatula HDACs. However, inhibiting HDACs activity does not correlate directly with histone acetylation since non-histone proteins can also be subjected to their deacetylase activity44. Due to this, the level of histone acetylation was also assessed.

Inhibition of HDAC enzymatic activity. Amount of deacetylated product without and with SAHA. Error bars indicate S.E.M. for n = 3. *p value ≤ 0.05.
Analysis of histone H3 acetylation levels
The level of histone acetylation was determined by immunoblotting. Cell extracts were recovered along growth after the addition of SAHA at different days and the level of histone H3 acetylation was assessed.
Histone H3 was chosen to study acetylation levels since it has been described as the main target of acetylation in plants, in contrast with mammalian cells and fungi where acetylation is mainly observed on histone H443,44. Since HDACs have different specificity for different lysines45, total histone H3 acetylation levels were studied and normalized to histone H3 protein levels.
An increase of histone H3 acetylation levels was observed 2 to 3 days after addition of SAHA in both experiments. When SAHA was added on day 3 of growth, a 1.6-fold increase of acetylation was observed. This increase was maintained throughout growth, with a 1.4-fold increase of acetylation levels observed on day 11 (see Fig. 6A). Addition of SAHA on the 7th day of growth resulted in a 3.1-fold increase of acetylation levels on day 11 (see Fig. 6A).

Histone H3 relative acetylation levels. (A) Relative acetylation level of histone H3 with and without 2.5 µM SAHA throughout Medicago culture growth obtained from (B) western blot analysis of cell extract samples treated with SAHA on day 3 of growth and (C) on day 7 of growth, with anti-acetyl-histone H3 and anti-histone H3 antibodies. Control experiment lanes: 3, C5, C7 and C11. *p value ≤ 0.05; **p value ≤ 0.01; ***p value ≤ 0.001. Error bars indicate S.E.M. for n = 3. Full-length gels and immunoblots are shown in Supplementary Figure S4.
The analysis of histone H3 acetylation levels confirms that SAHA is able to inhibit HDAC activity in vitro (Fig. 5) and this inhibition leads to the accumulation of acetyl groups in histone H3 lysines.
Conclusion
This report describes for the first time the use of a histone deacetylase inhibitor for the improvement of recombinant protein production in plant cell suspension cultures.
The effect of SAHA addition to Medicago truncatula cell culture was assessed at different growth stages. Although some degree of cell toxicity was observed, in a dose dependent manner, SAHA addition at day 3 and day 7 resulted in an increase of recombinant protein accumulation. When SAHA was added on day 7, protein was found to accumulate inside of the cells, rather than being secreted to the medium. This diversion from the targeted destination can result from an ER stress response, triggered by the synthesis of high amounts of recombinant proteins, and can lead to unexpected intracellular protein sorting46,47.
This report also demonstrates for the first time, to our knowledge, the use of digital PCR with plant cell culture samples. With this technique, it was possible to observe that addition of SAHA had a stimulatory effect on L-PGDS transgene expression and associated protein production, resulting in the accumulation of protein up to 5-fold higher than the control experiment. To prove the hypothesis that the addition of SAHA increases L-PGDS protein production by inhibiting HDACs and, therefore, increasing histone acetylation levels, the percentage of HDAC inhibition by SAHA was assessed. It was demonstrated for the first time that SAHA inhibits the activity of Medicago truncatula HDACs, which results in increased Histone H3 acetylation. Furthermore, the increase of histone H3 acetylation is concurrent with an increase in gene expression and of protein accumulation.
This work lays the first stone for the use of epigenetic modulation as a biotechnology tool to increase the yield of recombinant protein production in plant cell suspension cultures.
Material and Methods
Plant material, growth conditions and treatment with HDAC inhibitor
Medicago truncatula cell suspension cultures producing human lipocalin-type prostaglandin D2 synthase (L-PGDS)28 were grown in Murashige and Skoog medium (Duchefa, The Netherlands) supplemented with 30 g/L sucrose (Duchefa, The Netherlands), 4.65 μM kinetin (Duchefa, The Netherlands), 4.52 μM 2.4-D (Sigma-Aldrich, USA) and 65 μM dithiothreitol (Sigma-Aldrich, USA) at pH 5.8, in an orbital shaker (24 °C, 125 rpm) in the dark.
Experiments were established starting by subculturing a two-week old culture. Then, different SAHA concentrations were added at different time points (day 0, 3 and 7) of the culture growth and 2.5 μM SAHA was used for the following experiments. SAHA was added and for each time point biologic triplicates were prepared. SAHA solution was prepared in DMSO (Sigma-Aldrich, USA), which we previously verified did not affect cell growth. Spent medium was collected by vacuum filtration on days 3, 5, 7 and 11 from cultures with and without SAHA and stored at −20 °C.
Cell viability tests
Cell viability was assessed by dye exclusion (indicative of an intact membrane) using 0.4% trypan blue in PBS-T (Sigma-Aldrich, USA) (1 volume of cells: 1 volume of trypan blue). Live cells with intact membranes are distinguished by their ability to exclude dyes that easily penetrate dead or damaged cells. For each experiment, biologic replicates of all time points were analyzed (six for control experiments and three for SAHA experiments) and the ratio between live and total cells was determined by hemacytomer cell count distributions. A minimum of 50 cells were obtained for cell counting of each replicate. The average percentage of live cells and S.E.M. were calculated.
Screening of recombinant L-PGDS production
Western blot analysis was performed with spent medium and cell extract samples collected during cell culture growth. Medium samples were 10-fold concentrated using centrifugal filtration units with 10 kDa cut-off Amicon Ultra-15 (Millipore, USA). Cells were ground using liquid nitrogen and a mortar. Extraction buffer (100 mM ascorbic acid, 500 mM NaCl, 5 mM β-mercaptoethanol, pH 8.0) was added to ground cells in 1:1 ratio (g of cells: mL of extraction buffer). Samples were resolved in a 12.5% SDS-PAGE gel and proteins were transferred to a nitrocellulose membrane (0.2 µm, Amersham, UK). The membrane was blocked with 5% skimmed milk powder (Nestlé, Switzerland) and 3% BSA (NZYTech, Portugal) in PBS-T, for 1 hour at room temperature with gentle shaking, followed by incubation with 1:500 anti-L-PGDS (Ab 61866, Abcam, UK) in PBS-T, overnight at 4 °C. Then, the membrane was incubated with 1:8000 HRP conjugated anti-rabbit (A0545, Sigma-Aldrich, USA) in PBS-T, for 2 hours at room temperature with gentle shaking. Signal was detected by chemiluminescence using Amersham ECL Prime reagent (GE Healthcare Life Sciences, UK) in ChemiDoc™ XRS + System (Bio-Rad, USA). Three biological replicates were analyzed and western blots repeated three times.
Analysis of Histone H3 acetylation levels
Cell extracts were taken from cells recovered by vacuum filtration at day 3, 5, 7 and 11 of growth, as already described. Total protein extracts were resolved in a 15% SDS-PAGE gel and proteins were transferred to a nitrocellulose membrane (0.2 µm, Amersham, UK). The membrane was blocked with 5% BSA (NZYTech, Portugal) in PBS-T for one hour at room temperature, with gentle shaking, followed by an overnight incubation with 1:5000 anti-acetyl-histone H3 (06–599, Merck Millipore, USA) in PBS-T, at 4 °C with gentle shaking. Then, the membrane was incubated in 1:8000 anti-rabbit HRP conjugated (A0545, Sigma-Aldrich, USA) for two hours in agitation at room temperature. Acetyl-histone H3 bands were detected using Amersham ECL Prime reagent (GE Healthcare Life Sciences, UK) and Hyperfilm ECL (GE Healthcare Life Sciences, UK) in a dark room. Then, the membrane was stripped, by washing five times with PBS-T buffer for 30 minutes each. Before reprobing with anti-histone H3 antibody, the membrane was incubated with the secondary antibody anti-rabbit-HRP in order to ensure that the stripping procedure was correctly performed. If no signal was detected, the membrane was washed three times for 5 minutes each with PBS-T and incubated with 1:25000 anti-histone H3 (07–690, Merck Millipore, USA) in PBS-T, at 4 °C, overnight, with gentle shaking. Following incubation for two hours in agitation at room temperature with 1:8000 anti-rabbit HRP conjugated, histone H3 was detected as already described for acetyl-histone H3. Western blots were repeated three times.
Relative quantification of recombinant L-PGDS and levels of acetylated histone H3 was performed using FIJI software48 based on the relative intensity of bands49.
Immunoblots of L-PGDS, acetylated histone H3 and histone H3 were performed using the protocol described in50 in which several SDS-PAGE gel strips are organized in a single nitrocellulose membrane. This allows a correct quantitative comparison between samples on different gels or immunoblots by simultaneous transfer and detection of several samples in the same membrane.
Nuclei isolation and extraction
Control Medicago cells were harvested by vacuum filtration at day 11 of growth as described above. Cells were ground with liquid nitrogen in a mortar. Ground cells were homogenized with cytoplasmic extraction buffer (25 mM Tris-HCl pH 6.5, 0.45 M sucrose, 5 mM MgCl2, 5 mM β-mercaptoethanol, 0.5 mM PMSF, 0.364 µM Pepstatin A, 1.2 µM E64, 0.1% Triton X-100) in a 1:4 ratio (g of cells: mL of cytoplasmic extraction buffer). The homogenate was filtered with a nylon mesh, followed by several filtration steps with decreasing pore size filters (150 µm, 100 µm, 50 µm and 30 µm, Sysmex-Partec, Germany). To pellet the nuclei, the 30 µm filtrate was centrifuged for 5 minutes at 4000 g and the pellet washed with cytoplasmic extraction buffer without 0.1% Triton X-100 (adapted from51). The pellet was then resuspended in 100 µL of nuclei extraction buffer (50 mM HEPES pH 5.7, 420 mM NaCl, 0.5 mM EDTANa2, 0.1 mM EGTA, 10% glycerol) and sonicated for 30 seconds (Bioruptor® Plus Sonication System, Diagenode, Belgium). After incubation on ice for 30 minutes, the homogenate was centrifuged at 11000 g for 10 minutes and the pellet was discarded. Total soluble protein was determined using the Bradford Protein Assay (Bio-Rad, USA) following the manufacturer’s instructions. Nuclear extractions were stored at −80 °C until use.
Histone deacetylase (HDAC) activity assay
The inhibition of HDACs activity by 2.5 µM SAHA was assessed with EpiQuick HDAC Activity/inhibition Assay Kit (Epigentek, USA), according to the manufacturer’s instructions. Briefly, nuclear extracts, prepared as described above, were incubated for 90 minutes, at 37 °C, with a HDAC specific substrate, in the presence or absence of SAHA. Capture antibody was added and incubated for one hour at room temperature. Then, detection antibody was added and incubated at room temperature for 30 minutes. Technical triplicates were analyzed. Total HDAC inhibition was determined by reading the absorbance at 450 and 655 nm and calculated using the following formula (equation (1)):
RNA isolation from Medicago cell samples
Total RNA was isolated from cell samples, with and without 2.5 µM SAHA treatment, recovered at days 3, 5, 7 and 11 of the cell growth, with biological triplicates. Cell samples were obtained by vacuum filtration, flash-frozen in liquid nitrogen and stored at −80 °C until RNA isolation was performed. Cell samples were ground with eppendorf pestles in the presence of 500 µL of TRIzol reagent (NZYTech, Portugal). Next, Direct-zolTM RNA MiniPrep (Zymo Research, USA) was used to perform RNA isolation as indicated by manufacturer’s instructions. RNA sample concentrations were measured in a NanoDrop 2000c UV-Vis Spectrophotometer (Thermo Scientific, USA) and run in a 0.8% agarose gel to check for RNA integrity. Genomic DNA contamination in RNA samples was assessed by performing a PCR with primers designed for promoter and terminator of L-PGDS gene. RNA samples were negative for genomic DNA contamination. Samples were flash-frozen in liquid nitrogen and stored at −80 °C until use.
cDNA synthesis
500 ng of isolated RNA was used to synthesize cDNA using the ImProm-IITM Reverse Transcription System (Promega, USA), according to the manufacturer’s instructions. 0.5 µg of oligo(dT) and random primers (Promega, USA) were used for each reaction. Briefly, RNA and primers were mixed and incubated at 70 °C for 5 minutes and placed on ice for a further 5 minutes. Then, the master mix was added to the previous reaction, to a final volume of 20 µL. The reactions were transferred to a thermocycler (Mastercycler, Eppendorf, Germany) and incubated as follows: 25 °C for 5 minutes, 42 °C for one hour, 70 °C for 15 minutes and then on hold at 4 °C. cDNA samples were stored at −20 °C until use.
QuantStudioTM 3D Digital PCR
The QuantStudioTM 3D Digital PCR (QS3D; Applied Biosystems, ThermoFischer Scientific, USA) was used to quantify L-PGDS gene expression in the cell samples in the presence and absence of 2.5 µM SAHA. 0.005 ng to 0.25 ng of cDNA were used (so that final copies/µL value for each sample was within the range of 200 and 2000, as recommended by the manufacturer) and a TaqMan® probe (Hs00168748_m1 for human L-PGDS, ThermoFischer Scientific, USA) labeled with FAM was added to the QS3D master mix. Briefly, each QS3D chip was loaded with 14.5 µL of reaction (one chip per replicate) and sealed using a QS3D Chip Loader (ThermoFischer Scientific, USA), following the manufacturer’s instructions. The QS3D chips were loaded into a Dual Flat Bloc GeneAmpTM PCR System 9700 thermal cycler to perform amplification, with the following conditions: 96 °C for 10 minutes, 98 °C for 30 seconds and 60 °C for 2 minutes (both for 40 cycles) and hold at 25 °C (to avoid chip condensation). After amplification, chips were imaged on QS3D Instrument (ThermoFischer Scientific, USA) and data analysis was carried out in QS3D Analysis SuiteTM Software, which takes in account the samples dilution factor. The software applies a quantification algorithm based on the Poisson model and estimates copy number/µL mean values with a 95% confidence interval.
Statistical data analysis
All results obtained from cell viability, from relative quantification of proteins (L-PGDS, acetylated histone H3 and histone H3) and from the histone deacetylase activity assay were statistically analyzed by multiple t-tests (multiple comparisons using the Holm-Šídák method) with α = 0.05 (significance level). All statistical analysis was performed with GraphPad Prism version 6.0c for MacOS52. Statistical significance was assumed for p values ≤0.05.
References
Hellwig, S., Drossard, J., Twyman, R. M. & Fischer, R. Plant cell cultures for the production of recombinant proteins. Nat Biotech. 22, 1415–1422 (2004).
Dirk, B. et al. N-Glycosylation of Plant-produced RecombinantProteins. Curr. Pharm. Des. 19, 5503–5512 (2013).
Santos, R. B. et al. Putting the Spotlight Back on Plant Suspension Cultures. Front. Plant Sci. 7, 297 (2016).
Hiatt, A., Caffferkey, R. & Bowdish, K. Production of antibodies in transgenic plants. Nature. 342, 76–78 (1989).
Dalton, A. C. & Barton, W. A. Over-expression of secreted proteins from mammalian cell lines. Protein Sci. 23, 517–525 (2014).
Wulhfard, S., Baldi, L., Hacker, D. L. & Wurm, F. Valproic acid enhances recombinant mRNA and protein levels in transiently transfected Chinese hamster ovary cells. J. Biotechnol. 148, 128–132 (2010).
Yang, W. C. et al. Addition of Valproic Acid to CHO Cell Fed-Batch Cultures Improves Monoclonal Antibody Titers. Mol. Biotechnol. 56, 421–428 (2014).
Fomina-Yadlin, D. et al. Transcriptome analysis of a CHO cell line expressing a recombinant therapeutic protein treated with inducers of protein expression. J. Biotechnol. 212, 106–115 (2015).
Liu, Y., Zhou, X., Song, Z. & Zhang, Y. Sodium butyrate enhances the acidic isoform content of recombinant human erythropoietin produced by Chinese hamster ovary cells. Biotechnol. Lett. 36, 907–911 (2014).
Lee, S. M., Kim, Y.-G., Lee, E. G. & Lee, G. M. Digital mRNA profiling of N-glycosylation gene expression in recombinant Chinese hamster ovary cells treated with sodium butyrate. J. Biotechnol. 171, 56–60 (2014).
Allen, M. J. et al. Identification of novel small molecule enhancers of protein production by cultured mammalian cells. Biotechnol. Bioeng. 100, 1193–1204 (2008).
Pietrocola, F. et al. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 21, 805–821 (2015).
Seto, E. & Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. 6, a018713 (2014).
Gregoretti, I., Lee, Y.-M. & Goodson, H. V. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J. Mol. Biol. 338, 17–31 (2004).
Venturelli, S. et al. Plants Release Precursors of Histone Deacetylase Inhibitors to Suppress Growth of Competitors. Plant Cell. 27, 3175–3189 (2015).
Lee, J. Y.-C. et al. Inhibition of HDAC3- and HDAC6-Promoted Survivin Expression Plays an Important Role in SAHA-Induced Autophagy and Viability Reduction in BreastCancer Cells. Front. Pharmacol. 7, 81 (2016).
Xu, W. S., Parmigiani, R. B. & Marks, P. A. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 26, 5541–5552 (2011).
Finnin, M. S. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 401, 188–193 (1999).
Marks, P. A. et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 1, 194–202 (2001).
Marks, P. A. & Breslow, R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90 (2007).
Zhang, Q. et al. Histone acetylation and reactive oxygen species are involved in the preprophase arrest induced by sodium butyrate in maize roots. Protoplasma. 254, 167–179 (2017).
Arfmann, H.-A. & Haase, E. Effect of sodium butyrate on the modification of histones in cell cultures of Nicotiana tabacum. Plant Sci. Lett. 21, 317–324 (1981).
Santos, A. P., Abranches, R., Oliveira, M. & Shaw, P. Plasticity of Chromatin Organization in the Plant Interphase Nucleus in Nuclear Functions in Plant Transcription, Signaling and Development (eds O. Pontes & H. Jin) 57–79 (Springer New York, 2015).
Rosa, S. et al. Cell Differentiation and Development in Arabidopsis Are Associated with Changes in Histone Dynamics at the Single-Cell Level. Plant Cell. 26, 4821–4833 (2014).
Santos, A. P. et al. The architecture of interphase chromosomes and gene positioning are altered by changes in DNA methylation and histone acetylation. J. Cell Sci. 115, 4597–4605 (2002).
Patanun, O. et al. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Alleviates Salinity Stress in Cassava. Front. Plant Sci. 7, 2039 (2017).
Pires, A. S. et al. High levels of stable phytase accumulate in the culture medium of transgenic Medicago truncatula cell suspension cultures. Biotechnol. J. 3, 916–923 (2008).
Pires, A. S., Santos, R. B., Nogueira, A. C. & Abranches, R. Production of human lipocalin-type prostaglandin D synthase in the model plant Medicago truncatula. In Vitro Cell. Dev. Biol. Plant. 50, 276–281 (2014).
Pires, A. S. et al. Expression of a recombinant human erythropoietin in suspension cell cultures of Arabidopsis, tobacco and Medicago. Plant Cell Tiss. Org. Cult. 110, 171–181 (2012).
Urade, Y. & Hayaishi, O. Biochemical, structural, genetic, physiological, and pathophysiological features of lipocalin-type prostaglandin D synthase. Biochim. Biophys. Acta. 1482, 259–271 (2000).
Nicolson, S. C. et al. Identification and Validation of Small Molecules That Enhance Recombinant Adeno-associated Virus Transduction following High-Throughput Screens. J. Virol. 90, 7019–7031 (2016).
Milavec, M. et al. GMO quantification: valuable experience and insights for the future. Anal. Bioanal. Chem. 406, 6485–6497 (2014).
Santos, C. et al. Expression Profiling of Castanea Genes during Resistant and Susceptible Interactions with the Oomycete Pathogen Phytophthora cinnamomi Reveal Possible Mechanisms of Immunity. Front. Plant Sci. 8, 515 (2017).
Kadam, S. et al. Genomic-assisted phylogenetic analysis and marker development for next generation soybean cyst nematode resistance breeding. Plant Sci. 242, 342–350 (2016).
Pratap, J. et al. The Histone Deacetylase Inhibitor, Vorinostat, Reduces Tumor Growth at the Metastatic Bone Site and Associated Osteolysis, but Promotes Normal Bone Loss. Mol. Cancer Ther. 9, 3210–3220 (2010).
Frenzel, A., Hust, M. & Schirrmann, T. Expression of Recombinant Antibodies. Front. Immunol. 4, 217 (2013).
Joseph, M. et al. Proteomic characterisation of endoplasmic reticulum-derived protein bodies in tobacco leaves. BMC Plant Biol. 12, 36 (2012).
Vergara, M. et al. Differential Effect of Culture Temperature and Specific Growth Rate on CHO Cell Behavior in Chemostat Culture. PLOS ONE. 9, e93865 (2014).
Chen, F. et al. The combined effect of sodium butyrate and low culture temperature on the production, sialylation, and biological activity of an antibody produced in CHO cells. Biotechnol. Bioprocess Eng. 16, 1157–1165 (2011).
Kantardjieff, A. et al. Transcriptome and proteome analysis of Chinese hamster ovary cells under low temperature and butyrate treatment. J. Biotechnol. 145, 143–159 (2010).
Jiang, Z. & Sharfstein, S. T. Sodium butyrate stimulates monoclonal antibody over-expression in CHO cells by improving gene accessibility. Biotechnol. Bioeng. 100, 189–194 (2008).
Waterborg, J. H. Evolution of histone H3: emergence of variants and conservation of post-translational modification sites. Biochem. Cell Biol. 90, 79–95 (2011).
Waterborg, J. H. Sequence analysis of acetylation and methylation in two histone H3 variants of alfalfa. J. Biol. Chem. 265, 17157–17161 (1990).
Lusser, A., Kölle, D. & Loidl, P. Histone acetylation: lessons from the plant kingdom. Trends Plant Sci. 6, 59–65 (2001).
Ma, X., Lv, S., Zhang, C. & Yang, C. Histone deacetylases and their functions in plants. Plant Cell Rep. 32, 465–478 (2013).
Arcalis, E. et al. The dynamic behavior of storage organelles in developing cereal seeds and its impact on the production of recombinant proteins. Front. Plant Sci. 5, 439 (2014).
González-Melendi, P., Pires, A. S. & Abranches, R. Cell-line-dependent sorting of recombinant phytase in cell cultures of Medicago truncatula. Funct. Plant Biol. 36, 431–441 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9, 676–682 (2012).
Miller, L. Analyzing gels and western blots with ImageJ. lukemiller.org http://lukemiller.org/index.php/2010/11/analyzing-gels-and-western-blots-with-image-j/ (2017).
Aksamitiene, E., Hoek, J. B., Kholodenko, B. & Kiyatkin, A. Multi-strip Western blotting to increase quantitative data output. Electrophoresis. 28, 3163–3173 (2007).
Shen, W. H. & Gigot, C. Protein complexes binding to cis elements of the plant histone gene promoters: multiplicity, phosphorylation and cell cycle alteration. Plant Mol. Biol. 33, 367–379 (1997).
GraphPad Software GraphPad Prism version 6.0c for Mac OS X. GraphPad Software, Inc., La Jolla CA, USA. www.graphpad.com.(2013)
Acknowledgements
This work was supported by Portuguese national funds through Fundação para a Ciência e a Tecnologia (FCT, Portugal) grants PTDC/BIA-PLA/2411/2012 and UID/Multi/04551/2013. Authors are grateful to Carmen Santos for advice on Digital PCR; Rita Ventura for useful discussions; and James Yates for revising the manuscript.
Author information
Authors and Affiliations
Contributions
R.B.S. and R.A. designed the experiments. R.B.S. performed the experiments and prepared the figures. R.B.S., A.S.P. and R.A. analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Santos, R.B., Pires, A.S. & Abranches, R. Addition of a histone deacetylase inhibitor increases recombinant protein expression in Medicago truncatula cell cultures. Sci Rep 7, 16756 (2017). https://doi.org/10.1038/s41598-017-17006-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17006-9
This article is cited by
-
Generation of transgenic cell suspension cultures of the model legume Medicago truncatula: a rapid method for Agrobacterium mediated gene transfer
Plant Cell, Tissue and Organ Culture (PCTOC) (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
