
Abstract
Although largemouth bass Micropterus salmoides has shown its extremely economic, ecological, and aquacultural significances throughout the North American and Asian continents, systematic evaluation of genetic variation and structure of wild and cultured populations of the species is yet to be documented. In this study, we investigated the genetic structure of M. salmoides from 20 wild populations and five cultured stocks across the United States and China using eight microsatellite loci, which are standard genetic markers for population genetic analysis. Our major findings are as follows: (1) the result of Fst showed largemouth bass had high genetic differentiation, and the gene flow indicated the genetic exchange among wild populations is difficult; (2) AMOVA showed that 14.05% of the variation was among populations, and 85.95% of the variation was within populations; (3) The majority of largemouth bass populations had a significant heterozygosity excess, which is likely to indicate a previous population bottleneck; (4) Allelic richness was lower among cultured populations than among wild populations; (5) Effective population size in hatcheries could promote high levels of genetic variation among individuals and minimize loss of genetic diversity; China’s largemouth bass originated from northern largemouth bass of USA. The information provides valuable basis for development of appropriate conservation policies for fisheries and aquaculture genetic breeding programs in largemouth bass.
Similar content being viewed by others

Introduction
Largemouth bass Micropterus salmoides (Lacépède) is native to South-eastern United States1,2,3. As one of the top predatory species, it has shown its extremely economic and ecological significances throughout the North American Continent4,5,6,7,8. This freshwater bass had gained the loyalty of the estimated 50 million U.S. anglers at the beginning of this century9; thus, fingerlings and juveniles of this species are widely cultivated in public and private hatcheries to supply recreational fishing waters10. It has also been regarded as one of the dominated non-salmonid freshwater food fishes11.
It is universally acknowledged that the divergence of lineages within largemouth bass occurred due to geological changes during some geologic time12,13,14; and two subspecies of this fish species, i.e. the Florida largemouth bass M. s. floridanus and the northern largemouth bass M. s. salmoides, occupied their native habitats from peninsular “Florida to throughout northeastern Mexico”, southeastern Canada, and the U.S. corridor in between15. The further divergence of the latter subspecies at northern and southern latitudes has been identified through genetic analysis16,17,18,19. Growth differences and environmental adaptability of these populations has been focused on by many researchers20; and the perceived superior growth characteristics of M. s. floridanus resulted in its fast and wide introduction to far north of its native range almost all over the United States21,22.
Largemouth bass with uncertain lineages have also been introduced to many countries in Europe23,24,25,26, Asia27,28,29, and Africa1,30 since the 19th century and gained access to a large area of habitats outside its native range; and it is playing greater role in freshwater fisheries and ecological systems of these regions.
A trio of factors, including arbitrary introduction, intraspecific hybrid, and environmental stress, makes it difficult to detect the impacts of human activity, especially aquaculture and recreational fishing, on the genetic diversity of largemouth bass. Philipp revealed the genetic distinctions among some largemouth bass groups were continually being diluted by introgression16. Studies on four cultured populations of largemouth bass in Guangdong, China indicated that the stocks are northern largemouth bass depleted in genetic variation28. Actually, short-term allelic changes like this had been a concern for some populations within United States decades ago31,32,33. Recently, attention has been paid to allele persistence of largemouth bass. Johnson and Fulton finished comparison of the allele frequencies of three discriminate allozyme loci between Florida and northern largemouth bass and inferred that genes persisted through many generations20. What puzzles ecologists and culturists who are working with largemouth bass is that they are not sure about the taxonomy and genetic background when they are confronted with either a wild or hatchery produced largemouth bass population.
Microsatellite loci are standard genetic markers for population genetic analysis, and have many advantages, including codominant alleles, extensive genome coverage (present in both coding and noncoding regions), high allelic diversity at the loci, and the ability to determine genotypes from small samples of tissue using polymerase chain reaction (PCR)34. Although single nucleotide polymorphisms (SNPs) are becoming new tools in genetics and genomics studies, comparative analyses of SNP and microsatellite marker data for population genetic analysis in Diabrotica virgifera virgifera resulted in similar conclusions with respect to population structure35. Therefore, microsatellite should still be an invaluable tool for various population genetics studies36.
Lutz-Carrillo37 examined microsatellite DNA variation at 11 loci in populations of Florida largemouth bass M. s. floridanus and northern largemouth bass M. s. salmoides from northern and southern latitudes in North America and found despite the systematic introduction of Florida largemouth bass into Texas, genetic integrity of a number of populations had not been or were only minimally affected. But they suggested that a full assessment of the variation throughout the range of each subspecies should be conducted to identify minimally impacted environments and quantify the levels of gene flow from impacted to intact water bodies to continue to restart the necessary work performed by Philipp16 over 20 years ago. Lutz-Carrillo subsequently developed 52 novel microsatellite markers from M. s. floridanus, for use in population genetic studies and expected them to be helpful for the resolution of hatchery-released individuals in the wild, introgression among Micropterus species, and gene flow following stocking and restoration efforts38.
In this study, we investigated the population genetic structure of hatchery stocks and wild populations in the largemouth bass based on eight polymorphic microsatellite loci. Samples encompassed both populations from wild habitats and hatcheries or domestic fish ponds. Our objectives were as follows: (1) to determine genetic diversity of wild largemouth bass populations in North America; (2) to evaluate genetic variation in hatchery largemouth bass and identify differential strain contribution affecting the genetic variability (3) to provide valuable information for the fishery management and conservation implication of largemouth bass.
Results
Genetic diversity
Across all 25 populations sampled, the w96 locus had the highest number of alleles (18.8) while the mdo6 locus had the least (6.6). The total number of alleles at each locus ranged from 3 to 53. The average number of alleles across all loci was the highest (20.8) in the RBRMS and lowest (7.3) in the SJFL. No significant genetic associations among loci were observed. The value of observed heterozygosities per population was detected from 0.0213 to 1. The expected heterozygosities were from 0.1158 to 0.9905 (Table 1).
The test for fit to Hardy-Weinberg proportions revealed that most of loci were found to be deviated from Hardy-Weinberg because of heterozygote deficiencies or excess (after bonferroni correction).
Gene flow
The result of Fst was 0.19 (among 25 populations), showing that largemouth bass had high genetic differentiation, and the gene flow indicated the genetic exchange among wild populations is difficult (Table 2).
Genetic variation
Genetic variation statistics obtained by AMOVA were significant at all hierarchical levels. When all populations were grouped together and all loci were considered, 14.05% of the variation was among populations, and 85.95% of the variation was within populations (Table 3).
Population structure
The UPGMA tree built from the matrix of pairwise allele-sharing distance among 25 populations (Fig. 1) and the assignment tests revealed the multilocus microsatellite genotypes to discriminate populations of largemouth bass. The phylogeny based on eight microsatellites revealed a clear distinction between northern and southern populations, although samples from 25 populations were different.

UPGMA tree of 25 populations based on the allele-sharing distance.
The software STRUCTURE was used to examine how the sampled populations clustered based on the genetic data. The preliminary STRUCTURE run indicated that the most likely number of clusters was two; thus, the second and more robust simulation was run with k = 26. However, in the latter analysis, the results suggested that the most likely number of clusters was four (Fig. 2).

Structure analysis of populations (the inferred clusters, k = 4). The sampling location abbreviations (see Table 4) are indicated along the X-axis. Each vertical line represents one individual, and Y-coordinate denotes each individual’s percentage assignment to each of these seven genetic clusters.
Population bottlenecks
The majority of largemouth bass populations had a significant heterozygosity excess, which is likely to indicate a previous population bottleneck. However, under the SMM and TPM models, there is no bottleneck signature; under the IAM model, PLWI (P = 0.01) and LKTX (P = 0.008) suggested that there had been a recent population bottleneck.
Discussions
This study is the first to use microsatellite DNA markers to examine the genetic variation within and among wild and hatchery populations of largemouth bass across America and China. Other freshwater fish species using microsatellites revealed thenumber of alleles per locus (5.4)39 was lower than we observed (18.1), which may have been due to the geographic we sampled, the number of populations we sampled, or the loci we selected. Some loci failed to be amplified in cultured populations, which might be due to high sensitivity of these loci under environmental pressure and selective breeding. The highest values of the mean number of alleles and private alleles were detected in the RBRMS population. In the past, hatchery procedures often had overlooked genetic differences between populations, as well as the consequences of reducing heterogeneity through inbreeding16. In the present study, the value of observed heterozygosities per population was detected from 0.0213 to 1. The expected heterozygosities were from 0.1158 to 0.9905. Thus, the populations we collected can provide a wide range of selection to avoid reducing heterogeneity resulting from inbreeding. The inspection of genetic information provides fishery managers with a means for understanding and predicting stocking population variation for regulations and other management programs in largemouth bass fisheries.
Genetic diversity is increasingly declining as a result of various factors, including direct human impact and structural alteration of ecosystems resultingfrom changes in human life styles, bottleneck experienced, inbreeding, etc40. Genetic diversity values are a better indicator of the genetic polymorphism within a population, because estimates of the mean number of alleles are influenced by sample size. In the present study, the majority of largemouth bass populations had a significant heterozygosity excess, which is likely to indicate a previous population bottleneck. However, comparing Southern and Northern populations of largemouth bass, the South Carolina populations showed higher values of gene diversity and allele richness. The populations have maintained their genetic integrity and been only minimally affected. Nevertheless, the high genetic diversity of invasive species may be caused by hybridization and variation. In this study, the results showed that the largemouth bass had high genetic differentiation. In addition, according to AMOVA analysis, overall genetic differentiation among largemouth bass from the 25 populations was significant (P < 0.05), suggesting significant genetic differentiation among localities or populations. However, overall, the genetic variations within populations are higher than those among populations. In the natural environment, the long-term success of stocked largemouth bass might be the greatest for cohorts with the highest levels of genetic variability16. Microsatellite allelic frequencies and Fst analyses showed that all 25 populations were genetically unique. These genetic differences have resulted from long-term exposure to natural selective pressures and provide thespecies with enough variation to deal with the Barge range of different environmental conditions in which it exists41.
The largemouth bass has two subspecies, the Florida largemouth bass M. s. floridanus and the northern largemouth bass M. s. salmoides. They occupy their native habitats from peninsular Florida to throughout northeastern Mexico, southeastern Canada, and the U.S. corridor in between15. The genetic difference is particularly pronounced for the two recognized subspecies, but pertains to populations within each subspecies as well. The UPGMA tree built from the matrix of pairwise allele-sharing distance among 25 populations across America and the assignment tests revealed the multilocus microsatellite genotypes to discriminate populations of largemouth bass. The phylogeny based on eight microsatellites revealed a clear distinction between northern and southern populations. The China population and Minneapolis populations were clustered in a group, indicating that China’s largemouth bass originated fromnorthern largemouth bass, since China introduced largemouth bassonly one time from California, USA before our sampling time.
In conclusion, there is relatively high genetic diversity of wild largemouth bass populations in North America, and significant genetic differentiation among localities or populations in the largemouth bass, but obviously lower genetic diversity in hatchery stocks. The study suggested that China’s largemouth bass originated from northern largemouth bass of USA. This study provided valuable information for the fishery management and conservation implication of largemouth bass, and indicated that the differentiation in largemouth bass was caused by the founder effect. Besides the founder effect, geographicaldistance might also be responsible for the significant genetic differentiation among wild populations. These analyses clearly reveal substantial genetic differences among populations in the United States. This study demonstrates the need for incorporating genetic information and principles into current and future fisheries management programs. However, the effectiveness of the stocking populations should be evaluated and the long-term effects on the genetic composition of specified largemouth bass populations should be monitored.
Methods
Populations studied
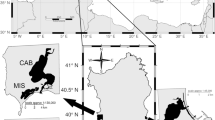
A total of 1,045 individuals were collected by trapping and/or electro-fishing from 25 different localities and coordinated by geographic sampling coordinates throughout North America and China (Fig. 1 and Supplementary Table S1). Twenty wild populations and five hatchery populations were analyzed. Details of the samples are given in Table 4, and the approximate geographical location of the sampled populations is indicated in Fig. 3.

Sampling sites for 20 wild and 5 hatchery M. salmoides populations. Abbreviations are as listed in Table 4.
Fin clips were the sources of nuclear DNA. They were collected from the freshly caught fish and immediately preserved in 95% ethanol.
Extraction of DNA and microsatellite amplification
Total DNA was extracted from an optimum volume of fin tissue following a modified version of the pure gene protocol for extraction from fish tissue37. Amplification of microsatellite loci were performed using eight primers selected from those isolated and characterized by Lutz-Carrillo (Table 5)16,37,38. Each locus was amplified with a three-primer system in which only the M13 and CAG primers were fluorescently labeled with FAM, HEX, or NED.
Polymerase chain reaction of 6 μL contains 3 μL of JumpStart RedMix (Sigma), 1.5 pmol of both non-tailed and labelled primers, and 0.1 pmol of the tailed primer, 25 ng DNA, in the presence of 100 μm spermidine. Amplification was be conducted in PTC-100 thermal cyclers (MJ Research) using an initial denaturation at 94 °C for 2 min, followed by 35 cycles of 30 s denaturation at 94 °C, 30 s annealing at a locus-specific temperature (Table 5), 30 s extension at 72 °C, and a final 5-min extension at 72 °C. Amplification products were separated using an ABI 3130 Prism DNA genetic analyzer.
Data analyses
Analyses of genetic variation and genetic differentiation
Deviations from Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were being tested for all locus-population and locus-locus combinations using ARLEQUIN version 2.042. Significance levels were be modulated for multiple comparisons using the sequential Bonferroni method43. Genetic variation was evaluated according to observed heterozygosity and the number of alleles per locus within populations and regions. Private alleles among populations were computed using GenAlEx 644.
Analyses of molecular variance
Analyses of molecular variance (AMOVA) were conducted with Arlequin. One AMOVA was performed under the null hypothesis of no genetic structure. AMOVAS will examine population structure after combining populations in various ways to test for geographical structure.
Assignment tests
Genetic structure among populations was analyzed based on a chord distance according to Cavalli-Sforza and Edwards45. A UPGMAtree was constructed using the programs NEIGHBOR and CONSENSE in PHYLIP version 3.6 in terms of Felsenstein46. Genetic structure among individuals was assessed using the model-based Bayesian clustering method according to Pritchard47 and Lutz-Carrillo37. Assignment tests were performed with geneclass version 2.048. The program STRUCTURE was used to identify genetic clusters without using any prior information of the sampling location of the individuals47. Two separate analyses were carried out in STRCTURE, both times with allele frequencies as correlated and the admixture model. The first analysis was used to infer the most likely range for K. Here, we used a burn-in of 10000 and a MCMC length of 5000 iterations and the simulated number of populations from K = 1–25. The upper limit of 25 was chosen as this corresponds to the number of sampled populations. Twelve independent simulations were performed of each K to check for consistency across runs. The preliminary results were assessed using the Evanno method where the most likely K was determined by the distribution of △K.
Bottlenecks analyses
Allelic richness was calculated with FSTAT version 2.9.3.249. All these tests were adjusted for multiple simultaneous comparisons using a sequential Bonferroni correction. Means are reported as ±SE. We used bottleneck version 1.2.0250 to test for evidence consistent with recent population bottlenecks or expansions by recognizing significant heterozygosity excess or deficiency for each population using a Wilcoxon sign–rank test. We conducted the tests using 1,000 iterations using the SMM of microsatellite evolution, as well as a two-phased mutation model (TPM). Allele frequency data was tested for evidence of a “heterozygosity excess” (HE) using the program bottleneck48,50. Three statistical tests (sign test, standardized differences test, and Wilcoxen signed-ranks test) were conducted in order to determine whether there is significant HE, which may indicate that a recent bottleneck has occurred.
Ethics
This study and all experimental procedures involving animals were performed according to the protocol approved by the Ohio State University Institutional Animal Care and Use Committee. Permissions for field studies were granted by: Ohio Department of Natural Resources, South Carolina Department of Natural Resources, Texas Parks & Wildlife Department, Minnesota Department of Natural Resources, Mississippi Wildlife Fisheries & Parks, Florida Blackwater Fisheries Research and Development Center, Wisconsin Genoa Federal Fish Hatchery (Supplementary Table S1).
References
Hickley, P., North, R., Muchiri, S. M. & Harper, D. M. The diet of largemouth bass, Micropterus salmoides, in Lake Navaisha, Kenya. J. Fish Biol. 44, 607–619, https://doi.org/10.1111/j.1095-8649.1994.tb01237.x (1994).
Azuma, M. & Motomura, Y. Feeding habits of largemouth bass in a non-native environment: the case of a small lake with bluegill in Japan. Environ. Biol. Fish 52, 379–389, https://doi.org/10.1023/A:1007476104352 (1998).
Martyniuk, C. J., Sanchez, B. C., Szabo, N. J., Denslow, N. D. & Sepúlveda, M. S. Aquatic contaminants alter genes involved in neurotransmitter synthesis and gonadotropin release in largemouth bass. Aquat. Toxicol. 95, 1–9, https://doi.org/10.1016/j.aquatox.2009.06.009 (2009).
Savino, J. F. & Stein, R. A. Behavior of fish predators and their prey: habitat choice between open water and dense vegetation. Environ. Biol. Fishes 24, 287–293, https://doi.org/10.1007/BF00001402 (1989).
Williamson, J. H. & Carmichael, G. J. An aquacultural evaluation of Florida, Northern, and hybrid largemouth bass, Micropterus salmoides. Aquaculture 85, 247–257, https://doi.org/10.1016/0044-8486(90)90024-H (1990).
Tidwell, J. H., Webster, C. D. & Coyle, S. D. Effects of dietary protein level on second year growth and water quality for largemouth bass (Micropterus salmoides) raised in ponds. Aquaculture 145, 213–223, https://doi.org/10.1016/S0044-8486(96)01356-7 (1996).
Tidwell, J. H., Webster, C. D., Coyle, S. D. & Schulmeister, G. Effects of stocking density on growth and water quality for largemouth bass Micropterus salmoides grow out in ponds. J. World Aquac. Soc. 29, 79–83, https://doi.org/10.1111/j.1749-7345.1998.tb00302.x (1998).
Lowe, M. R., DeVries, D. R., Wright, R. A., Ludsin, S. A. & Fryer, B. J. Coastal largemouth bass (Micropterus salmoides) movement in response to changing salinity. Can. J. Fish Aquat. Sci. 66, 2174–2188, https://doi.org/10.1139/F09-152 (2009).
Tomalin, T. BASS could turn Florida into the big fish. St. Petersburg Times (Florida), South Pinellas Edition, April 26, Tuesday, 1C. (2005).
Arslan, T., Phelps, R. P. & Osborne, J. A. Effects of oestradiol-17b or 17a-methyltestosterone administration on gonadal differentiation of largemouth bass Micropterus salmoides (Lacépède). Aquac. Res. 40, 1813–1822 (2009).
Williamson, J. H., Carmichael, G. J., Graves, K. G., Simco, B. A. & Tomasso, J. R. Centrarchids in Culture of Nonsalmonid Freshwater Fishes (ed. Stickney, R. R.) 145–197 (Chemical Rubber Company, 1986).
Bailey, R. M. & Hubbs, C. L. The black basses (Micropterus) of Florida, with description of a new species. Univ. Mich. Mus. Zool. Occas. Pap. 516, 1–40, http://hdl.handle.net/2027.42/56954 (1949).
Nedbal, M. A. & Philipp, D. P. Differentiation of mitochondrial DNA in largemouth bass. Trans. Am. Fish Soc. 123, 460–468, doi: 10.1577/1548-8659(1994)123<0460:DOMDIL>2.3.CO;2 (1994).
Near, T. J., Kassler, T. W., Koppelman, J. P., Dillman, K. B. & Philipp, D. P. Speciation in North American black basses Micropterus (Actinopterygii: Centrarchidae). Evolution 57, 1610–1621, https://doi.org/10.1111/j.0014-3820.2003.tb00368.x (2003).
MacCrimmon, H. R., Robbins, W. H., Stroud, R. H. & Clepper. H. Black Bass Biology and Management. Washington, D.C., Sport Fishing Institute (1975).
Philipp, D. P., Childers, W. F. & Whitt, G. S. A biochemical genetic evaluation of the northern and Florida subspecies of largemouth bass. Trans. Am. Fish Soc. 112, 1–20, doi:10.1577/1548-8659(1983)112<1:ABGEOT>2.0.CO;2 (1983)
Addison, J. H. & Spencer, S. L. Preliminary evaluation of three strains of largemouth bass, Micropterus salmoides (Lacépède), stocked in ponds in south. Proc. SE Assoc. Game Fish Comm. 25, 366–374 (1971).
Fields, R., Lowe, S. S., Kaminski, C., Whitt, G. S. & Philipp, D. P. Critical and chronic thermal maxima of northern and Florida largemouth bass and their reciprocal F1 and F2 hybrids. Trans. Am. Fish Soc. 116, 856–863, doi:10.1577/1548-8659(1987)116<856:CACTMO>2.0.CO;2 (1987).
Philipp, D. P. & Whitt, G. S. Survival and growth of northern, Florida, and reciprocal F1 hybrid largemouth bass in central Illinois. Trans. Am. Fish Soc. 120, 156–178, doi:10.1577/1548-8659(1991)120<0058:SAGONF>2.3.CO;2 (1991).
Johnson, R. L. & Fulton, T. Incidence of Florida largemouth bass alleles in two northern Arkansas populations of largemouth bass, Micropterus salmoides Lacépède. Am. Midl. Nat. 152, 425–429, doi:10.1674/0003-0031(2004)152[0425:IOFLBA]2.0.CO;2 (2004).
Gilliland, E. R. Experimental stocking of Florida largemouth bass into small Oklahoma reservoirs. Proc. Ann. Conf. SE Assoc. Fish Wildl. Agen. 46, 487–494 (1992).
Hobbs, B., Horton, C., Claybrook, L., Shinn, B. & Swann, D. Arkansas Game and Fish Commission largemouth management plan. Arkansas Game and Fish Commission. Little Rock, Ark: 34 (2002).
Lever, C. Naturalized Fishes of the World. London: Academic Press. 436 p. (1996).
Lorenzoni, M. et al. Growth and reproduction of largemouth bass (Micropterus salmonides Lacépède, 1802) in Lake Trasimeno (Umbria, Italy). Fish Res. 56, 89–95 (2002).
Petit, G., Beauchaud, M., Attia, J. & Buisson, B. Food intake and growth of largemouth bass (Micropterus salmoides) held under alternated light/dark cycle (12L:12D) or exposed to continuous light. Aquaculture 228, 397–401, https://doi.org/10.1016/S0044-8486(03)00315-6 (2003).
Ribeiro, F., Elvira, B., Collares-Pereira, M. J. & Moyle, P. B. Profiling alien fishes in Iberian watersheds across several invasion stages. Biol. Invas. 10, 89–102, https://doi.org/10.1007/s10530-007-9112-2 (2008).
Nakai, K. Recent faunal changes in Lake Biwa, with particular reference to the bass fishing boom in Japan: Ancient lakes, their cultural and biological diversity (eds Kawanabe, H., Coulter, G. W. & Roosevelt, A. C.) 227–244 (Kenobi Productions, 1999).
Bai, J., Lutz-Carrillo, D. J., Quan, Y. & Liang, S. Taxonomic status and genetic diversity of cultured largemouth bass Micropterus salmoides in China. Aquaculture 278, 27–30, https://doi.org/10.1016/j.aquaculture.2008.03.016 (2008).
Lee, W. O., Park, J. Y., Yoon, S. W. & Kim, C. H. A hermaphroditic specimen of the introduced largemouth bass, Micropterus salmoides (Perciformes; Centrarchidae), from Cheongpyeong Lake, South Korea. J. Appl. Ichthyol. 26, 145–146, https://doi.org/10.1111/j.1439-0426.2009.01341.x (2010).
Gratwicke, B. & Marshall, B. E. The relationship between the exotic predators Micropterus salmoides and Serranochromis robustus and native stream fishes in Zimbabwe. J. Fish Biol. 58, 68–75, https://doi.org/10.1111/j.1095-8649.2001.tb00499.x (2001).
Maceina, M. J., Murphy, B. R. & Isely, J. J. Factors regulating Florida largemouth bass stocking success and hybridization with northern largemouth bass in Aquilla Lake, Texas. Trans. Am. Fish Soc. 117, 221-231, doi:10.1577/1548-8659(1988)117<0221:FRFLBS>2.3.CO;2 (1988).
Gilliland, E. R. & Whittaker, J. Introgression of Florida largemouth bass introduced into northern largemouth bass populations in Oklahoma reservoirs. Proc. Ann. Conf. SE Assoc. Fish Wildl. Agen. 39, 56–64 (1989).
Brown, M. L. & Murphy, B. R. Temporal genetic structure of an intergrade largemouth bass (Micropterus salmoides) population. Ecol. Fresh Fish 3, 18–24, https://doi.org/10.1111/j.1600-0633.1994.tb00103.x (1994).
Zane, L., Bargelloni, L. & Patarnello, T. Strategies for microsatellites isolation: a review. Mol. Ecol. 11, 1–16, https://doi.org/10.1046/j.0962-1083.2001.01418.x (2002).
Coates, B. S. et al. Comparative Performance of Single Nucleotide Polymorphism and Microsatellite Markers for Population Genetic Analysis. J. Hered. 100, 556–564, https://doi.org/10.1093/jhered/esp028 (2009).
Li, Q. et al. Characterization of genic microsatellite markers derived from expressed sequence tags in Pacific abalone (Haliotis discus hannai). Chinese Journal of Oceanology and Limnology 28, 46–54, https://doi.org/10.1007/s00343-010-9233-9 (2010).
Lutz-Carrillo, D. J., Nice, C. C., Bonner, T. H., Forstner, M. R. J. & Fries, L. T. Admixture analysis of Florida largemouth bass and northern largemouth bass using microsatellite loci. Trans. Am. Fish Soc. 135, 779–791, https://doi.org/10.1577/T04-221.1 (2006).
Lutz-Carrillo, D. J., Hagen, C., Dueck, L. A. & Glenn, T. C. Isolation and characterization of microsatellite loci for Florida largemouth bass, Micropterus salmoides floridanus, and other micropterids. Mol. Ecol. Resour. 8, 178–184, https://doi.org/10.1111/j.1471-8286.2007.01917.x (2010).
DeWoody, J. A. & Avise, J. C. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J. Fish Biol. 56, 461–473, https://doi.org/10.1111/j.1095-8649.2000.tb00748.x (2000).
Yamazaki, Y., Sasaki, M., Nakamura, T., Nakano, S. & Nishio, M. Decreasing genetic diversity in wild and captive populations of endangered Itasenpara bittering (Acheilognathus longipinnis) in the Himi region, central Japan, and recommendations for conservation. Conserv. Genet. 15, 921–932, https://doi.org/10.1007/s10592-014-0589-9 (2014).
Philipp, D. P., Childers, W. F. & Whitt, G. S. Management Implications for Different Genetic Stocks of Largemouth Bass (Micropterus salmoides) in the United States. Can. J. Fish Aquat. Sci. 38, 1715–1723, https://doi.org/10.1139/f81-220 (1981).
Schneider, S., Roessli, D., Excoffier, L. ARLEQUIN: a software for population genetics data analysis, version 2.000. Department of Anthropology, University of Geneva, Geneva, Switzerland (2000).
Rice, W. R. Analyzing tables of statistical tests. Evolution 43, 223–225 (1989).
Peakall, R. & Smouse, P. E. GenAlEx 6: genetic analysis in excel-Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295, https://doi.org/10.1111/j.1471-8286.2005.01155.x (2005).
Cavalli-Sforza, L. L. & Edwards, A. W. F. Phylogenetic analysis: models and estimation procedures. Evolution 32, 550–570, https://doi.org/10.1111/j.1558-5646.1967.tb03411.x (1967).
Felsenstein, J. PHYLIP: Phylogeny inference package (version 3.2). Cladistics 5, 164–166, https://doi.org/10.1111/j.1096-0031.1989.tb00562.x (1989).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Piry, S. et al. Geneclass2: a software for genetic assignment and first-generation migrant detection. J. Hered 95, 536–539, https://doi.org/10.1093/jhered/esh074 (2004).
Goudet, J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2), http://www2.unil.ch/popgen/softwares/fstat.htm.
Cornuet, J. M. & Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014, https://doi.org/10.0000/PMID8978083 (1997).
Acknowledgements
We thank Ohio Department of Natural Resources (Mr. Scott Hale and Dr. Richard Zweifel), South Carolina Department of Natural Resources (Dr. James Bulak), Texas Parks & Wildlife Department (Dr. Dijar Lutz), Minnesota Department of Natural Resources, Mississippi Wildlife Fisheries & Parks, Florida Blackwater Fisheries Research and Development Center (Mr. David Yeager), Wisconsin Genoa Federal Fish Hatchery (Mr. Doug Aloisi), Pear River Fishery Research Institute (Junjie Bai and Shenjie Li) for their coordination of collection of finclip samples. We thank Dean Rapp and Paul O’Bryant for their assistance throughout the experiment, and Joy Bauman for her comments on the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Han-Ping Wang and Dan Wang. Performed the experiments: Dan Wang, Hong Yao, Yong-Jiang Xu and Xu-Fa Ma. Analyzed the data: Dan Wang and Yan-He Li. Contributed reagents/materials/analysis tools: Han-Ping Wang and Hong Yao. Wrote the paper: Dan Wang, Han-Ping Wang and Yan-he Li.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, D., Yao, H., Li, YH. et al. Global diversity and genetic landscape of natural populations and hatchery stocks of largemouth bass micropterus salmoides across American and Asian regions. Sci Rep 9, 16697 (2019). https://doi.org/10.1038/s41598-019-53026-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53026-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
