
Abstract
Parabolic flight maneuvers of Novespace’s Airbus A310 ZERO-G produce subsequent phases of hypergravity (about 20 s), microgravity (about 22 s) and another 20 s hypergravity on experiments located in the experiment area of the aircraft. The 29th DLR parabolic flight campaign consisted of four consecutive flight days with thirty-one parabolas each day. Euglena gracilis cells were fixed with TRIzol during different acceleration conditions at the first and the last parabola of each flight. Samples were collected and analyzed with microarrays for one-color gene expression analysis. The data indicate significant changes in gene expression in E. gracilis within short time. Hierarchical clustering shows that changes induced by the different accelerations yield reproducible effects at independent flight days. Transcription differed between the first and last parabolas indicating adaptation effects in the course of the flight. Different gene groups were found to be affected in different phases of the parabolic flight, among others, genes involved in signal transduction, calcium signaling, transport mechanisms, metabolic pathways, and stress-response as well as membrane and cytoskeletal proteins. In addition, transcripts of other areas, e.g., DNA and protein modification, were altered. The study contributes to the understanding of short-term effects of microgravity and different accelerations on cells at a molecular level.
Similar content being viewed by others


Introduction
Euglena gracilis is a unicellular freshwater flagellate, which is distantly related to kinetoplastids, a group of parasitic protozoans, such as Trypanosoma or Leishmania1,2. It shows distinct movement behavior with respect to external stimuli, such as light, oxygen, and acceleration3,4,5 and has been a model organism for biological clocks6,7, photosynthesis8,9, and movement physiology10,11. In the absence of light, cells show negative gravitactic behavior, adjusting the swimming direction against the vector of acceleration. Both gravitaxis and the light-directed phototaxis in E. gracilis are based on active physiological mechanisms10,12,13,14,15,16,17,18. However, a large part of the underlying signal transduction chain remains to be elucidated. So far, a distinct calmodulin, as well as a particular protein kinase A, were found to be involved in graviorientation12,13. In addition, a flagellar protein has been implicated in the process19. Furthermore, the contribution of mechanosensitive ion channels, changing membrane potentials and calcium seems very likely16,17.
Previous parabolic flight campaigns studied the movement and physiological parameters of E. gracilis: in the 45th ESA parabolic flight campaign, a change in the beating pattern of the E. gracilis’ flagellum during the transition from hypergravity (hyper-g) to microgravity (μg) and vice-versa was observed18. Earlier, in the 29th ESA parabolic flight campaign16, changes in membrane potential, and the cytosolic calcium concentration were measured during different accelerations. Furthermore, movement analysis revealed a fast and precise adaptation of the swimming behavior depending on the prevailing acceleration17. Negative gravitaxis was more pronounced during hyper-g, while µg led to a random movement as compared to a 1 g control sample. While these studies confirmed the physiological nature of the gravitactic orientation, no studies on differential gene expression in E. gracilis had been performed in the course of parabolic flights. This was in large part due to the lack of complete genomic and transcriptomic datasets of E. gracilis. However, very recent advances in the genome and transcriptome sequencing of E. gracilis allowed the analysis of global gene expression pattern induced by different stimuli2,20,21,22. In this study, we used this knowledge to investigate changes in E. gracilis gene expression in parabolic flights using a large-scale microarray analysis system. We show that short time accelerations or microgravity is sufficient to cause significant changes in gene expression in E. gracilis.
Results
Hierarchical clustering
To correct for any adaptational effects, individual accelerations in the first and last parabolas were analyzed independently. After removal of all outliers, at least 3 samples for each acceleration were employed for hierarchical clustering. Cell cultures subjected to the same acceleration within a parabola showed the highest degree of similarity independent of the flight day (Fig. 1). For parabola 1, samples of the µg phase showed a close resemblance to the second hyper-g phase (1.8 g-2), while 1 g and the first hyper-g phase (1.8 g-1) exhibited closer relations. In parabola 31, these similarity clusters changed since 1.8 g-2 and 1 g, as well as µg and 1.8 g-1, appear to relate to each other.

Hierarchical clustering of gene expression data of parabolic flight samples from parabola 1 (A) and 31 (B). Independent samples cluster together based on the subjected acceleration irrespective of the flight day for both parabolas. (A) For parabola 1, µg (grey bars) and 1.8 g-2 (green) show close resemblance as well as 1 g (blue) and 1.8 g-1 (red). (B) In parabola 31, clusters appear for 1.8 g-2 (green) and 1 g (blue) as well as µg (grey) and 1.8 g-1 (red). Hierarchical clustering was performed based on normalized intensity values using Pearson uncentered correlation and Wards linkage rules. At least three independent samples for each acceleration from different flight days were employed for hierarchical clustering.
Comparison of gene expression changes between parabola 1 and 31
Comparison of gene expression changes between parabola 1 and 31 showed very distinct differences. Overall, 260 genes were found to be significantly regulated during the first and last parabola by a fold change of ≥1.5 and a p-value ≤ 0.05. About 130 genes met these criteria during parabola 1, while 143 transcripts were regulated in parabola 31 (Fig. 2). Only 13 transcripts were found to be regulated during both parabolas (Table 1). However, these regulations occurred in different phases within the parabolas (1.8 g-1, µg, 1.8 g-2) and showed contrary fold changes in parabola 1 and 31. Four of the transcripts were in good accordance, when their protein sequences were compared in the BLAST database to sequences of other organisms, like carboxyl-terminal-processing peptidase chloroplastic-like isoform X2 (#7821, E-value 2.4E-60), molybdopterin adenylyltransferase (#14765, E-value 3.1E-85), GPI-anchor surface protein (#627, E- value 1.8E-109), and mitogen-activated kinase kinase kinase NPK1 (#450, E- value 4.3E-50). An adenylate guanylate cyclase domain-containing protein (#1113, E-value 9.2E-07), as well as an acyl-binding domain-containing protein 5-like (#30616, E-value 1.6E-09), showed low similarity. Seven of the regulated transcripts showed no similarities to other known proteins or only weak similarities to hypothetical proteins. Therefore, no annotation was possible.

Overall change of gene expressions between all significantly (p < 0.05) differentially expressed genes (FC > 1.5) in the first (#1, orange circle) and last parabolas (#31, blue circle), respectively. Differentially expressed genes of all phases (1 g, µg and 1.8 g) during parabolas were pooled. The intersection between the two circles show the number of genes that were affected in both the parabolas 1 and 31.
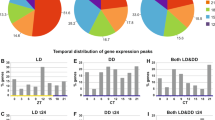
The overall distribution of significantly regulated genes in parabola 1 and 31 is shown in Table 2. The changes were relatively small but distinct. In the course of the first parabola, no changes beyond 2-fold were detected and the number of regulated transcripts was similar for the different accelerations. Lower numbers of genes showed regulation more than 2-fold during various accelerations in the last parabola. These included BMP family ABC transporter substrate-binding protein (EG_35058), Slh1p interacting factor (EG_15691) and ubiquitin-conjugating enzyme E2 Z-like (EG_3282). For further analysis, fold changes above 1.5 were closely investigated. During the first hyper-g phase (1.8 g-1), 51 transcripts showed significant changes in expression, while in the subsequent µg and second hyper-g (1.8 g-2) phases, 42 and 46 transcripts were altered, respectively. In the last parabola, the number of regulated genes increased with the duration of the parabola, starting from 29 transcripts in the first hyper-g phase to 51 during the µg phase to 81 transcripts in the second hyper-g (1.8 g-2) phase.
Gene expression changes in parabola 1
In total, 130 transcripts were significantly altered under various accelerations in parabola 1 (Fig. 3), indicating that changes are acceleration- as well as time-dependent. In this regard, only very few genes were regulated in more than one phase. For instance, among all significantly regulated transcripts, only a single one was regulated in both hyper-g phases. Furthermore, during µg and the second hyper-g (1.8 g-2) phase, up-regulations of transcripts were more pronounced than down-regulations. During µg phase, 26 transcripts were up-regulated and 16 down-regulated and in the second hyper-g phase, 29 transcripts showed up-regulation, while only 17 exhibited down-regulation.

Gene expression changes during different acceleration phases of the first parabola. Most of the gene expression changes are specific for a particular acceleration phase. Each circle represents an acceleration phase (orange = first 1.8 g, blue = µg, and red = second 1.8 g phase). Intersections between circles show the number of genes that changed in more than one acceleration. A fold change cut-off of 1.5-fold and a p-value of 0.05 were applied using moderated t-test.
Table 3 summarizes the annotation of domains or functions of all altered transcripts in parabola 1. For some transcripts, Blast2GO found no similarities to any of the known proteins, which were marked as not applicable (NA). For others, it was not possible to assign a function by means of Gene Ontology (GO) annotation or manual inspection (marked as no GO) due to poor annotation. Therefore, sequences were checked manually and revised if necessary. The conserved domains or gene ontologies were determined for about 50% of all transcripts. During the first parabola, most altered transcripts were involved in signal transduction processes and transport mechanisms. Furthermore, nuclear processes including DNA binding, nuclear localization, and DNA replication, recombination and repair as well as transcription were also regulated throughout the entire parabola. Interestingly, photosynthesis-associated transcripts, as well as a blue-light using FAD (BLUF)-domain proteins, were also affected although samples were fixed in the dark. Gene set enrichment analysis (GSEA) found only one enriched GO for DNA metabolic processes (GO:0006259) during the microgravity phase in parabola 1.
A list of transcripts found to be significantly regulated during the different acceleration phases of parabola 1 as well as their corresponding sequence description, organism and similarity measure (E-value) as determined by Blast2GO or manual BLAST search are shown in Table S1. The affected signal transduction elements included different protein kinases, adenylate and guanylate cyclases, and a single phosphodiesterase. Furthermore, possible prokaryotic sensor histidine kinases (E. gracilis transcript numbers EG_15442 and EG_836) were up-regulated during both hyper-g phases. Affected transport mechanisms included elements involved in the intracellular transport as well as the transport of macromolecules and ions across membranes (Table S1). Interestingly, a BLUF domain protein was down-regulated (EG_31756), showing similarities to E. gracilis PACα subunit, involved in the light-directed movement called phototaxis. Furthermore, a transcript similar to E. gracilis chloroplast light-harvesting complex II precursor Lhcbm3 (EG_41739) was also down-regulated. A very high similarity of the transcript EG_627 was found to a GPI-anchor surface protein, which was down-regulated in the first hyper-g and up-regulated in the subsequent µg period. During the second hyper-g, an up-regulation of a possible fructose-1,6-bisphosphate aldolase (EG_43521) was found.
Gene expression changes in parabola 31
In parabola 31, 143 transcripts significantly regulated. The number of altered transcripts increased with the duration of the parabola. In the initial hyper-g phase, 29 transcripts were regulated, while the µg and the last hyper-g phase affected 51 and 81 transcripts, respectively (Fig. 4). Furthermore, up- and down-regulations were uniformly distributed between different phases.

Gene expression changes during different acceleration phases of parabola 31. Most of the gene expression changes are specific for a particular acceleration phase. Each circle represents an acceleration phase (orange = first 1.8 g, blue = µg, and red = second 1.8 g phase). Intersections between circles show the number of genes that changed in more than one acceleration. A fold change cut-off of 1.5-fold and a p-value of 0.05 were applied using moderated t-test.
Table 4 summarizes the annotation of transcripts for parabola 31. Protein domains or gene ontologies were found only for about 50% of the transcripts. However, none of those showed similarities to transcripts with annotations in E. gracilis. Thus, it was necessary to rely on similarities to protein descriptions found in other organisms (E-value). In addition, no significantly regulated Gene Ontology terms were found in the GSEA analysis and therefore, the analysis focused on single transcript validation (Table S2). Most pronounced effects were found for transcripts related to signal transduction elements, protein modification, membrane proteins, transport mechanisms, and calcium-binding proteins. Among the signal transduction elements were different kinases, phosphatases, cyclases, and a PAS sensor histidine kinase. Large subsets of transcripts were found to be involved in protein ubiquitination processes acting as ligases or hydrolases but were only regulated during both hyper-g phases. The analysis of transport mechanisms revealed that mainly active export mechanisms were affected as most transport proteins showed to have an ATP-binding cassette. Further, a possible ammonia transporter was found. Membrane proteins included a GPI-anchor surface protein (EG_627), with very high similarity scores in BLAST search. It was down-regulated in the second hyper-g phase. While in the 1.8 g-1 and µg phases, single stress proteins (EG_14035 and EG_1003) were found, no regulation for such mechanisms was seen during the second hyper-g phase. During µg and 1.8 g-2 phases, a NADP malic enzyme (EG_8836) functioning in energy production and conversion was up-regulated.
Discussion
Earlier experiments performed on parabolic flights with the model organism E. gracilis have shown that it can undergo a very fast adaptation of its swimming behavior during the course of a parabola, which is coupled to intracellular mechanisms16,17,18. In the recent 29th DLR parabolic flight campaign, this knowledge was broadened by the analysis of gene expression changes. The data obtained revealed that the short-term alterations of acceleration induce significant changes in gene expression in E. gracilis, in particular of transcripts involved in signal transduction, transport mechanism, metabolic pathways and stress-response. Changes in gene expression within seconds of microgravity obtained with parabolic flights have been reported previously in callus cultures of Arabidopsis thaliana23 as well as in human cell lines24,25.
Gene expression changes in E. gracilis correspond to observations in other organisms
The data partially accord with previously obtained gene expression changes on the spacecraft Shenzhou 8, where E. gracilis cells were fixed after 45 min of exposure to microgravity26. Quantitative PCR analysis revealed effects on the genes involved in signal transduction, calcium signaling, stress response, heat shock, and cell cycle proteins. In this study, regulation of different signal transduction elements, such as mitogen-activated kinase kinase kinase NPK1, hybrid sensor histidine kinase/response regulator, serine/threonine-protein kinase, adenylate guanylate cyclase domain-containing protein, cyclin-dependent kinase-like 2, PAS domain-containing sensor histidine kinase, etc., was observed in E. gracilis during different phases of a parabola. However, these changes cannot be directly linked to a distinct pathway, as the only recently accessible transcriptome data lack comprehensive annotations21. Interestingly, some of the transcripts found to be regulated in E. gracilis showed similarities to specific observations involved in hyper-g and μg experiments in other cell types. For example, effects on the primary metabolism were found to be induced by parabolic flights. In callus cultures of Arabidopsis thaliana, changes in the phosphorylation pattern of malate dehydrogenase were investigated23, which also showed altered transcriptional levels (EG_8836) in E. gracilis in the present study. Furthermore, a potential fructose-1,6-bisphosphate aldolase 1 (EG_transcript_43521), acting in glycolysis, was up-regulated in the second hyper-g phase during parabola 1. Interestingly, higher transcript levels and an increase in the activity of a product produced by this enzyme, dihydroxyacetone phosphate, which is an activator of pyruvate kinase was found during a sounding rocket campaign in Arabidopsis cell cultures27,28. This could emphasize the assumption of an increase in glycolytic turnover.
Another aspect well studied under altered gravitational conditions is the production of reactive oxygen species (ROS), which have been identified to function as a second messenger in the signal transduction of gravitational changes23,29,30,31. We also found transcripts involved in the production of ROS regulated during parabola 31 (EG_1003; EG_14035) indicated as stress proteins. However, the production of ROS and transcripts involved in the protection against damages induced by free oxygen radicals seems less pronounced and the latter was not found to be significantly regulated. During a space mission with samples fixed after 45 min, the ROS involved transcripts were found to be regulated in a more significant manner26.
The up-regulation of an ammonium transporter (EG_1810) during the second hyper-g phase is consistent with the data found for A. thaliana cell cultures during a TEXUS campaign and was associated with a stress-induced up-regulation of protein synthesis28. This could be in agreement with the pronounced regulation of elements (e.g. E3 ubiquitin-ligases and ubiquitin carboxyl-terminal hydrolases) of protein modification processes found in parabola 31 as well as transcripts associated with the transcription and translation in the first and last parabola.
Interestingly, transcripts involved in photosynthesis and transcripts containing a BLUF domain, which is associated with light-directed movement in Euglena, were regulated in the course of the first parabola. Although cells were kept in dark during the whole experiment, differences in regulation were found between the first sample (1 g control) and the following acceleration phases (1.8 g-1, µg, 1.8 g-2). This could indicate interconnectivity between the impact of changed accelerations and perception of light (13,19).
Different responses between parabola 1 and 31
In general, results from the first parabola vary significantly from transcripts regulated during parabola 31. Only 13 out of 271 transcripts were regulated during both parabolas. In the time between, cells experienced a further 29 parabolas during a 2 h period, which could possibly lead to an adaptation of their physiological state to the encountered acceleration changes, and could therefore explain the different responses. Although in-flight movement analysis was not performed in this campaign, previous parabolic flight campaigns have shown no differences in the swimming behavior during the various parabolas17. This would indicate that the adaption is limited to the transcriptional and most likely also to the protein level (unpublished data). Within the parabolas, the varying accelerations (1.8 g-1, µg, 1.8 g-2) also induced very specific effects with only a few transcripts being regulated under more than one phase. In parabola 1 and 31, no transcripts were regulated in all phases of acceleration. Most pronounced overlaps occur in subsequent phases during either of the parabolas, indicating a time-dependent effect. This could be emphasized by the fact that at least during parabola 31, the number of regulated transcripts increases with the progression of the parabola. Interestingly, most of the co-regulated transcripts showed the opposite direction of regulations in the phases.
Fast acceleration changes during a parabolic flight are sufficient to generate distinct gene expression changes
The measured gene expression changes occur within a timeframe of 20 s. Even though this seems to be too short for profound alterations, other studies have also found similar effects23,32,33,34. From numbers reported in literature, Thiel et al.35 concluded that these changes are plausible if the responsible signaling cascades only take seconds to transduce into the nucleus and the chromatin structure allows immediate binding of transcription factors. The mRNA synthesis by RNA polymerases II has been shown to be within the time frame of a single acceleration during a parabola36,37,38,39. A further relevant step towards a translatable mRNA is the pre-mRNA processing by splicing, which may be the most time-consuming step. However, recent reports about co-transcriptional splicing show that transcriptional speed also regulates splicing and that it can be conducted much faster than expected (reviewed in40,41). So far, time constants for splicing vary greatly between different cell types and methods used for measurement, making further investigations necessary, but one of the fastest splicing durations was found in HeLa cells with an average of 30 s42. Furthermore, measurements of pre-mRNA levels, as well as process other than the de novo synthesis of mRNA, have to be considered when analyzing rapid gene expression changes, as found during a parabolic flight.
As Thiel et al.35 concluded, a prerequisite for rapid gene expression changes is a fast mechanical transduction from the membrane, where acceleration changes are possibly perceived, into the nucleus. They referred to predictions from the tensegrity model, which describes a force propagation of 30 m s−1 along the pre-stressed cytoskeleton and studies linking changes in cell geometry with changes in nuclear and chromatin structure and gene expression changes (reviewed in43,44,45,46). At an acceleration of 1 g, elastic fibers of the cytoskeleton, such as microtubules, are under tensional loading. Sudden transfer to µg unloads the compressed fibers leading to cytoskeletal reorganization and changes in cell geometry47,48,49. The cytoskeleton in E. gracilis has been shown to be directly related to changes in cell geometry50. However, predictions from the tensegrity model for a free-swimming organism like E. gracilis need to be investigated further. In this study, we found changes in cell surface proteins (EG_10146, EG_627, EG_1870), the cytoskeleton and their interacting proteins (EG_800, EG_14051, EG_22888, EG_23041, EG_12478, EG_38089), as well as DNA-binding (EG_31054), and chromatin organization domains (EG_26357) during various acceleration phases, indicating a possible role of cytoskeletal components in transduction of mechanical forces in E. gracilis.
The conformity of the results in the independent cell samples taken at independent subsequent flight days presents a strong argument that the data are not just due to the random fluctuations or aberrations during sample preparation and measurement, flight vibrations, changes in light regime, subsequent phases of altered accelerations and/or mechanical stress of cells being transferred into the flight hardware. In addition, the similarity of gene expression changes of independent cell samples at distinct phases of a parabola was much closer than the conformity of gene expression changes of the same sample at different phases of acceleration.
Conclusion
It is known that E. gracilis reacts to changes in acceleration by adaptation of its swimming behavior. However, it was so far unknown whether these conditions induce changes at the transcriptional level. This study shows for the first time that gene expression in E. gracilis is capable of global dynamic alterations induced by parabolic flights. Results indicated significant changes in gene expression within a very small time. Changes in acceleration for ~20 s resulted in up to two-fold up- or down-regulations of particular genes in Euglena. In addition, the transcription differed between the first and the last parabolas indicating adaptation effects in the course of the flight. It was also observed that different phases of the parabolic flight affected different gene groups, which corresponds to data found in other organisms such as those involved in signal transduction, calcium signaling, transport mechanisms, metabolic pathways, stress-response, membrane and cytoskeletal proteins, and DNA and protein modification. Taken together, this study contributes to the understanding of gene expression changes under altered accelerations and microgravity and can be used as a reference data set for further on-ground and space flight related studies.
Material and Methods
Cells and growth conditions
E. gracilis KLEBS strain Z (obtained from the SAG, Göttingen) was grown in 150 mL complex medium51 in 300 mL Erlenmeyer flasks. A fresh culture was inoculated for every flight day with 3 × 106 cells per 150 mL of medium. Cell number was determined using a Thoma chamber (VWR, Radnor, Pennsylvania, USA). To generate a controlled growth condition, a portable, isolated plastic container was used as a culture chamber. The culture was grown at 20 °C and defined constant light of 25 W/m2 was achieved with mixed warm-white and cold-white illumination (Solarox, Stendal, Germany; same mix and intensity as in the cell culture room used for stock cultures in the laboratory). The experiments were performed with eight days old cultures showing pronounced negative gravitaxis.
Parabolic flight campaign
Experiments were conducted in the course of the 29th DLR parabolic flight campaign in September 2016 in Bordeaux, France. A modified Airbus A310 performed flight maneuvers resulting in short periods (22 s) of µg, flanked by two hyper-g periods (each about 20 s) before and after the µg phase (Fig. 5). Detailed descriptions about parabolic flight campaigns are provided in Pletser et al.52. The schedule of the campaign was as follows: Ten days sample preparation as well as validation of the experiments in terms of safety followed by four subsequent flight days with 31 parabolas each day.

Schematic representation of the acceleration levels of a typical parabola. Grey triangles indicate the sampling events at the end of 1 g, at the end of the first hyper-g phase of 20 s, at the end of the µg phase of 22 s, and at the end of the second hyper-g phase of 20 s. These sampling events were repeated for the 31st parabola as well.
Experimental rack, filling and fixation procedure
For inflight fixation of samples, a syringe-based fixation unit was used, allowing manual release of a fixative into syringes containing E. gracilis cells. During preflight preparations, 2 mL of cells from the cell culture flask were carefully filled into 20 mL syringes (Henke-Sass Wolf GmbH, Tuttlingen, Germany). Each syringe was sealed with parafilm and connected by serological tubing (Extension line type Heidelberger, B. Braun Melsungen AG, Melsungen Germany) to another 20 mL syringe containing 13 mL TRIzol® (Life Technologies, Karlsbad, USA) as a fixative. The syringe pairs were mounted into a custom-made holder, which provided space for 16 syringe-pairs in total (Fig. S1), allowing fixation of eight independent samples in duplicates. The syringe holder was integrated into the experimental rack, constructed according to the safety requirements. For fixation, TRIzol® was released from one syringe into the counterpart containing cells. The applied pressure destroyed the parafilm membrane. Due to safety requirements, the pistons of the syringes were activated by sliders, which connect the inside of the box mechanically with the outside. The sliders were pushed manually in defined time intervals during the first and the last parabola. The fixation time points for each of the parabolas were chosen as follows: first fixation step: 1 g phase ten seconds before onset of hyper-g, second step: end of hyper-g phase (20 s 1.8 g), third step: end of µg phase (20 s of µg) and fourth step: end of second hyper-g phase (20 s hyper-g) (Fig. S1). Each fixation step was performed in duplicate. All four consecutive flight days were used to collect sample material. Samples were stored at -80 °C and shipped on dry ice from Bordeaux, France to Erlangen, Germany for further analysis.
RNA isolation
For the isolation of total RNA from cells, 2.6 mL of chloroform (Merck, Darmstadt, Germany) were added to all parabolic flight samples. Samples were shaken vigorously for 15 s and incubated for 3 min at room temperature. Following this, the upper aqueous phase obtained after centrifugation at 14,000 × g and 4 °C for 40 min was transferred into a fresh tube and RNA was precipitated by the addition of 2 mL isopropanol (Sigma-Aldrich, St. Louis, USA). Samples were carefully inverted and incubated at room temperature for 10 min, followed by centrifugation at 12,000 × g and 4 °C for 30 min. Subsequently, the RNA pellet was washed twice with 75% ethanol (VWR, Radnor, USA), dried at 50 °C and resuspended in 20 µL of nuclease-free water (Thermo Fischer Scientific, Waltham, USA). RNA concentration and quality were checked using the NanoDrop™ lite spectrophotometer (Thermo Fischer Scientific, Waltham, USA) and the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA) according to the Agilent Technologies RNA 6000 Nano Assay protocol.
cDNA-synthesis and synthesis of fluorescent complementary RNA (cRNA) for microarray analysis
For hybridization on a microarray, fluorescent cRNA was generated using the Low Input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, USA) according to manufacturer’s instruction. In brief, 100 ng of total RNA was complemented with 2 µL spike-in RNA (One-color RNA Spike-In Kit, Agilent Technologies, Santa Clara, USA), and a T7 primer mix. Primer and template RNA were denaturized for 10 min at 65 °C, followed by 5 min incubation on ice. For reverse transcription, 5x first-strand buffer, 0.1 M DTT, 10 mM dNTP mix, and Affinity Script RNase Block Mix were added to the RNA mix and the reaction was performed at 40 °C for 2 h, followed by heat inactivation of the reverse transcriptase at 70 °C for 15 min. In a subsequent in-vitro transcription step, the cDNA was used as a template in order to produce Cy3-labeled cRNA. The cDNA was mixed with T7 polymerase, a transcription master mix, and Cy3-labelled CTP and incubated for 2 h at 40 °C. Resulting cRNA was column purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and eluted in 30 µL nuclease-free water. Concentration and labeling efficacy of cRNA was determined with a NanoDrop ND-1000 UV-VIS spectrophotometer version 3.2.1 (NanoDrop Technologies, Inc., USA).
Custom-made Agilent-microarrays for E. gracilis
Large-scale microarray analysis was performed with the Agilent microarray platform for one-color gene expression analysis. The microarray was designed with the online design tool eArray for SurePrint G3 Custom GE 8 × 60 K microarrays (Design ID 084219, G4102A, Agilent, Santa Clara, USA) on the basis of the new E. gracilis transcriptome data2. Specific 60-mer oligonucleotide probes were designed in sense orientation for 20,396 different transcripts. Slides were printed by Agilent Technologies in a non-contact inkjet printing process with each slide containing eight identical arrays with a total of 62,976 spots. All transcripts under investigation were plotted at least in triplicates with randomized distribution in order to exclude spatial artifacts during hybridization. In addition, 330 Euglena-specific replicate probes (control of array reproducibility) as well as 1319 Agilent Technologies controls (spike-in controls, dark and bright corners, and different positive controls) were plotted on the chip.
Microarray hybridization and wash
Hybridization reactions were performed using the Gene Expression Hybridization Kit (Agilent Technologies, Santa Clara, USA) according to the manufacturer’s instructions for 8-pack microarrays. In brief, 600 ng cRNA was fragmented with 25× fragmentation buffer, mixed with 10× gene expression blocking agent and incubated for 30 min at 60 °C. Subsequently, 2× Hi-RPM hybridization buffer was added to the samples. After centrifugation (13000 rpm, 1 min), samples were loaded onto the array. For every single array on the slide, 40 µL were pipetted into individual gaskets of an Agilent gasket slide and the microarray was placed on top and sealed in a SureHyb hybridization chamber (G2534, Agilent, Santa Clara, USA). The slide was incubated in a rotator rack (G2530-60029, Agilent, USA) in a hybridization oven (G2545A, Agilent Technologies, Santa Clara, USA) for a minimum of 17 h at 65 °C, at a rotational speed of 10 rpm. After overnight incubation, the hybridization chamber was disassembled and the microarray was immediately subjected to stringency washes using the Gene Expression Wash Buffer kit (Agilent, Santa Clara, USA) as recommended by the manufacturer. Slides were washed with wash buffer 1 at room temperature for 1 min, followed by another 1 min wash in wash buffer 2 at approximately 37 °C. Slides were then dried in an oil-free air stream and immediately used for scanning of the microarrays.
Scanning of the microarrays
Microarray slides were scanned with an Agilent C microarray scanner (G2565CA, Agilent Technologies, Santa Clara, USA). Excitation of cyanin-3-labeled samples was performed with a SHG-YAG laser (emission: 532 nm) in combination with an emission filter (550–610 nm). Fluorescence signals were converted into electrical signals, which were subsequently digitized to 16-bit TIFF files. Settings of the scanner were as follows: Channel Green, resolution 3 µm, scan region Agilent HD 61 × 21.6 mm, TIFF 16-bit, dynamic range 100–10%.
Evaluation of the microarray scans
The images obtained with the microarray scanner were analyzed with Agilent Feature Extraction software (version 11.5.1.1, Agilent Technologies, Santa Clara, USA). The settings of the extraction protocol were as follows: Extraction protocol: GE1_1105_0ct12, Grid 084219_D_F_20160803, Background method: No Background, Background detrend: On, Multiplicative detrend: True.
Agilent microarray data analysis and transcript annotation
For normalization, quality assessment and fold change analysis, the microarray data were further processed with GeneSpring 14.8 GX (Agilent Technologies, Santa Clara, USA) software. All samples were normalized by quantile normalization with a baseline set to the median of all samples. Compromised spots as well as outlier samples, as identified by principal component analysis, were removed from further analysis53. For hierarchical clustering, ANOVA (one-way) statistical analysis was performed and displayed using the Pearson uncentered (absolute) correlation and Wards linkage rule. The calculation of fold changes was done by the moderated t-test and the p-value computation was performed asymptotically as data were assumed to show normal distribution. Cut-off values for the fold change and p-value were set to 1.5-fold and 0.05, respectively. Each condition was compared to the one before, resulting in the investigation of acceleration-dependent effects: 1 g vs 1.8 g-1, 1.8 g-1 vs µg, µg vs 1.8 g-2. At least three independent samples for each acceleration step from different flight days were used for fold change analysis and hierarchical clustering.
For automated transcript annotation, Blast2GO pro (version 4.1.9, BioBam Bioinformatics, Spain) software was used54,55,56,57, which remotely performs BLAST and InterProScan searches, extracts Gene Ontology (GO) terms as well as enzyme codes (EC)58,59,60,61,62. The initial BLAST search was performed based on the protein sequence of the transcript, showing results with an expectation value smaller than 1.0E-3. During mapping and annotation, the BLAST and InterProScan results were linked to their corresponding GO terms from the non-redundant reference protein database (PIR). EC assignment was performed based on GO annotation and visualized via KEGG pathway analysis63,64,65. Furthermore, the gene set enrichment analysis (GSEA) was performed to find regulated functional groups66.
Data Availability
The large-scale data of microarray analysis have been deposited into ArrayExpress. All raw data and detailed experimental protocols can be accessed under the link https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-8064.
References
Adl, S. M. et al. The Revised Classification of Eukaryotes. Journal of Eukaryotic Microbiology 59, 429–514, https://doi.org/10.1111/j.1550-7408.2012.00644.x (2012).
Ebenezer, T. E., Carrington, M., Lebert, M., Kelly, S. & Field, M. C. Euglena gracilis Genome and Transcriptome//Euglena gracilis Genome and Transcriptome: Organelles, Nuclear Genome Assembly Strategies and Initial Features. Organelles, Nuclear Genome Assembly Strategies and Initial Features. Advances in experimental medicine and biology 979, 125–140, https://doi.org/10.1007/978-3-319-54910-1_7 (2017).
Häder, D.-P. Polarotaxis, gravitaxis and vertical phototaxis in the green flagellate, Euglena gracilis. Arch Microbiol 147, 179–183, https://doi.org/10.1007/BF00415281 (1987).
Porterfield, D. M. Orientation of Motile Unicellular Algae to Oxygen: Oxytaxis in Euglena. Biol Bull 193, 229–230, https://doi.org/10.1086/BBLv193n2p229 (1997).
Richter, P., Ntefidou, M., Streb, C., Lebert, M. & Häder, D.-P. Physiological characterization of gravitaxis in Euglena gracilis. J Gravitat Physiol 9, 279–280 (2002).
Pfleging, P., Morawietz, G. & Hardeland, R. Differences in oscillation frequencies between two key enzymes of aromatic amino acid catabolism in Euglena: Circadian rhythmicity of tryptophan 2,3‐dioxygenase vs. predominance of ultradian periods in tyrosine aminotransferase. Biol Rhythm Res 25, 59–66, https://doi.org/10.1080/09291019409360275 (1994).
Tong, J. & Edmunds, L. N. Role of cyclic GMP in the mediation of circadian rhythmicity of the adenylate cyclase-cyclic AMP-phosphodiesterase system in Euglena. Biochemical Pharmacology 45, 2087–2091, https://doi.org/10.1016/0006-2952(93)90020-W (1993).
Doege, M., Ohmann, E. & Tschiersch, H. Chlorophyll fluorescence quenching in the alga Euglena gracilis. Photosyn. Res. 63, 159–170, https://doi.org/10.1023/A:1006356421477 (2000).
Ekelund, N. G. A. Interactions between photosynthesis and ‘light-enhanced dark respiration’ (LEDR) in the flagellate Euglena gracilis after irradiation with ultraviolet radiation. Journal of Photochemistry and Photobiology B: Biology 55, 63–69, https://doi.org/10.1016/S1011-1344(00)00029-4 (2000).
Ntefidou, M., Iseki, M., Watanabe, M., Lebert, M. & Häder, D.-P. Photoactivated adenylyl cyclase controls phototaxis in the flagellate Euglena gracilis. Plant physiology 133, 1517–1521, https://doi.org/10.1104/pp.103.034223 (2003).
Häder, D.-P., Ntefidou, M., Iseki, M. & Watanabe, M. In Light Sensing in Plants, edited by Wada, M., Shimazaki, K. & Iino, M. (Springer, Tokyo, Berlin, Heidelberg, New York), pp. 223–229 (2005).
Daiker, V., Lebert, M., Richter, P. & Häder, D.-P. Molecular characterization of a calmodulin involved in the signal transduction chain of gravitaxis in Euglena gracilis. Planta 231, 1229–1236, https://doi.org/10.1007/s00425-010-1126-9 (2010).
Daiker, V., Häder, D.-P., Richter, P. R. & Lebert, M. The involvement of a protein kinase in phototaxis and gravitaxis of Euglena gracilis. Planta 233, 1055–1062, https://doi.org/10.1007/s00425-011-1364-5 (2011).
Häder, D.-P., Richter, P. R., Schuster, M., Daiker, V. & Lebert, M. Molecular analysis of the graviperception signal transduction in the flagellate Euglena gracilis: Involvement of a transient receptor potential-like channel and a calmodulin. Advances in Space Research 43, 1179–1184, https://doi.org/10.1016/j.asr.2009.01.029 (2009).
Lebert, M., Richter, P. & Häder, D.-P. Signal perception and transduction of gravitaxis in the flagellate Euglena gracilis. Journal of plant physiology 150, 685–690, https://doi.org/10.1016/S0176-1617(97)80284-2 (1997).
Richter, P. R., Schuster, M., Wagner, H., Lebert, M. & Hader, D.-P. Physiological parameters of gravitaxis in the flagellate Euglena gracilis obtained during a parabolic flight campaign. Journal of plant physiology 159, 181–190, https://doi.org/10.1078/0176-1617-00605 (2002).
Richter, P. R., Schuster, M., Meyer, I., Lebert, M. & Häder, D.-P. Indications for acceleration-dependent changes of membrane potential in the flagellate Euglena gracilis. Protoplasma 229, 101–108, https://doi.org/10.1007/s00709-006-0209-8 (2006).
Strauch, S. M., Richter, P., Schuster, M. & Häder, D.-P. The beating pattern of the flagellum of Euglena gracilis under altered gravity during parabolic flights. J Plant Physiol 167, 41–46, https://doi.org/10.1016/j.jplph.2009.07.009 (2010).
Nasir, A. et al. Identification of a flagellar protein implicated in the gravitaxis in the flagellate Euglena gracilis. Scientific reports 8, 7605, https://doi.org/10.1038/s41598-018-26046-8 (2018).
Ebenezer, T. E. et al. Transcriptome, proteome and draft genome of Euglena gracilis. BMC biology 17, 11, https://doi.org/10.1186/s12915-019-0626-8 (2019).
O’Neill, E. C. et al. The transcriptome of Euglena gracilis reveals unexpected metabolic capabilities for carbohydrate and natural product biochemistry. Molecular bioSystems 11, 2808–2820, https://doi.org/10.1039/c5mb00319a (2015).
Yoshida, Y. et al. De novo assembly and comparative transcriptome analysis of Euglena gracilis in response to anaerobic conditions. BMC genomics 17, 182, https://doi.org/10.1186/s12864-016-2540-6 (2016).
Hausmann, N. et al. Cytosolic calcium, hydrogen peroxide and related gene expression and protein modulation in Arabidopsis thaliana cell cultures respond immediately to altered gravitation. Parabolic flight data. Plant Biol 16, 120–128, https://doi.org/10.1111/plb.12051 (2014).
Ma, X. et al. Differential gene expression profile and altered cytokine secretion of thyroid cancer cells in space. FASEB J 28, 813–835, https://doi.org/10.1096/fj.13-243287 (2014).
Nassef, M. Z. et al. Real Microgravity Influences the Cytoskeleton and Focal Adhesions in Human Breast Cancer Cells. IJMS (International journal of molecular sciences 20, 3156, https://doi.org/10.3390/ijms20133156 (2019).
Nasir, A. et al. The influence of microgravity on Euglena gracilis as studied on Shenzhou 8. Plant Biol 16(Suppl 1), 113–119, https://doi.org/10.1111/plb.12067 (2014).
Maier, R.-M. et al. Anaplerotic responses of Arabidopsis thaliana cell cultures to altered gravitational field strengths - NLM Catalog - NCBI. In: Proceedings of the 16th ESA symposium on European Rocket and Ballon Programmes and Related Research, St. Gallen, Switzerland (2003).
Martzivanou, M., Babbick, M., Cogoli-Greuter, M. & Hampp, R. Microgravity-related changes in gene expression after short-term exposure of Arabidopsis thaliana cell cultures. Protoplasma 229, 155–162, https://doi.org/10.1007/s00709-006-0203-1 (2006).
Adrian, A. et al. The oxidative burst reaction in mammalian cells depends on gravity. Cell communication and signaling: CCS 11, 98, https://doi.org/10.1186/1478-811X-11-98 (2013).
Joo, J. H. Role of Auxin-Induced Reactive Oxygen Species in Root Gravitropism. Plant physiology 126, 1055–1060, https://doi.org/10.1104/pp.126.3.1055 (2001).
Mugnai, S. et al. Oxidative stress and NO signalling in the root apex as an early response to changes in gravity conditions. BioMed research international 2014, 834134, https://doi.org/10.1155/2014/834134 (2014).
Fengler, S. et al. Changes in Gene Expression of Arabidopsis Thaliana Cell Cultures Upon Exposure to Real and Simulated Partial-g Forces. Microgravity sci. Technol. 28, 319–329, https://doi.org/10.1007/s12217-015-9452-y (2016).
Thiel, C. S. et al. Dynamic gene expression response to altered gravity in human T cells. Sci Rep 7, 35, https://doi.org/10.1038/s41598-017-05580-x (2017).
Wehland, M. et al. Differential gene expression of human chondrocytes cultured under short-term altered gravity conditions during parabolic flight maneuvers. CCS (Cell communication and signaling) 13, 18, https://doi.org/10.1186/s12964-015-0095-9 (2015).
Thiel, C. S. et al. Rapid coupling between gravitational forces and the transcriptome in human myelomonocytic U937 cells. Scientific reports 8, 13267, https://doi.org/10.1038/s41598-018-31596-y (2018).
Danko, C. G. et al. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Molecular cell 50, 212–222, https://doi.org/10.1016/j.molcel.2013.02.015 (2013).
Darzacq, X. et al. In vivo dynamics of RNA polymerase II transcription. Nature structural & molecular biology 14, 796–806, https://doi.org/10.1038/nsmb1280 (2007).
Maiuri, P. et al. Fast transcription rates of RNA polymerase II in human cells. EMBO reports 12, 1280–1285, https://doi.org/10.1038/embor.2011.196 (2011).
Milo, R. & Phillips, R. Cell biology by the numbers. (Garland Science Taylor & Francis Group, New York, NY, Abingdon, 2016).
Alpert, T., Herzel, L. & Neugebauer, K. M. Perfect timing. Splicing and transcription rates in living cells. Wiley interdisciplinary reviews. RNA 8, https://doi.org/10.1002/wrna.1401 (2017).
Herzel, L., Ottoz, D. S. M., Alpert, T. & Neugebauer, K. M. Splicing and transcription touch base. Co-transcriptional spliceosome assembly and function. Nature reviews. Molecular cell biology 18, 637–650, https://doi.org/10.1038/nrm.2017.63 (2017).
Huranová, M. et al. The differential interaction of snRNPs with pre-mRNA reveals splicing kinetics in living cells. The Journal of cell biology 191, 75–86, https://doi.org/10.1083/jcb.201004030 (2010).
Ramdas, N. M. & Shivashankar, G. V. Cytoskeletal control of nuclear morphology and chromatin organization. Journal of molecular biology 427, 695–706, https://doi.org/10.1016/j.jmb.2014.09.008 (2015).
Tajik, A. et al. Transcription upregulation via force-induced direct stretching of chromatin. Nature materials 15, 1287–1296, https://doi.org/10.1038/nmat4729 (2016).
Versaevel, M., Grevesse, T. & Gabriele, S. Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nature communications 3, 671, https://doi.org/10.1038/ncomms1668 (2012).
Wang, N., Tytell, J. D. & Ingber, D. E. Mechanotransduction at a distance. Mechanically coupling the extracellular matrix with the nucleus. Nature reviews. Molecular cell biology 10, 75–82, https://doi.org/10.1038/nrm2594 (2009).
Ingber, D. How cells (might) sense microgravity. FASEB J 13, S3–S15 (1999).
Ingber, D. E. In Cellular Mechanotransduction, edited by Mofrad, M. R. K. & Kamm, R. D. (Cambridge University Press, Cambridge), pp. 196–219 (2009).
Ingber, D. E., Wang, N. & Stamenovic, D. Tensegrity, cellular biophysics, and the mechanics of living systems. Reports on progress in physics. Physical Society (Great Britain) 77, 46603, https://doi.org/10.1088/0034-4885/77/4/046603 (2014).
Lonergan, T. A. Regulation of Cell Shape in Euglena gracilis. I. Involvement of the Biological Clock, Respiration, Photosynthesis, and Cytoskeleton. Plant physiology 71, 719–730, https://doi.org/10.1104/pp.71.4.719 (1983).
Starr, R. C. The culture collection of algae at Indiana University. Am J Bot 51, 1013–1044 (1964).
Pletser, V. et al. The First European Parabolic Flight Campaign with the Airbus A310 ZERO-G. Microgravity Sci. Technol 28, 587–601, https://doi.org/10.1007/s12217-016-9515-8 (2016).
Raychaudhuri, S., Stuart, J. M. & Altman, R. B. In Pacific Symposium on Biocomputing 2000, edited by Altman, R. B. et al. (World Scientific Pub. Co, Singapore, River Edge, N.J), pp. 455–466 (1999).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676, https://doi.org/10.1093/bioinformatics/bti610 (2005).
Conesa, A. & Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. International journal of plant genomics 2008, 619832, https://doi.org/10.1155/2008/619832 (2008).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucl Acids Res 36, 3420–3435, https://doi.org/10.1093/nar/gkn176 (2008).
Götz, S. et al. B2G-FAR, a species-centered GO annotation repository. Bioinformatics 27, 919–924, https://doi.org/10.1093/bioinformatics/btr059 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29, https://doi.org/10.1038/75556 (2000).
Gene Ontology Consortium: going forward. Nucl Acids Res 43, D1049–56; https://doi.org/10.1093/nar/gku1179 (2015).
Gish, W. & States, D. J. Identification of protein coding regions by database similarity search. Nat Genet 3, 266–272, https://doi.org/10.1038/ng0393-266 (1993).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl Acids Res 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucl Acids Res 44, D457–62, https://doi.org/10.1093/nar/gkv1070 (2016).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucl Acids Res 45, D353–D361, https://doi.org/10.1093/nar/gkw1092 (2017).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550, https://doi.org/10.1073/pnas.0506580102 (2005).
Acknowledgements
The work was funded by the DLR on behalf of the BMWi (grant 50WB1128). S.M.S.was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES).
Author information
Authors and Affiliations
Contributions
J.K.: conduction and analysis of experiments, draft preparation, P.R.: conduction and analysis of experiments, draft preparation, S.M.S.: hardware construction, data evaluation, draft preparation, B.P.: data evaluation, M.K.: data evaluation, draft preparation, V.D.: molecular analysis of data, J.S.: data evaluation, draft preparation, S.S.: microarray analysis, S.R.: microarray analysis, M.L.: project administration, funding acquisition, project management.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krüger, J., Richter, P., Stoltze, J. et al. Changes of Gene Expression in Euglena gracilis Obtained During the 29th DLR Parabolic Flight Campaign. Sci Rep 9, 14260 (2019). https://doi.org/10.1038/s41598-019-50611-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50611-4
This article is cited by
-
Development of a New Microgravity Experiment Facility with Electromagnetic Launch
Microgravity Science and Technology (2021)
-
Guest Edited Collection: Gravitational biology and space medicine
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
